A Disjunctive Marginal Edge of Evergreen Broad-Leaved Oak (Quercus gilva) in East Asia: The High Genetic Distinctiveness and Unusual Diversity of Jeju Island Populations and Insight into a Massive, Independent Postglacial Colonization

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material Sampling and DNA Extraction
2.2. Loci Isolation for Microsatellite Markers’ Development and Genotyping
2.3. Statistical Data Analysis
2.4. Ecological Niche Modeling
3. Results
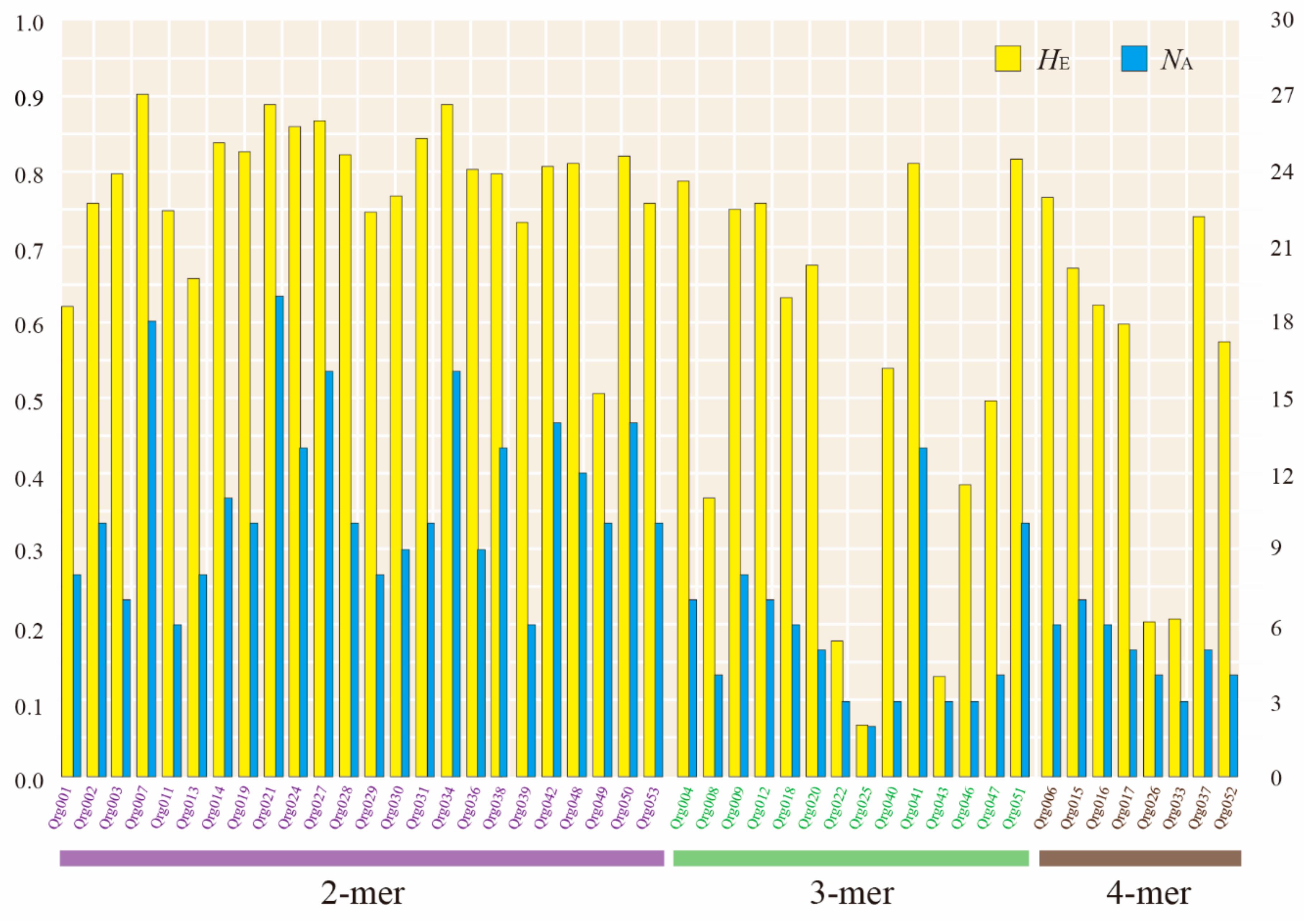
3.1. Development of Polymorphic Microsatellite Markers
3.2. Genetic Diversity
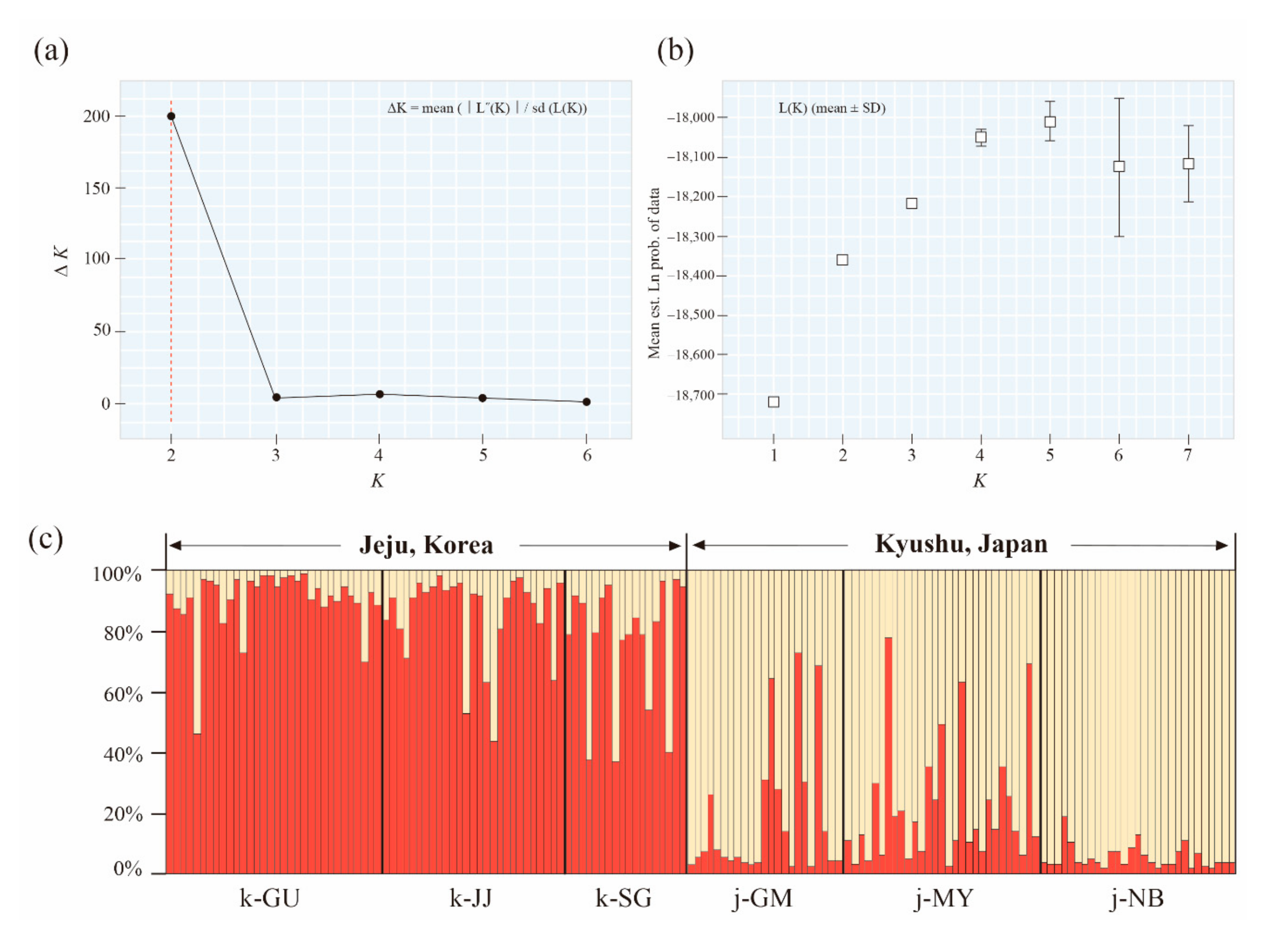
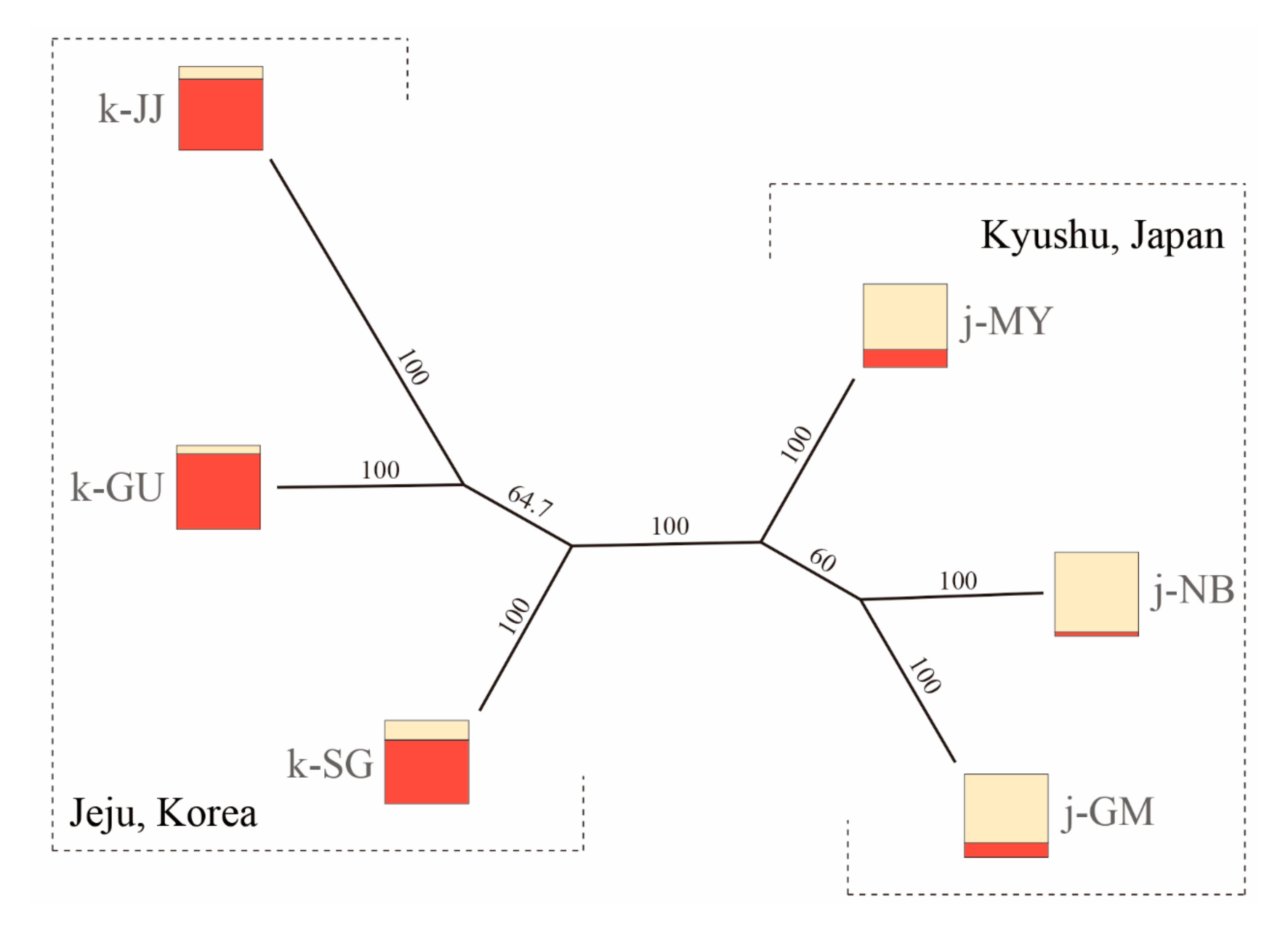
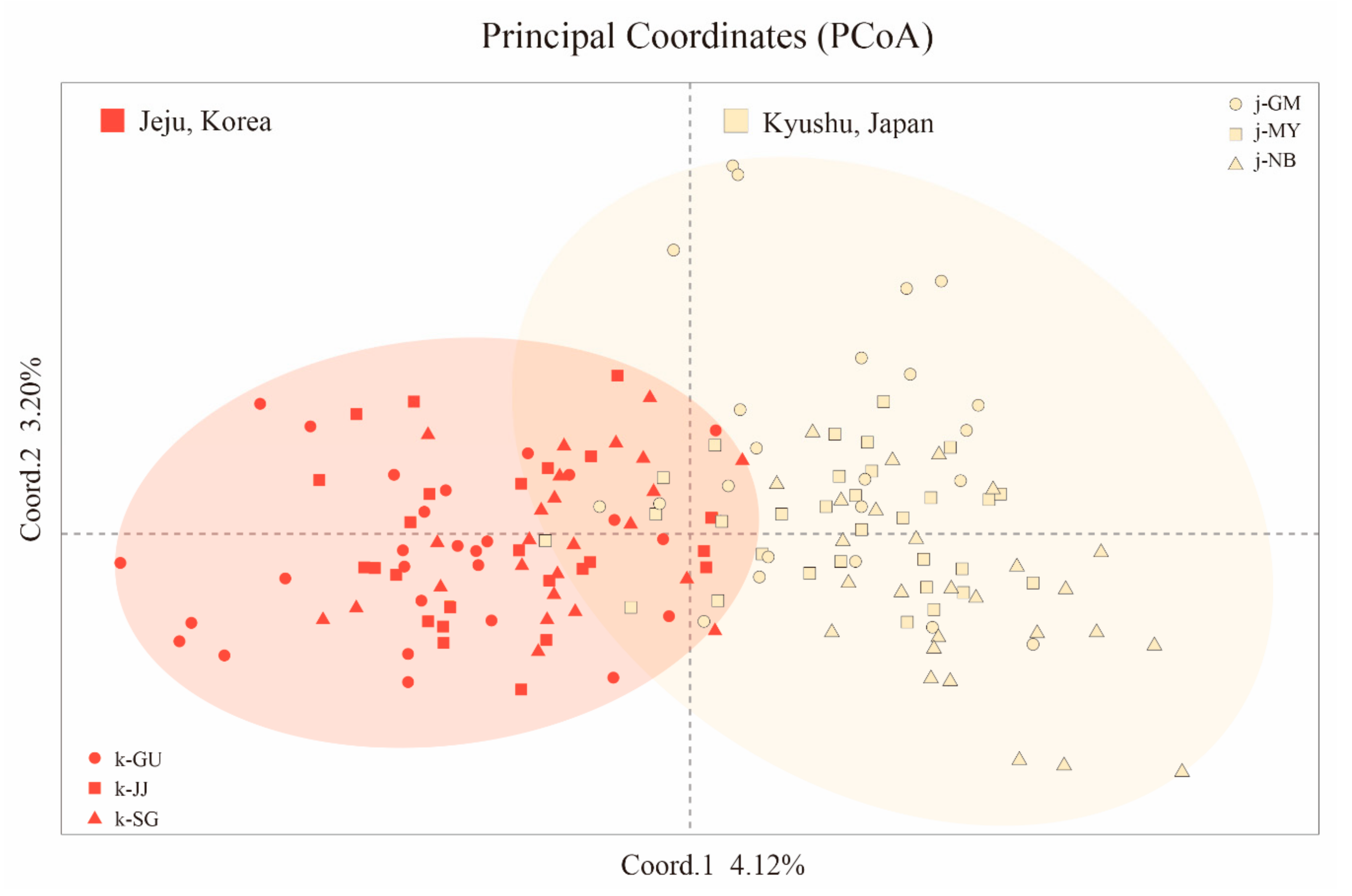
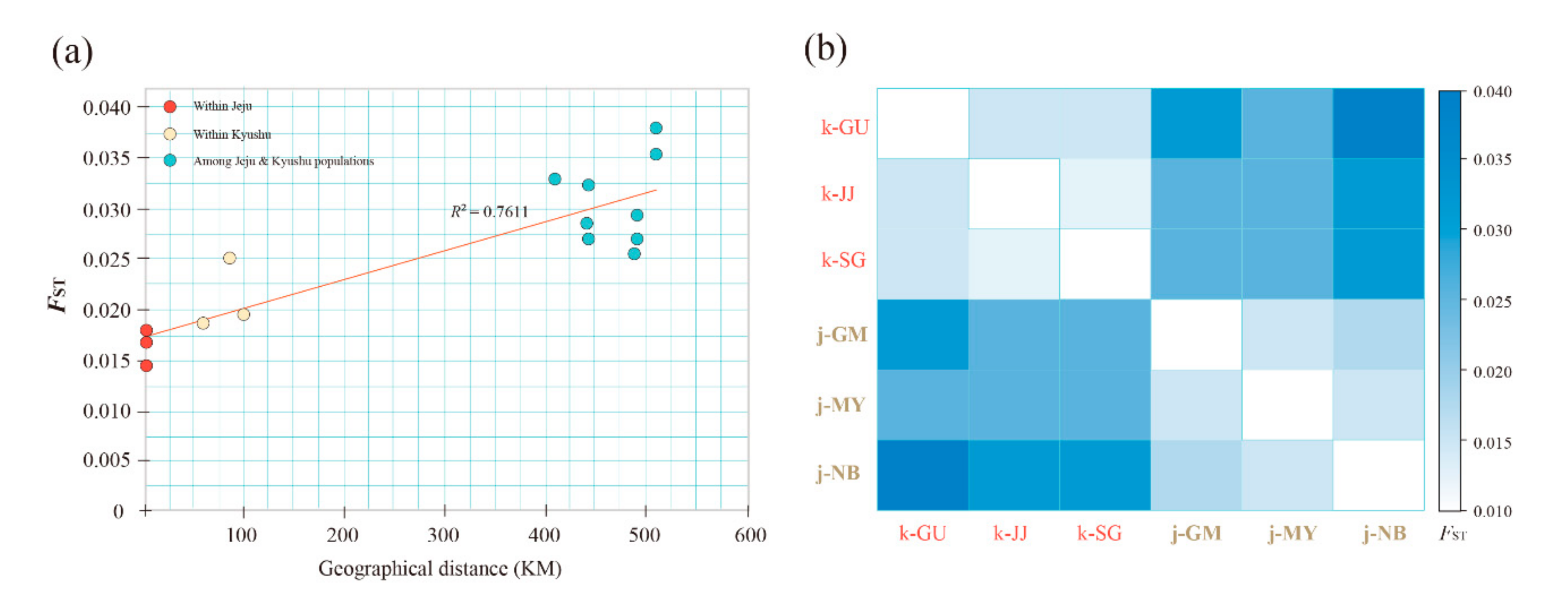
3.3. Population Structure
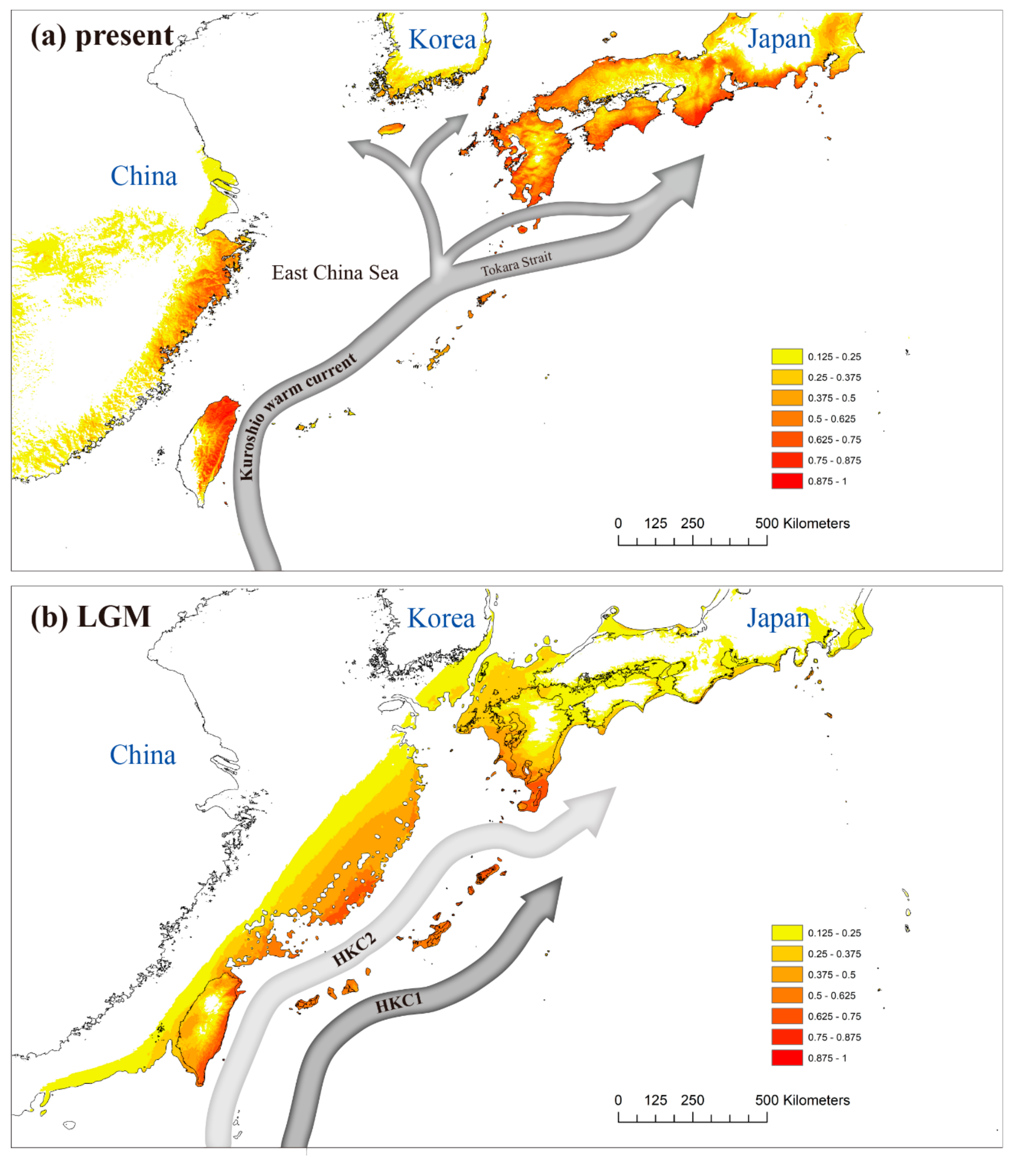
3.4. Ecological Niche Modeling
4. Discussion
Data Available
Author Contributions
Funding
Acknowledgments
Conflicts of Interest:
References
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.J.; Aguinagalde, I.; De Beaulieu, J.-L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial Refugia: Hotspots but Not Melting Pots of Genetic Diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Qiu, Y.-X.; Liu, Y.-H.; Qi, X.-S.; Kim, S.-H.; Han, J.; Takeuchi, Y.; Worth, J.R.P.; Yamasaki, M.; Sakurai, S.; et al. Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol. Ecol. 2012, 21, 3823–3838. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, D.H.; Choi, B.H. Phylogeography and genetic diversity of East Asian Neolitsea sericea (Lauraceae) based on variations in chloroplast DNA sequences. J. Plant Res. 2013, 126, 193–202. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, D.H.; Choi, I.S.; Choi, B.H. Genetic diversity and historical migration patterns of an endemic evergreen oak, Quercus acuta, across Korea and Japan, inferred from nuclear microsatellites. Plant Syst. Evol. 2014, 300, 1913–1923. [Google Scholar] [CrossRef]
- Jin, D.P.; Lee, J.H.; Xu, B.; Choi, B.H. Phylogeography of East Asian Lespedeza buergeri (Fabaceae) based on chloroplast and nuclear ribosomal DNA sequence variations. J. Plant Res. 2016, 129, 793–805. [Google Scholar]
- Tamaki, I.; Kawashima, N.; Setsuko, S.; Lee, J.-H.; Itaya, A.; Yukitoshi, K.; Tomaru, N. Population genetic structure and demography of Magnolia kobus: Variety borealis is not supported genetically. J. Plant Res. 2019, 132, 741–758. [Google Scholar] [CrossRef]
- Harrison, S.P.; Yu, G.; Takahara, H.; Prentice, I.C. Palaeovegetation: Diversity of temperate plants in East Asia. Nature 2001, 413, 129–130. [Google Scholar] [CrossRef]
- Alleaume-Benharira, M.; Pen, I.R.; Ronce, O. Geographical patterns of adaptation within a species’ range: Interactions between drift and gene flow. J. Evol. Biol. 2006, 19, 203–215. [Google Scholar] [CrossRef]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic variation across species’ geographical ranges: The central–marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef] [PubMed]
- Myking, T.; Vakkari, P.; Skrøppa, T. Genetic variation in northern marginal Taxus baccata L. populations. Implications for conservation. Forestry 2009, 82, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Villellas, J.; Ehrlén, J.; Olesen, J.M.; Braza, R.; Garcia, M. Plant performance in central and northern peripheral