Claudins in Renal Physiology and Pathology

1
Centre de Recherche des Cordeliers, INSERM, Sorbonne Université, Université de Paris, F-75006 Paris, France
2
Service de Physiologie, Hôpital Européen Georges Pompidou, Assistance Publique-Hôpitaux de Paris, F-75015 Paris, France
3
Centre de Référence des Maladies Rénales Héréditaires de l’Enfant et de l’Adulte (MARHEA), F-75015 Paris, France
4
Centre de Référence des Maladies Rares du Calcium et du Phosphate, F-75015 Paris, France
5
CNRS, ERL8228, F-75006 Paris, France
*
Author to whom correspondence should be addressed.
Genes 2020, 11(3), 290; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030290
Submission received: 15 January 2020
/
Revised: 24 February 2020
/
Accepted: 24 February 2020
/
Published: 10 March 2020
(This article belongs to the Special Issue Genetics of Tubulopathies)
Abstract
:Claudins are integral proteins expressed at the tight junctions of epithelial and endothelial cells. In the mammalian kidney, every tubular segment express a specific set of claudins that give to that segment unique properties regarding permeability and selectivity of the paracellular pathway. So far, 3 claudins (10b, 16 and 19) have been causally traced to rare human syndromes: variants of CLDN10b cause HELIX syndrome and variants of CLDN16 or CLDN19 cause familial hypomagnesemia with hypercalciuria and nephrocalcinosis. The review summarizes our current knowledge on the physiology of mammalian tight junctions and paracellular ion transport, as well as on the role of the 3 above-mentioned claudins in health and disease. Claudin 14, although not having been causally linked to any rare renal disease, is also considered, because available evidence suggests that it may interact with claudin 16. Some single-nucleotide polymorphisms of CLDN14 are associated with urinary calcium excretion and/or kidney stones. For each claudin considered, the pattern of expression, the function and the human syndrome caused by pathogenic variants are described.
1. Introduction
Almost every epithelium in multicellular organisms separates two compartments (apical and basolateral) with differing ion composition. Depending on the nature of the compartments, human epithelia can behave as barriers (e.g., the skin epithelium) or allow selective transport of solutes and/or solvent from apical to basolateral compartment or vice-versa (e.g., intestinal or renal tubular epithelia). The asymmetry of composition between apical and basolateral compartment is created and maintained by active transport (with the notable exception of passive transport that occurs as a result of Gibbs-Donnan effect). Active transport can occur transcellularly only, at the expense of energy release; for example hydrolysis of ATP releases energy used to actively pump ions across the plasma membrane and to build transmembrane electrochemical gradients. Passive diffusion between 2 compartments may be defined as the flow of solute that occurs in response to the difference in electrochemical potentials of the considered solute between both compartments. In an epithelium, diffusion can occur across plasma membranes or along the paracellular pathway. Because of the existing transmembrane electrochemical potential differences, sodium (Na+), calcium (Ca2+) and magnesium (Mg2+) cannot cross passively both apical and basolateral membranes: therefore, at some step, transcellular transport of Na+, Ca2+ and Mg2+ requires energy expenditure. By contrast, passive paracellular diffusion of Na+, Ca2+ and Mg2+ allows both transepithelial transport and energy saving [1]. The present review is a summary of our knowledge of normal and abnormal paracellular ion transport in the mammalian renal tubule. Specifically, it focuses on the role of claudin proteins, which are highly specialized proteins expressed at the tight junction, in health and disease.