populations of the widespread Plantago coronopus. Ecography 2013, 36, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Mimura, M.; Aitken, S.N. Local adaptation at the range peripheries of Sitka spruce. J. Evol. Biol. 2010, 23, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Safriel, U.N.; Volis, S.; Kark, S. Core and peripheral populations and global climate change. Isr. J. Plant Sci. 1994, 42, 331–345. [Google Scholar] [CrossRef]
- Gibson, S.Y.; Van Der Marel, R.C.; Starzomski, B.M. Climate change and conservation of leading-edge peripheral populations. Conserv. Biol. 2009, 23, 1369–1373. [Google Scholar] [CrossRef]
- Milad, M.; Schaich, H.; Bürgi, M.; Konold, W. Climate change and nature conservation in Central European forests: A review of consequences, concepts and challenges. Ecol. Manag. 2011, 261, 829–843. [Google Scholar] [CrossRef]
- Koskela, J.; Lefèvre, F.; Schueler, S.; Kraigher, H.; Olrik, D.C.; Hubert, J.; Yrjänä, L.; Alizoti, P.; Rotach, P. Translating conservation genetics into management: Pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol. Conserv. 2013, 157, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.S.; Lee, J.H.; Choi, B.H. Isolation and characterization of 12 microsatellite loci from Maackia fauriei (Fabaceae), a large tree endemic to Jeju Island. Conserv. Genet. Resour. 2014, 6, 1027–1029. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, E.J.; Lee, E.P.; Lee, S.Y.; Cho, K.; Lee, Y.K.; Chung, S.; Jeong, H.; You, Y.H. Characteristics of vegetation succession on the Pinus thunbergii forests in warm temperate regions, Jeju Island, South Korea. J. Ecol. Environ. 