2. Structure and Function of Tight Junction in the Kidney
The properties of the paracellular pathway along the renal tubular epithelium have been less intensively studied than the properties of the transcellular pathway. Nevertheless, epithelia have been classified as leaky or tight, according to the value of transepithelial resistance, RT. Because transcellular and paracellular pathways are organized in parallel, the reciprocal of transepithelial resistance equals the sum of the reciprocal of transcellular resistance (RC) and of the reciprocal of paracellular resistance (RP), according to Ohm’s law,
The direct consequence, assuming that RC is high and almost constant, is that «leaky» epithelia (low RT) must have low RP and «tight» epithelia (high RT) have high RP. Several investigators have measured RT in the various segments of the renal tubule of rodents and rabbits; they consistently found that the proximal tubule has the lowest RT of all segments and that RT increases to a maximal value in the inner medullary collecting duct (for review, see Reference [2]). Besides electrical resistance, every paracellular pathway is also characterized by its selectivity to ion charge and size [3,4,5]. The size selectivity of paracellular pathways was studied by several investigators, mostly in cells lines or intestinal epithelia. Most studies reported that at least two populations of pores are present in the paracellular pathway, the most restrictive having a pore size of 4–8 Å [6,7,8,9,10]. The diameter of the major cations in extracellular fluid is 6.62–8.60 Å when they are in their hydrated form and 1.44–2.98 Å when they are in their unhydrated form [11]. The selectivity to ion charge vary from epithelium to epithelium: some are cation selective whereas other are anion selective. Most of the resistance and charge selectivity of the paracellular pathway are determined at the tight junction. Tight junction is formed by a complex of multiple proteins, subdivided into transmembrane proteins and cytoplasmic plaque proteins, including signaling proteins and scaffolding proteins (adapters that link the tight junction complex to the actin cytoskeleton) [12,13]; tight junction contacts and their selective permeation properties are created by a large family of transmembrane proteins called claudins [14,15,16]. In mammals, the claudin gene family is composed of at least 27 members [2,17,18]. Claudins are relatively short transmembrane proteins that possess between 207 and 305 amino acids. The calculated molecular mass is between 21 and 34 kDa. The general structure of all claudins is similar, consisting in a short intracellular NH2 terminus, a longer intracellular COOH terminus, two extracellular segments (ECS1 and ECS2) (which form a β-sheet of five β-strands (4 β-strands in ECS1 and 1 β-strand in ECS2)), one intracellular loop and four transmembrane domains (helices). The first extracellular segment ECS1 contains the common motif W-LW-C-C and positively and negatively charged amino acid residues that determine the charge selectivity. This motif is critical for folding ECS1 in a region close to the plasma membrane and to form the characteristic β-sheet fold of ECS1 and ECS2 [19]. Claudins polymerize in tight junctions strands through cis (between two claudin within the same cell membrane) and trans (between claudins on opposing cell membranes) interactions [17] (Cis-polymerization would involve a short extracellular helix (ECH) at the end of ECS1 and the extracellular extension of the third transmembrane domain TM3 of a neighboring claudin [17]. Trans-interaction of claudins between neighboring cells is presumed to involve the intercellular interaction of the variable regions V1 (in ECS1) and V2 (in ECS2) [17,20,21]. An hypothetical model has been proposed: tight junction strands would consist of anti-parallel double-rows of claudins and the paracellular channel would be achieved by the interaction of the four claudins coming from double-rows [22]. A trans-interacting octamer model with a double pore has also been proposed [21,23]).
Among the claudin family, a highly conserved PDZ -binding domain is present near the end of the carboxy-terminal end, which binds to the homologous domain of scaffolding proteins such as ZO-1, which is important for the localization of claudins at the tight junction [24,25]. The COOH terminus also contains a number of potential phosphorylation sites, reported to play a role in the localization of claudins at the tight junction.
Many claudins are expressed along the mammalian renal tubule and collecting duct. Although some controversies remain regarding the exact localization of a specific claudin, the overall pattern is that every tubular segment expresses a specific association of claudins, likely accounting for the specific properties of the paracellular pathway of every nephron segment (Figure 1).
So far, only a few claudins have been causally involved in human monogenic diseases; they are claudin (CLDN) 10b, CLDN16 and CLDN19 and will be considered below.
3. Claudin 10b and the HELIX Syndrome
The human CLDN10 gene is located on chromosome 13q31-q34 and contains five exons [32]. There are two claudin 10 splice variants differing in the first exon that encode two main claudin 10 isoforms, claudin 10a and claudin 10b [33].
3.1. Phenotype
In 2017, three independent groups described the clinical consequences of biallelic mutations of the CLDN10 gene.