2019, 43, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Han, E.K.; Cho, W.B.; Choi, G.; Yang, S.; Choi, H.J.; Song, G.P.; Lee, J.H. New polymorphic microsatellite markers for Sarcandra glabra (Chloranthaceae), an evergreen broad-leaved shrub endangered in South Korea. J. For. Res. 2020, 25, 364–368. [Google Scholar] [CrossRef]
- Noshiro, S.; Sasaki, Y. Identification of Japanese species of evergreen Quercus and Lithocarpus (Fagaceae). IAWA J. 2011, 32, 383–393. [Google Scholar] [CrossRef]
- Tanouchi, H.; Yamamoto, S. Structure and regeneration of canopy species in an old-growth evergreen broad-leaved forest in Aya district, southwestern Japan. Vegetatio 1995, 117, 51–60. [Google Scholar] [CrossRef]
- Zeng, Q.-M.; Liu, B.; Lin, R.-Q.; Jiang, Y.-T.; Liu, Z.-J.; Chen, S.-P. The complete chloroplast genome sequence of Quercus gilva (Fagaceae). Mitochondrial DNA Part B 2019, 4, 2493–2494. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.U.; Jang, K.S.; Lim, H.; Kim, E.H.; Lee, K.H. Genetic Diversity of Quercus gilva in Je-ju Island. J. Korean Soc. For. Sci. 2018, 107, 151–157. [Google Scholar]
- Sugiura, N.; Kurokochi, H.; Tan, E.; Asakawa, S.; Sato, N.; Saito, Y.; Ide, Y. Development of 13 polymorphic chloroplast DNA markers in Quercus gilva, a regionally endemic species in Japan. Conserv. Genet. Resour. 2014, 6, 961–965. [Google Scholar] [CrossRef]
- Hyun, H.J.; Song, K.M.; Choi, H.S.; Kim, C.S. Dynamics and Distribution of Quercus gilva Blume Population in Korea. Korean J. Environ. Ecol. 2014, 28, 385–392. [Google Scholar] [CrossRef]
- Ministry of the Environment of Korea. Korean Red List of Threatened Species; National Institute of Biological Resources: Incheon, Korea, 2012.
- Suh, M.H.; Koh, K.S.; Ku, Y.B.; Kil, J.H.; Choi, T.B.; Suh, S.U.; Qiu, Y.X.; Liu, Y.H.; Oh, J.G. Research on the Conservation Strategy for the Endangered and Reserved Plants Based on the Ecological and Genetic Characteristics (Ⅰ); National Institute of Environmental Research: Incheon, Korea, 2001. [Google Scholar]
- Chung, M.Y.; López-Pujol, J.; Maki, M.; Kim, K.J.; Chung, J.M.; Sun, B.Y.; Chung, M.G. Genetic diversity in the common terrestrial orchid Oreorchis patens and its rare congener Oreorchis coreana: Inference of species evolutionary history and implications for conservation. J. Hered. 2012, 103, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Haig, S.M.; Miller, M.P.; Bellinger, R.; Draheim, H.M.; Mercer, D.M.; Mullins, T.D. The conservation genetics juggling act: Integrating genetics and ecology, science and policy. Evol. Appl. 2016, 9, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Stojnić, S.; Avramidou, E.; Fussi, B.; Westergren, M.; Orlović, S.; Matović, B.; Trudić, B.; Kraigher, H.; Aravanopoulos, F.A.; Konnert, M. Assessment of Genetic Diversity and Population Genetic Structure of Norway Spruce (Picea abies (L) Karsten) at Its Southern Lineage in Europe. Implications for Conservation of Forest Genetic Resources. Forests 2019, 10, 258. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.Y.; López-Pujol, J.; Chung, M.G. The role of the Baekdudaegan (Korean Peninsula) as a major glacial refugium for plant species: A priority for conservation. Biol. Conserv. 2017, 206, 236–248. [Google Scholar] [CrossRef]
- Aoki, K.; Tamaki, I.; Nakao, K.; Ueno, S.; Kamijo, T.; Setoguchi, H.; Murakami, N.; Kato, M.; Tsumura, Y. Approximate Bayesian computation analysis of EST-associated microsatellites indicates that the broadleaved evergreen tree Castanopsis sieboldii survived the Last Glacial Maximum in multiple refugia in Japan. Heredity 2019, 122, 326–340. [Google Scholar] [PubMed]
- Park, J.S.; Takayama, K.; Suyama, Y.; Choi, B.-H. Distinct phylogeographic structure of the halophyte Suaeda malacosperma (Chenopodiaceae/Amaranthaceae), endemic to Korea–Japan region, influenced by historical range shift dynamics. Plant Syst. Evol. 2019, 305, 193–203. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Kang, H.; Sun, X.; Yin, S.; Du, H.; Yamanaka, N.; Gapare, W.; Wu, H.X.; Liu, C. Phylogeography of Quercus variabilis based on chloroplast DNA sequence in East Asia: Multiple glacial refugia and mainland-migrated island populations. PLoS ONE 2012, 7, e47268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.Q.; Matsui, T.; Ohashi, H.; Dong, Y.-F.; Momohara, A.; Herrando-Moraira, S.; Qian, S.; Yang, Y.; Ohsawa, M.; Luu, H.T.; et al. Identifying long-term stable refugia for relict plant species in East Asia. Nat. Commun. 2018, 9, 4488. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.B.; So, S.K.; Han, E.K.; Myeong, H.H.; Park, J.S.; Hwang, S.H. Rear-edge, low-diversity, and haplotypic uniformity in cold-adapted Bupleurum euphorbioides interglacial refugia populations. Ecol. Evol. 2020, in press. [Google Scholar]
- Chung, M.Y.; López-Pujol, J.; Chung, M.G. Genetic homogeneity between Korean and Japanese populations of the broad-leaved evergreen tree Machilus thunbergii (Lauraceae): A massive post-glacial immigration through the Korea Strait or something else? Biochem. Syst. Ecol. 2014, 53, 20–28. [Google Scholar] [CrossRef]
- Cho, W.B.; Choi, I.S.; Choi, B.H. Development of microsatellite markers for the endangered Pedicularis ishidoyana (Orobanchaceae) using next-generation sequencing. Appl. Plant Sci. 2015, 3, 1500083. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.P.; Knaus, B.J.; Mullins, T.D.; Haig, S.M. SSR_pipeline: A Bioinformatic Infrastructure for Identifying Microsatellites from Paired-End Illumina High-Throughput DNA Sequencing Data. J. Hered. 2013, 104, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Cooper, A.; Markowitz, S.; Drummond, A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [PubMed]
- Luikart, G. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Manni, F.; Guérard, E.; Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: How barriers can be detected by using Monmonier’s algorithm. Hum. Biol. 2004, 76, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Monmonier, M.S. Maximum-Difference Barriers: An Alternative Numerical Regionalization Method. Geogr. Anal. 1973, 5, 245–261. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F. Maximum Likelihood Estimation of the Number of Nucleotide Substitutions from Restriction Sites Data. Genetics 1983, 105, 207–217. [Google Scholar]
- Dieringer, D.; Schlötterer, C. Two distinct modes of microsatellite mutation processes: Evidence from the complete genomic sequences of nine species. Genome Res. 2003, 13, 2242–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package) Version 3.6; Department of Genome Sciences, University of Washington: Washington, DC, USA, 2004. [Google Scholar]
- Merow, C.; Smith, M.J.; Silander, J.A. A practical guide to MaxEnt for modeling species’ distributions: What it does, and why inputs and settings matter. Ecography 2013, 36, 1058–1069. [Google Scholar] [CrossRef]