The first report described the phenotype of two unrelated patients bearing compound heterozygous mutations (Table 1, [34]). One adult female patient had hypokalemic metabolic alkalosis, chronic kidney disease (CKD) stage 3, polyuria, low urinary Mg2+ excretion and a tendency toward hypermagnesemia; the second patient displayed similar abnormalities associating hypokalemic metabolic alkalosis and a tendency toward hypermagnesemia. The second report described the phenotype of 13 affected patients from 2 distantly related families, all carrying the homozygous missense mutation in exon 1b (Table 1, [35]). All affected patients had anhidrosis, heat intolerance, inability to produce tears (alacrimia) and xerostomia. A biological assessment was available in 6 among the 13 patients, aged 23–47 years. All had normal plasma potassium concentration and plasma Mg2+ level was either high or in the upper range of normal values. Estimated glomerular filtration rate (eGFR) was higher than 60 mL/min/1.73 m2. Plasma renin and aldosterone concentrations were not available. The third report described the phenotype of 6 patients from 2 unrelated families [28]. All patients were bearing one homozygous missense mutation of the CLDN10 gene. All patients had hypohidrosis, renal loss of NaCl with secondary hyperaldosteronism and hypokalemia, as well as hypolacrimia, ichthyosis, xerostomia and severe enamel wear. Biological assessment showed plasma Mg2+ levels either high or at the upper limit of reference range and a blunted natriuretic and chloruretic response to furosemide infusion. The watery component of saliva was severely reduced in patients. Heterozygous family members were asymptomatic. Finally, the most recent report to date describes a teenager carrying a biallelic mutation of CLDN10 [36]. The patient had anhidrosis, xerostomia, alacrimia and hypokalemia; he developed hypermagnesemia and CKD over a 4 years follow-up period. The acronym HELIX (Hypohidrosis, Electrolyte disturbances, hypoLacrimia, Ichthyosis, Xerostomia) has been coined to name this syndrome (Online Mendelian Inheritance in Man (OMIM) #617671).
The similarities and differences between the various reports are summarized in Table 1. All patients with the HELIX syndrome have a functional defect of sweat, salivary and lacrimal glands; most of them have blood electrolyte abnormalities, hypermagnesemia and/or hypokalemia; 25% have CKD stage 3 and none has nephrocalcinosis.
3.2. Variants/Pathogenesis
Seven pathogenic, or likely to be pathogenic, nucleotide abnormalities have been reported in patients with HELIX syndrome, so far (Table 2). Three of them are located into exon 1b and affect exclusively the protein CLDN10b; 4 are into exon 2, 3 or 4 and affect both CLDN10a and CLDN10b protein. However, no obvious difference can be seen between the phenotypes of patients bearing only mutated CLDN10b and patients carrying mutated CLDN10a and CLDN10b, which does not help to understand the specific role of the former.
The regular pattern of expression of claudin 10 isoforms in the mammalian kidney is a matter of controversy. In in situ hybridization experiments, Van Itallie and coworkers showed that Cldn10a transcripts were predominantly expressed by cortical tubules, whereas Cldn10b transcripts were predominantly expressed in tubular segments located in the outer medulla [33]. A more detailed pattern was reported by Günzel and collaborators [38]—they reported that Cldn10a transcripts were expressed in the proximal convoluted tubule, cortical thick ascending limb (C-TAL) and cortical collecting duct, contrasting with Cldn10b transcripts tubular expression that was restricted to the medullary thick ascending limb (M-TAL) and inner and outer medullary collecting duct. Hadj-Rabia et al. and Breiderhof et al. reported partially different results—Cldn10a transcripts were exclusively found in convoluted and straight proximal tubule and Cldn10b transcripts in the M- and C-TAL; none was significantly detectable in more distal segments [28,39].
No antibody is available that helps to distinguish between claudin 10a and claudin 10b proteins. Van Itallie et al. reported that Cldn10 was strongly expressed at the tight junction of M- and C-TAL, thin ascending limb and medullary collecting duct and, to a lesser extent, at the tight junction of proximal tubule and cortical collecting duct [33].
Both in vitro (Table 3) and in vivo experiments have been conducted to unravel the function of Cldn 10b.
When heterologously expressed in MDCK-C7 or LLC-PK1 cells, both human and mouse claudin 10b decrease transepithelial resistance [33,38,40]; both human and mouse claudin 10b increase permeability (P) ratios PNa/PCl, PMg/PCl and PCa/PCl in MDCK-C7 cells [38]. By contrast, mouse Cldn10b affects neither transepithelial resistance nor PNa/PCl, when expressed in MDCK-II cells that form a low resistance epithelial layer [33].