- GBIF Secretariat. GBIF Backbone Taxonomy Checklist dataset. GBIF Backbone Taxon. 2019. [Google Scholar] [CrossRef]
- Brown, J.L.; Bennett, J.R.; French, C.M. SDMtoolbox 2.0: The next generation Python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. PeerJ 2017, 5, e4095. [Google Scholar] [CrossRef] [Green Version]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-Auza, R.W.; Zimmermann, N.E.; Linder, H.P.; Kessler, M. Climatologies at high resolution for the earth’s land surface areas. Sci. Data 2017, 4, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Danielson, J.J.; Gesch, D.B. Global Multi-Resolution Terrain Elevation Data 2010 (GMTED2010). In Geological Survey; Springer: Garretson, SD, USA, 2011. [Google Scholar]
- Gent, P.R.; Danabasoglu, G.; Donner, L.J.; Holland, M.M.; Hunke, E.C.; Jayne, S.R.; Lawrence, D.M.; Neale, R.B.; Rasch, P.J.; Vertenstein, M.; et al. The Community Climate System Model Version 4. J. Clim. 2011, 24, 4973–4991. [Google Scholar] [CrossRef]
- Watanabe, S.; Hajima, T.; Sudo, K.; Nagashima, T.; Takemura, T.; Okajima, H.; Nozawa, T.; Kawase, H.; Abe, M.; Yokohata, T.; et al. MIROC-ESM 2010: Model description and basic results of CMIP5-20c3m experiments. Geosci. Model Dev. 2011, 4, 845–872. [Google Scholar] [CrossRef] [Green Version]
- Ujiié, Y.; Ujiié, H.; Taira, A.; Nakamura, T.; Oguri, K. Spatial and temporal variability of surface water in the Kuroshio source region, Pacific Ocean, over the past 21,000 years: Evidence from planktonic foraminifera. Mar. Micropaleontol. 2003, 49, 335–364. [Google Scholar] [CrossRef]
- Kao, S.J.; Wu, C.R.; Hsin, Y.C.; Dai, M. Effects of sea level change on the upstream Kuroshio Current through the Okinawa Trough. Geophys. Res. Lett. 2006, 33. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Li, A.; Kao, S.; Gong, X.; Frank, M.; Kuhn, G.; Cai, W.; Yan, H.; Wan, S.; Zhang, H.-H.; et al. Synchronicity of Kuroshio Current and climate system variability since the Last Glacial Maximum. Earth Planet. Sci. Lett. 2016, 452, 247–257. [Google Scholar] [CrossRef]
- Vogt-Vincent, N.S.; Mitarai, S. A persistent Kuroshio in the glacial East China Sea and implications for coral paleobiogeography. Paleoceanogr. Paleoclimatol. 2020, e2020PA003902. [Google Scholar] [CrossRef]
- Janzen, D.H. Seed predation by animals. Annu. Rev. Ecol. Syst. 1971, 2, 465–492. [Google Scholar]
- Xiao, Z.; Zhibin, Z.; Wang, Y. Dispersal and germination of big and small nuts of Quercus serrata in a subtropical broad-leaved evergreen forest. Ecol. Manag. 2004, 195, 141–150. [Google Scholar] [CrossRef]
- Tsukada, M. Vegetation and Climate during the Last Glacial Maximum in Japan. Quat. Res. 1983, 19, 212–235. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, B.-H. Distribution of broad-Leaved evergreen plants on islands of Incheon, middle part of Yellow Sea. Korean J. Plant Taxon. 2008, 38, 315–332. [Google Scholar] [CrossRef]
- Aoki, K.; Suzuki, T.; Hsu, T.W.; Murakami, N. Phylogeography of the component species of broad-leaved evergreen forests in Japan, based on chloroplast DNA variation. J. Plant Res. 2004, 117, 77–94. [Google Scholar]
- Aoki, K.; Matsumura, T.; Hattori, T.; Murakami, N. Chloroplast DNA phylogeography of Photinia glabra (Rosaceae) in Japan. Am. J. Bot. 2006, 93, 1852–1858. [Google Scholar] [CrossRef]
- Liu, H.-Z.; Takeichi, Y.; Kamiya, K.; Harada, K. Phylogeography of Quercus phillyraeoides (Fagaceae) in Japan as revealed by chloroplast DNA variation. J. Res. 2013, 18, 361–370. [Google Scholar] [CrossRef]
- Litkowiec, M.; Lewandowski, A.; Rączka, G. Spatial Pattern of the Mitochondrial and Chloroplast Genetic Variation in Poland as a Result of the Migration of Abies alba Mill. From Different Glacial Refugia. Forests 2016, 7, 284. [Google Scholar] [CrossRef]
- Hou, Z.; Wang, Z.; Ye, Z.; Du, S.; Liu, S.; Zhang, J. Phylogeographic analyses of a widely distributed Populus davidiana: Further evidence for the existence of glacial refugia of cool-temperate deciduous trees in northern East Asia. Ecol. Evol. 2018, 8, 13014–13026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pasquale, G.; Saracino, A.; Bosso, L.; Russo, D.; Moroni, A.; Bonanomi, G.; Allevato, E. Coastal Pine-Oak Glacial Refugia in the Mediterranean Basin: A Biogeographic Approach Based on Charcoal Analysis and Spatial Modelling. Forests 2020, 11, 673. [Google Scholar] [CrossRef]
- Ito, S.; Ohtsuka, K.; Yamashita, T. Ecological distribution of seven evergreen Quercus species in southern and eastern Kyushu, Japan. Veg. Sci. 2007, 24, 53–63. [Google Scholar]
- Nakashizuka, T.; Iida, S. Composition, dynamics and disturbance regime of temperate deciduous forests in Monsoon Asia. Vegetatio 1995, 121, 23–30. [Google Scholar] [CrossRef]
- Nakao, K.; Matsui, T.; Horikawa, M.; Tsuyama, I.; Tanaka, N. Assessing the impact of land use and climate change on the evergreen broad-leaved species of Quercus acuta in Japan. Plant Ecol. 2011, 212, 229–243. [Google Scholar]
- Moritz, C. Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef]
- Crandall, K.A.; Bininda-Emonds, O.R.; Mace, G.M.; Wayne, R.K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Fraser, D.J.; Bernatchez, L. Adaptive evolutionary conservation: Towards a unified concept for defining conservation units. Mol. Ecol. 2001, 10, 2741–2752. [Google Scholar] [CrossRef]
- Palsbøll, P.J.; Bérubé, M.; Allendorf, F.W. Identification of management units using population genetic data. Trends Ecol. Evol. 2006, 22, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Lindley, S.T. Genomics and conservation units: The genetic basis of adult migration timing in Pacific salmonids. Evol. Appl. 2018, 11, 1518–1526. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Primer Sequence (5′–3′) | Repeat Motif | Number of Alleles | Size Range (bp) | Fluorescent Label | GenBank Accession No. |
|---|---|---|---|---|---|---|
| Multiplex mix A | ||||||
| Qrg001 | F: TCTGATGAGGTGCTGGAA R: TTGTTATCCAATTCTCTCCCT | (TC)12 | 7 | 100–118 | 6-FAM | MT811115 |
| Qrg002 | F: TGAGCTTGTTGATTGGAGAA R: CTTCAAGACGTACTACAGCA | (CA)12 | 6 | 158–172 | 6-FAM | MT811116 |
| Qrg003 | F: TTGGTGGAAGAGATTGTGAG R: CTCTTTGGGTTCTCTGTTGT | (CT)14 | 7 | 213–225 | 6-FAM | MT811117 |
| Qrg004 | F: TGGCTTCCTGACCATACATA R: GACTAACCCTGCCCTCAA | (GAA)6 | 6 | 107–122 | VIC | MT811118 |
| Qrg006 | F: CTCAATGGCGAAATCATCAG R: TCTATAGAGGCAGCAAACAC | (TTAG)8 | 5 | 220–236 | VIC | MT811119 |
| Qrg007 | F: GTTGGATTGGATTCTGTTGC R: TTCCCTCCTTGTCACGTT | (AG)12 | 15 | 103–135 | NED | MT811120 |
| Qrg008 | F: ATCGGAGCAAGAAATCAAAT R: CCACCAACTCTAATGCTGTA | (AAG)8 | 3 | 159–168 | NED | MT811121 |
| Qrg009 | F: CACTCTCTTCGACCTTCTTT R: TTCTGGGTTCTTGCTTATCG | (TCA)9 | 6 | 225–240 | NED | MT811122 |
| Multiplex mix B | ||||||
| Qrg011 | F: CGTTCAGATCAGGGTACAAA R: ATAAGCAAAGCACCCATGTA | (CA)14 | 5 | 160–170 | 6-FAM | MT811123 |
| Qrg012 | F: ATTAATGGAGAACTGCCCTC R: AGGATCATGAACTTCGACTG | (CTT)11 | 5 | 223–235 | 6-FAM | MT811124 |
| Qrg013 | F: TCTCAAACGGACCCATTTAA R: TCCTGTGATTACTGTCTATGC | (CT)13 | 5 | 108–120 | VIC | MT811125 |
| Qrg014 | F: GTCAGTATAGCATGTGGTGT R: TTGGTGAGTTGAGATTGCAA | (GA)14 | 8 | 159–189 | VIC | MT811126 |
| Qrg015 | F: TTCCCATTTCAGACAAGAGG R: GATTCGAACCCTCCTACAAA | (TAAC)7 | 7 | 209–237 | VIC | MT811127 |
| Qrg016 | F: CTCTACCATCAACATCCTGC R: AATTCCAGTTTTGCAGTCCA | (AGAC)6 | 6 | 124–148 | NED | MT811128 |
| Qrg017 | F: ACACCAAACAAAGCAAACAA R: TACGAACACAATCCAAACCT | (AACA)6 | 3 | 163–171 | NED | MT811129 |
| Qrg018 | F: CAACCACAATGTGTAAAGACA R: GCAAAAGAGTGTATGTGCTC | (ACA)10 | 4 | 218–236 | NED | MT811130 |
| Multiplex mix C | ||||||
| Qrg019 | F: AACTCTTGCTCCATTCATTT R: GGGTCTACAATTGAATTATGGC | (AG)13 | 8 | 133–149 | 6-FAM | MT811131 |
| Qrg020 | F: AGGATTTGTAGCTGACCCTA R: GCCAAGTAATCAAATTGACTGA | (GTT)8 | 4 | 166–178 | 6-FAM | MT811132 |
| Qrg021 | F: ACAAAGACTACGTGTGCATA R: TTTCTATGAAACGCAACAGC | (CT)14 | 10 | 229–253 | 6-FAM | MT811133 |
| Qrg022 | F: GGATGACATGGCTGATCTTC R: ATAACTGGAATGGCATGGAG | (AAG)7 | 3 | 123–135 | VIC | MT811134 |
| Qrg024 | F: CCTAAGAAGCACAGGTAAGG R: AGAGCAAGTGAGAAAGAGTC | (CT)14 | 11 | 237–263 | VIC | MT811135 |
| Qrg025 | F: CATATAGCCGAGGAAGAAGT R: GAAGGCAGAGGTTGGTTAAA | (GAA)6 | 2 | 134–137 | NED | MT811136 |
| Qrg026 | F: GATGGGAATGCTCTTAGGTC R: TTGTGAAGTCGCCTACAATT | (ATAG)6 | 3 | 180–188 | NED | MT811137 |
| Qrg027 | F: TGGAAATGACATTGTTACCCT R: CCGATGACAAGAATCCCAAT | (GA)14 | 12 | 235–271 | NED | MT811138 |
| Multiplex mix D | ||||||
| Qrg028 | F: TAAAGGAGTGCATGGTGAAA R: AGTGAAGCCTCTTTCCTAGA | (CT)13 | 9 | 127–147 | 6-FAM | MT811139 |
| Qrg029 | F: AAGATAACTGCACGCTTGTA R: TCAGAAATCGCTCATACCTG | (TG)13 | 7 | 184–196 | 6-FAM | MT811140 |
| Qrg030 | F: CTATTCATGGACTCCTCTGT R: AATTGCAAGGCCTTAGAACT | (AG)15 | 7 | 235–249 | 6-FAM | MT811141 |
| Qrg031 | F: GGTTAGGGCTCTTTCCAAAT R: CTCTCCCTTTCTTTCACTGT | (GA)13 | 8 | 131–145 | VIC | MT811142 |
| Qrg033 | F: TCTTGCCAATCTAAATCCCA R: TGCATGATACAGAAACACCA | (AAGA)7 | 2 | 239–247 | VIC | MT811143 |
| Qrg034 | F: GGACATCTACAGCCTACAAA R: CGCAGACCAAATATCATTCTC | (CT)12 | 12 | 143–173 | NED | MT811144 |
| Qrg036 | F: TAACTTTGTTCTCGCCTGA R: AATGTAGAGCCTGTTTGCAT | (GA)13 | 7 | 239–259 | NED | MT811145 |
| Multiplex mix E | ||||||
| Qrg037 | F: TTCGAGATAGGACAGAGGAG R: TGTGTTTGATTAGCGGAGAA | (AAGA)8 | 5 | 128–144 | 6-FAM | MT811146 |
| Qrg038 | F: TGGCTATGATAATTGTGGGT R: CTCAACCCTGTTATCTCACC | (GA)17 | 8 | 182–204 | 6-FAM | MT811147 |
| Qrg039 | F: AAAGTGGATTTGCAGCCTAA R: GACAATGGAGAAGGGACAAT | (TC)14 | 6 | 244–260 | 6-FAM | MT811148 |
| Qrg040 | F: GCATTTCTCTCTCTGGTTCA R: AAGTACCCTCCATCTACGTT | (AAG)6 | 3 | 128–146 | VIC | MT811149 |
| Qrg041 | F: CTTCCTCGTCAATAGTCCAC R: AGTGAGTTTGATACGCTTGT | (AAG)12 | 9 | 186–228 | VIC | MT811150 |
| Qrg042 | F: CCCACACATTATACCACGAA R: CTACTAACAACCGCAACTCT | (AG)17 | 8 | 227–253 | VIC | MT811151 |
| Qrg043 | F: CATACATCCTAGTGCAGCAG R: GGTAGCTCAAGTTCACAGTT | (CAA)6 | 2 | 149–155 | NED | MT811152 |
| Multiplex mix F | ||||||
| Qrg046 | F: CTGCCCCTAACTAATCTGTT R: GTAGATGATGAGGTTGTGGG | (TGT)6 | 2 | 149–152 | 6-FAM | MT811153 |
| Qrg047 | F: AGACCAGTAGATGCTTCAAA R: ATTCATGACCCTCCTTCTCA | (AAG)9 | 3 | 208–217 | 6-FAM | MT811154 |
| Qrg048 | F: TCCATCGTCAACAAAGGATT R: AACCAGTTCTCACTCTCTCT | (AG)17 | 7 | 235–269 | 6-FAM | MT811155 |
| Qrg049 | F: CAACTACTGTAGCCTTGTGT R: TATGCCTCCAGTGTACTACA | (CA)12 | 7 | 146–166 | VIC | MT811156 |
| Qrg050 | F: GGGACCATAGCAGTGTTAAT R: AGCCCTCCCTTATTTATTCC | (TC)21 | 8 | 192–216 | VIC | MT811157 |
| Qrg051 | F: CTCCTCTTGGCTATGACATC R: TCTTGTTTGAGGAAGTTGACA | (TTC)14 | 10 | 235–259 | VIC | MT811158 |