Inconsistent results on permeability to Ca2+, Mg2+, Na+ and chloride may result from the endogenous expression of distinct claudins in the cell lines used in experiments. Overexpression of any claudin in a cell line with a claudin background different from that normally co-expressed with the claudin under investigation can induce variable/conflicting interaction and results [42].
Mice (10 weeks old) with a deletion of Cldn10b in the TAL (Cldn10b cKO) show marked hypermagnesemia with low fractional excretion of Mg2+ in urine, mild hyperphosphatemia, mild polyuria with inability to concentrate urine upon drinking water deprivation, elevated fractional excretion of potassium and medullary nephrocalcinosis without overt impairment in GFR [39]. Fractional excretion of Na+ was not consistently elevated in Cldn10b cKO mice. Noteworthy, most of the in vivo effects of Cldn10b deletion, except polyuria and high fractional excretion of potassium, were corrected by simultaneous deletion of Cldn16 (see FHHNC, below) [43].
Several of the mutant proteins have been heterologously expressed in various cell lines. In HEK293 and MDCK-C7 cells, cell surface expression of p.P149R and p.D73N CLDN10b was not decreased, as compared to wild-type protein, whereas that of p.E157_T192del CLDN10b, p.N48K CLDN10b or CLDN10b ∆4 (exon 4 was deleted) was reduced [34,35]. When expressed in MKTAL cells [44], both p.M1? and p.S131L mutated CLDN10b proteins were weakly expressed at cell surface, as compared with wild-type protein [28].
Isolated perfused TALs from Cldn10b cKO mice had a higher transepithelial resistance and a lower paracellular permeability ratio PNa/PCl than TALs from normal littermates [39]. Based on the results described above, a consensual agreement is that the specific effect of claudin 10b is to increase the paracellular permeability to Na+. Actually, a lower paracellular PNa in TAL, sweat glands and salivary glands may account for most of the clinical consequences of CLDN10b loss-of-function mutations. In the M-TAL, fifty per cent of Na+ is passively reabsorbed along the paracellular pathway: a decrease in PNa impairs Na+ reabsorption, causing a renal loss of Na+, extracellular fluid depletion with secondary hyperaldosteronism and a renal loss of potassium; the high transepithelial voltage would elevate passive paracellular Mg2+ and Ca2+ reabsorption in the TAL. In salivary and sweat ducts, where chloride is secreted transcellularly, Na+ is passively secreted along the paracellular pathway. A loss-of-function mutation of CLDN10b would decrease Na+ secretion and the formation of saliva and sweat.
3.3. Prognosis and Treatment
The long-term prognosis is unknown; however, 25% of patients had CKD stage 3 at the time of presentation and close follow-up is advised.
No specific treatment is available to improve the condition of patients with HELIX syndrome. High NaCl and fluid intake is advised, with potassium supplements and drugs blocking aldosterone secretion and/or action when hypokalemia is present. Artificial tears and saliva can be used to relieve symptoms of eye and mouth dryness, respectively. Prolonged intense physical activity should be discouraged, particularly when the outside temperature is high, to prevent the risk of hyperthermia.
4. Claudin 16, Claudin 19 and Familial Hypomagnesemia with Hypercalciuria and Nephrocalcinosis (FHHNC)
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is an autosomal recessive disorder caused by variants of the CLDN16 (OMIM #248250) and CLDN19 (OMIM #248190) genes.
The human CLDN16 gene is located on chromosome 3q27 and contains five exons. Two potential start codons produce either a long (305 amino acids, 33 kDa) or short (235 amino acids, 27 kDa) CLDN16 protein isoform [45,46]. It is unknown which one of the two versions (long or short) is the physiologically relevant or whether both are functional. In vitro, Hou et al. described that the short CLDN16 is expressed at cell-cell borders whereas the long CLDN16 is targeted to endosomes and lysosomes [46]. However, in other studies, full length CLDN16 is expressed at the tight junction [47,48].
The human CLDN19 gene, located on chromosome 1p34.2, contains also fives exons, encoding CLDN19, a protein of 224 amino acids [32].
Because of the autosomal recessive pattern of inheritance, parental consanguinity is frequent [26,49]. The exact prevalence of FHHNC is unknown but it is a very rare disorder, likely to be under-diagnosed.
4.1. Phenotype
FHHNC is characterized by an excessive urinary loss of Ca2+ and Mg2+, resulting in hypomagnesemia and hypercalciuria. Patients have nephrocalcinosis (a parenchymal deposition of calcium–based crystal in the renal parenchyma) and renal failure that may progress toward end stage renal disease early in life [26,45,49,50,51]. Of note, hypomagnesemia can be absent [51,52].
Age at onset ranges from 0 to more to 30 years for CLDN16 [45,49,50,52,53,54,55,56] and CLDN19 mutations [50,51,57,58] and the diagnosis can be delayed.
Patients commonly present with recurrent urinary tract infections, hematuria and abacterial leukocyturia, nephrolithiasis, polyuria/polydipsia with nycturia from infanthood [50,51,52,59,60,61,62] due to impaired urinary concentrating ability [54]. Hypocitraturia is frequently mentioned [45,52,54,55,61,63,64,65,66,67] but urinary acidification capacity has seldom been assessed. A few reports mention distal defect of urinary acidification [45,54,59]: incomplete distal renal tubular acidosis may affect up to 80% of patients [45,66] but it remains unclear whether it is directly caused by CLDN16/19 mutation or by nephrocalcinosis. Hypocitraturia may also be linked to renal failure.
Parathyroid hormone levels seems to be higher than in control patients with chronic renal failure of other origin [49], which may be due to hypomagnesemia-related secondary hyperparathyroidism.
Patients with CLDN19 mutations frequently display severe ocular abnormalities (myopia, pigmentary retinitis, macular coloboma, strabismus, astigmatism, nystagmus, macular scars, macular degeneration, anisocoria, retinochoroiditis) [50,51,57,58,68], contrasting with milder and rarer ocular abnormalities reported with CLDN16 mutations (strabismus, myopia, astigmatism, hypermetropia) [45]. CLDN19 is expressed in human fetal retinal pigment epithelium; claudin 19 may be involved in the development and the function of retinal pigment epithelium and retinal neurogenesis [69,70].
Cldn16 and Cldn19 are also expressed in ameloblast tight junction and loss-of-function mutations of CLDN16 and CLDN19 genes are associated with amelogenesis imperfecta [71,72]. The lack of Cldn16 strongly impairs tight junction organization in secretory stage ameloblasts [71].
An ophthalmological and a dental evaluation are required.
Muscular-exercise intolerance with pain, weakness and electromyographical alterations [67] has been reported with CLDN19 mutation but whether this finding is specific of CLDN19 patients’ needs to be confirmed: hypomagnesemia can lead to non-specific symptoms (muscle weakness and tetany [62]) but Cldn19 is also expressed in murine Schwann cells [73]. Cldn19-deficient mice exhibit behavioral abnormalities that could be due to peripheral neuropathy [73]. Accurate assessment of neuromuscular status in CLDN16 patients has to be performed in order not to miss subtle abnormalities.
4.2. Prognosis and Treatment
The prognosis of FHHNC is poor as it leads frequently to renal failure in childhood. All patients are affected by nephrocalcinosis but the severity of renal failure is variable.
In order to establish a genotype-phenotype correlation, Konrad et al. studied the function of CLDN16 mutants overexpressed in vitro and classified mutants according to a partial or a total loss of function: the age at onset of symptoms was younger and the decline of GFR was faster in patients bearing CLDN16 mutations causing a complete loss of function in both alleles [49].
However, there is a clinical heterogeneity of disease severity in families even among patients bearing the same CLDN16 [45,62,77,78] and CLDN 19 mutation [58].
Patients with CLDN19 mutations progress to chronic kidney disease (defined as an eGFR < 60 mL/min/1.73 m2) earlier than patients with CLDN16 mutations and have poorer renal survival [50].
There is no specific therapy for this syndrome. Patients can be treated by thiazide diuretics in order to reduce urinary Ca2+ excretion, potassium citrate (as citrate is a crystallization inhibitor) and Mg2+ supplements. Additionally, high fluid intake and salt restriction are advised. The effectiveness of these treatments on the course of nephrocalcinosis and on the decline of GFR in FHHNC is unknown. Hydrochlorothiazide has been shown to be effective in correcting hypercalciuria due to CLDN16 mutations in some [52,79] but not all patients [50,51,52]. Mg2+ supplementation might also be ineffective to correct hypomagnesemia [50,51,52]. Hydrochlorothiazide could aggravate hypomagnesemia. Treatment by amiloride can be discussed as potassium-sparing diuretics may have Mg2+-sparing properties [80].