| Qrg052 | F: ACTTGTAACTAACCTGGCTC R: CTAGGAGGATGAAATGGCAA | (CTAA)8 | 4 | 150–162 | NED | MT811159 |
| Qrg053 | F: TGACAGTACATGGTAAAGCT R: TTCTTGGTCTTGAATGAGGA | (CT)14 | 7 | 204–228 | NED | MT811160 |
| Locus | NA | HO | HE | Null | Locus | NA | HO | HE | Null |
|---|---|---|---|---|---|---|---|---|---|
| Qrg001 | 8 | 0.703 | 0.619 | 0.0020 | Qrg027 | 16 | 0.829 | 0.865 | 0.0031 |
| Qrg002 | 10 | 0.741 | 0.755 | 0.0053 | Qrg028 | 10 | 0.842 | 0.820 | 0.0018 |
| Qrg003 | 7 | 0.741 | 0.794 | 0.0112 | Qrg029 | 8 | 0.677 | 0.743 | 0.0275 |
| Qrg004 | 7 | 0.778 | 0.786 | 0.0057 | Qrg030 | 9 | 0.253 | 0.765 | 0.2774 * |
| Qrg006 | 6 | 0.772 | 0.763 | 0.0033 | Qrg031 | 10 | 0.810 | 0.841 | 0.0091 |
| Qrg007 | 18 | 0.918 | 0.899 | 0.0012 | Qrg033 | 3 | 0.222 | 0.209 | 0.0048 |
| Qrg008 | 4 | 0.361 | 0.366 | 0.0058 | Qrg034 | 16 | 0.867 | 0.885 | 0.0038 |
| Qrg009 | 8 | 0.570 | 0.748 | 0.0868 * | Qrg036 | 9 | 0.627 | 0.800 | 0.0910 * |
| Qrg011 | 6 | 0.684 | 0.745 | 0.0071 | Qrg037 | 5 | 0.696 | 0.738 | 0.0118 |
| Qrg012 | 7 | 0.759 | 0.756 | 0.0026 | Qrg038 | 13 | 0.759 | 0.794 | 0.0076 |
| Qrg013 | 8 | 0.513 | 0.657 | 0.0728 * | Qrg039 | 6 | 0.753 | 0.731 | 0.0035 |
| Qrg014 | 11 | 0.861 | 0.835 | 0.0016 | Qrg040 | 3 | 0.468 | 0.539 | 0.0218 |
| Qrg015 | 7 | 0.620 | 0.671 | 0.0073 | Qrg041 | 13 | 0.823 | 0.808 | 0.0025 |
| Qrg016 | 6 | 0.608 | 0.622 | 0.0044 | Qrg042 | 14 | 0.734 | 0.805 | 0.0204 |
| Qrg017 | 5 | 0.589 | 0.597 | 0.0117 | Qrg043 | 3 | 0.133 | 0.131 | 0.0058 |
| Qrg018 | 6 | 0.532 | 0.631 | 0.0464 | Qrg046 | 3 | 0.348 | 0.384 | 0.0192 |
| Qrg019 | 10 | 0.759 | 0.824 | 0.0121 | Qrg047 | 4 | 0.475 | 0.496 | 0.0067 |
| Qrg020 | 5 | 0.633 | 0.674 | 0.0050 | Qrg048 | 12 | 0.595 | 0.808 | 0.0977 * |
| Qrg021 | 19 | 0.861 | 0.886 | 0.0062 | Qrg049 | 10 | 0.430 | 0.505 | 0.0353 |
| Qrg022 | 3 | 0.165 | 0.179 | 0.0099 | Qrg050 | 14 | 0.791 | 0.819 | 0.0044 |
| Qrg024 | 13 | 0.741 | 0.857 | 0.0355 | Qrg051 | 10 | 0.810 | 0.814 | 0.0036 |
| Qrg025 | 2 | 0.044 | 0.067 | 0.0316 | Qrg052 | 4 | 0.551 | 0.572 | 0.0106 |
| Qrg026 | 4 | 0.139 | 0.203 | 0.0607 * | Qrg053 | 10 | 0.741 | 0.756 | 0.0029 |
| ID | Location | Coordinates | EV | N | NA | AR | PA | Priv | HO (SE) | HE (SE) | FIS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Jeju Island | |||||||||||
| k-GU | Gueok-ri, Daejeong-eup, Jeju | 33°18′8.21″ N, 126°16′36.59″ E | 136 | 32 | 254 | 5.729 | 11 | 0.043 | 0.660 (0.037) | 0.645 (0.033) | − 0.024 |
| k-JJ | Jeoji-ri, Hangyeong-myeon, Jeju | 33°18′45.36″ N, 126°17′3.76″ E | 168 | 27 | 254 | 5.905 | 4 | 0.016 | 0.656 (0.040) | 0.640 (0.035) | 0.003 |
| k-SG | Seogwang-ri, Andeok-myeon, Jeju | 33°17′57.47″ N, 126°18′59.97″ E | 201 | 18 | 237 | 5.925 | 8 | 0.034 | 0.639 (0.039) | 0.651 (0.033) | 0.043 |
| Mean | 168.3 | 25.6 | 248.3 | 5.853 | 7.7 | 0.031 | 0.652 | 0.645 | 0.007 | ||
| Pooled populations | 77 | 301 | 7.525 | 33 | 0.110 | 0.641 | 0.657 | 0.018 | |||
| Kyushu | |||||||||||
| j-GM | Kuma-gun, Kumamoto Prefecture | 32°17′39.5″ N, 130°52′17.6″ E | 485 | 23 | 238 | 5.656 | 5 | 0.021 | 0.638 (0.039) | 0.634 (0.036) | −0.005 |
| j-MY | Miyakonojo-shi, Miyazaki Prefecture | 31°50′55.7″ N, 131°13′30.4″ E | 230 | 29 | 261 | 5.940 | 10 | 0.038 | 0.636 (0.034) | 0.647 (0.035) | 0.003 |
| j-NB | Nobeoka-shi, Miyazaki Prefecture | 32°39′15.8″ N, 131°41′14.3″ E | 38 | 29 | 224 | 5.201 | 4 | 0.018 | 0.613 (0.043) | 0.615 (0.038) | 0.023 |
| Mean | 251 | 27 | 241 | 5.600 | 6.3 | 0.026 | 0.629 | 0.632 | 0.007 | ||
| Pooled populations | 81 | 302 | 7.503 | 34 | 0.113 | 0.628 | 0.648 | 0.015 | |||
| Population | Wilcoxon Test | Mode Shift | |
|---|---|---|---|
| TPM | SMM | ||
| Jeju Island | |||
| k-GU | 0.476153 | 0.988818 | No |
| k-JJ | 0.465576 | 0.991747 | No |
| k-SG | 0.294988 | 0.925049 | No |
| Kyushu | |||
| j-GM | 0.383349 | 0.974584 | No |
| j-MY | 0.292696 | 0.982035 | No |
| j-NB | 0.135276 | 0.765987 | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, E.-K.; Cho, W.-B.; Park, J.-S.; Choi, I.-S.; Kwak, M.; Kim, B.-Y.; Lee, J.-H. A Disjunctive Marginal Edge of Evergreen Broad-Leaved Oak (Quercus gilva) in East Asia: The High Genetic Distinctiveness and Unusual Diversity of Jeju Island Populations and Insight into a Massive, Independent Postglacial Colonization. Genes 2020, 11, 1114. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101114
Han E-K, Cho W-B, Park J-S, Choi I-S, Kwak M, Kim B-Y, Lee J-H. A Disjunctive Marginal Edge of Evergreen Broad-Leaved Oak (Quercus gilva) in East Asia: The High Genetic Distinctiveness and Unusual Diversity of Jeju Island Populations and Insight into a Massive, Independent Postglacial Colonization. Genes. 2020; 11(10):1114. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101114
Chicago/Turabian StyleHan, Eun-Kyeong, Won-Bum Cho, Jong-Soo Park, In-Su Choi, Myounghai Kwak, Bo-Yun Kim, and Jung-Hyun Lee. 2020. "A Disjunctive Marginal Edge of Evergreen Broad-Leaved Oak (Quercus gilva) in East Asia: The High Genetic Distinctiveness and Unusual Diversity of Jeju Island Populations and Insight into a Massive, Independent Postglacial Colonization" Genes 11, no. 10: 1114. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101114