Inhibitors of endocytosis may provide novel therapeutic strategies [81,82]. Blocking clathrin-mediated endocytosis increases surface expression of some CLDN16 mutants in vitro. Primaquine, an antimalarial agent increases also cell surface localization of a CLDN16 mutant in vitro [83].
The only treatment of end-stage renal disease is renal replacement therapy. As the primary defect resides in the kidney, there is no recurrence of FHHNC after kidney transplant.
4.3. Variants/Pathogenesis
To date, sixty-nine CLDN16 disease-causing variants have been described including missense/nonsense variants (53), splice site variants (5), small deletions (5), small insertions (2), small indels (2), gross deletions (1), complex mutation (1) (Table 4).
CLDN16 mutations are located in the two ECSs but also affect the TMDs and the cytoplasmic regions. No link between the affected domain and the phenotype has been described.
The most frequent CLDN16 disease-causing variant, c.453G>T (p.L151F) is found in almost 50% of the German and Eastern European patients described so far [45,52,100].
Twenty-two CLDN19 disease-causing variants have been described so far including missense/nonsense variants (19), small deletions (2), gross deletions (1) (Table 5).
Most patients from Spain or southwestern France with CLDN19 mutation carry the CLDN19 p.(G20D) mutant that may reveal a founder effect [50,51,57,103].
Cldn16 and Cldn19 are expressed at the tight junction of the C-TAL of Henle’s loop in rodents and play a key role in the paracellular transport of Ca2+ and Mg2+. There, 25% of filtered Ca2+ and 70% of filtered Mg2+ are reabsorbed. The selective transport in the TAL of Ca2+ and of Mg2+ depends: (1) on a selective paracellular permeability to Ca2+ and Mg2+ and (2) on a driving force, the lumen-positive transepithelial voltage generated by the active transcellular transport of NaCl and at the end of the C-TAL by NaCl paracellular back flux as the epithelium is more permeable to Na+ than to chloride [107] (Figure 2).
The selective paracellular permeability to Ca2+ and Mg2+ is most likely conferred by the expression of Cldn16 and 19. In the TAL, Cldn3, 10b, 16 and 19 are expressed under basal conditions [127]. However TAL tight junctions show a mosaic expression of either Cldn10b or Cldn3/Cldn16/Cldn19 in the cortex and in the outer stripe of outer medulla (OS) in mice and rat [127] (Figure 3). Cldn16 is virtually absent from the inner stripe of outer medulla (IS) TAL whereas Cldn10b is highly expressed. Cldn19 is expressed in tight junction and detected intracellularly in OS- and C-TAL whereas it is only detected intracellularly in IS-TAL [127,128]. The Cldn16 proportion of total tight junction length in C- and OS-TAL ranges from 37 to 97% [127]. The permeability ratio PMg/PNa is higher in C- and OS-TAL than in IS-TAL [127].
In patients, bi allelic mutations of CLDN16 cause a selective defect in paracellular Mg2+ and Ca2+ reabsorption in the TAL, with intact NaCl reabsorption demonstrated by the normal response to furosemide infusion [54]. However the exact role of claudins 16 and 19 remain a matter of debate. In vitro permeability studies assessing the function of claudin 16 and/or claudin 19 expressed in cell lines have yielded contradicting results. Two hypotheses are been put forward. The first one is that claudin16/claudin19 might function as a divalent cation selective paracellular pore. The second is that claudin16/claudin19 increase PNa/PCl, a prerequisite for the diffusion potential in the C-TAL.
Some investigators found that claudin 16 increases transepithelial transport of Ca2+ [129] and of Mg2+ [83,130,131], when heterogously expressed in epithelial cells (Table 6). Others found an effect of CLDN16 only on Mg2+ (not Ca2+) permeability [48]. Still others found that CLDN16, when expressed in distinct cell lines, elicits only a small increase in transepithelial Mg2+ permeability but a greater increase in Na+ permeability [46,47].
The mechanism by which claudin 19 affects Mg2+ and Ca2+ reabsorption is also unclear according to in vitro heterologous expression studies [47,134] (Table 7).
CLDN 16 and CLDN 19 can interact with each other [47], forming a cis heterodimer at the cell membrane in vitro [135]. Their co expression increases cation selectivity of the tight junction [47]: the effects of CLDN16 and of CLDN19, when co-expressed in LLC-PK1 cells, are synergistic on the PNa/PCl permeability ratio [47]: CLDN19 decreases the permeability to chloride whereas CLDN16 increases the permeability to Na+ (Table 5 and Table 6).
The functional consequences of some human CLDN16 mutations has also been studied by heterologous expression in cells in vitro. Disease-causing mutations can alter intracellular trafficking of claudin 16 or disturb claudin16-claudin 19 interaction or result in a loss of paracellular transport despite its expression at tight junction. Some mutations retain residual function whereas other result in a complete loss of function [46,47,49,81,82,85,96,131]. Similar results have been found with claudin 19 mutants [47,57].
Different mouse models have been engineered to better understand the pathophysiology of FHHNC. Cldn 16 knock-out (KO) model has been engineered a few years ago [136]. Cldn 16 KO mice have hypomagnesemia and hypercalciuria [136]. Lack of Cldn16 decreases PCa/PNa and PMg/PNa in isolated, perfused CTAL whereas PNa/PCl is unaltered [43]. Cldn 16 knockdown (KD) and Cldn 19 KD mice models have been generated by RNA interference technology. Both have hypomagnesemia and significantly increased urinary excretion of Ca2+ without significant renal insufficiency [137,138,139]. NaCl absorption may be impaired as Cldn 16 KD mice have low blood pressure and elevated plasma aldosterone concentration. In isolated, perfused TAL from Cldn 16 KD, PNa/PCl is decreased and PNa/PMg is unaffected [139]. Hou et al. conclude that Cldn16 is a non-specific cation channel [139]. The hypothesis is that the mutated Cldn16 protein has an indirect effect on Mg2+ reabsorption by decreasing PNa/PCl, thereby reducing the diffusion potential/the transepithelial voltage and driving force for Mg2+ reabsorption [138,139]. These results are seemingly in conflict with those from Cldn10b cKO mice (see above). However, Cldn10b disruption also lowers PNa/PCl but affects IS-, OS- and C-TAL. In this model, the high transepithelial voltage may elevate passive paracellular Mg2+ and Ca2+ reabsorption in the C-TAL. Moreover, the mosaic pattern is abrogated and Cldn16 and Cldn19 are expressed in all tight junction in C- and OS- TAL and expression of Cldn 16 expands to IS- TAL, which likely increases the permeability to Mg2+ and Ca2+ [43,127].
Of note, in mice models (Cldn10 KO, Cldn16 KO, Cldn10/Cldn16 double KO and Cldn 16 KD), bi-ionic diffusion potential may have been measured in the presence of high peritubular MgCl2 or CaCl2 concentration that activate the basolateral calcium-sensing receptor and make the interpretation of the data more complex [39,43,139].
Renal abnormalities and electrolyte imbalances have not been investigated in Cldn19 KO mice [73].
Because of the described interaction between Cldn16 and Cldn19, one would have expected a loss of expression of Cldn19 in Cldn16 KO TAL. Surprisingly, the expression of Cldn19 is unaffected in Cldn16 KO mice. The mosaic pattern of expression of either Cldn10b or Cldn3/Cldn19 persisted [127]. In contrast, the majority of Cldn19 tight junctions immunostaining is lost in Cldn16 KD TAL and Cldn16 staining disappeared from tight junctions in Cldn19 KD TAL [137]. Whether the interaction of Cldn16 and Cldn19 is required for their assembly into tight junction is not clear in murine models. Unfortunately, no staining of renal biopsy from FHHNC patient has been performed to our best knowledge.
Renal tissue examination showed Ca2+ deposits along the basement membrane of medullary tubules of Cldn16 KD [139] but not of Cldn16 KO mice. None of the models faithfully recapitulate the human disease, they are not complicated by renal failure [136,139]. The pathogenesis of parenchymal deposition of Ca2+-containing crystal in the kidney (while Ca2+ is less reabsorbed) and of progressive renal failure in FHHNC remains unclear.
5. Claudin 14
The human CLDN14 gene, located on chromosome 21q22.3, contains 1 translated exon, encoding CLDN14, a protein of 239 amino acids [32].
Sixteen CLDN14 disease-causing variants have been described so far including missense/nonsense variants (12), regulatory substitutions (1) small deletions (2), small indels (1) (13 variants class «DM», 2 variants class «DM?», 1 variant class «Disease-associated polymorphisms with supporting functional evidence DFP») [32].
Mutations in CLDN14 cause autosomal recessive non-syndromic deafness-29 (DFNB29, OMIM #614035) [140,141], a phenotype reproduced in Cldn14 KO mice [142]. Heterozygous mutations of CLDN14 have been described in neonates with vein of Galen malformation [143].
No rare variant of the CLDN14 gene has been described in Humans with abnormal renal ion handling. However, claudin 14 may have a role in renal Ca2+ and Mg2+ handling.
Claudin 14 may interact with claudin 16 in TAL and decrease the cation selectivity of the claudin 16-claudin 19 heteromeric complex [144].
The expression of Cldn14 is regulated by changes in dietary Ca2+ [144,145]. Cldn14 is either not detected [144] or detected in few murine OS- and C-TAL with distinct location to the tight junction under control conditions; on a high Ca2+-containing diet, Cldn14 is highly expressed at murine tight junction of OS- and C-TAL but not in IS-TAL [128]. The expression of Cldn14 is also regulated by chronic changes in dietary Mg2+ content in mice and rat [146].
High Ca2+ diet and allosteric agonists of calcium-sensing receptor CaSR may trigger the expression of Cldn14 via the inhibition of the transcription of two microRNAs miR-9 and miR-374 suppressing Cldn14 gene expression [144,145,147].
Cldn14 KO mice have a higher plasma Mg2+ concentration with a lower fractional excretion rate of Mg2+ and of Ca2+ under high Ca2+ dietary condition [144]. On the other hand, overexpression of Cldn14 in TAL generates a phenotype with a lower plasma Mg2+ concentration and a higher fractional excretion rate of Mg2+ and of Ca2+ [147].
When overexpressed in cell culture models, Cldn 14 decreases cation permeability and Ca2+ flux [142,145] (Table 8).
Some single-nucleotide polymorphisms (SNPs) of CLDN14 are associated with 24 h urinary Ca2+ excretion and/or kidney stones [148,149,150,151,152,153]. In silico analysis and in vitro studies of the SNP rs78250838:C> T suggest that it may introduce a novel insulinoma-associated 1 (INSM1) transcription factor binding site, enhancing CLDN14 mRNA and protein expression [151]. Some of these SNPs are also associated with bone mineral density in women [148], serum total CO2 [148], Mg2+ [150], potassium [150] and PTH [148,150]. One the other hand, Corre et al. identified an SNP (rs172639) in a noncoding intergenic region that was associated with the urinary Mg2+ over Ca2+ concentration ratio in spot urine [146]. This SNP is part of a large linkage disequilibrium block spanning the 3’ CLDN14 gene region that contains two microRNA (miR-374 and miR-9) binding sites [144,146].
6. Conclusions
Our knowledge regarding claudins in the mammalian kidney and their critical role in ion homeostasis has considerably expanded within the past two decades. This has largely been driven by the need to understand the clinical consequences and provide care to patients affected by rare syndromes caused by genetic mutations of some claudins. The severity of FHHNC and HELIX syndromes underscores the importance of paracellular transport in ion homeostasis. Despite the progresses made in the past years, many questions are left unanswered. Out of the many claudins expressed along the renal tubule and collecting duct, only three have been unequivocally traced to rare renal clinical syndromes in humans. It is quite possible that as yet unidentified syndromes will be recognized as been caused by rare variants of other claudins in the coming years. Besides, a lot has to be understood regarding the physiology of claudins. For example, we do not know which factors determine which claudin(s) is/are expressed at a given tight junction. This is particularly important in the C-TAL where one given cell can make tight junctions with its neighboring cells that differ in claudin composition. Although we know that the properties of tight junctions and hence that of claudins, are tightly regulated [154], we ignored most of the hormones and mechanisms that are involved in the short- and long-term control of claudin function and expression. Finally, providing a specific and effective treatment to patients with claudin-related rare diseases requires that we are able to target mutant claudin to the right tight junction and/or to correct the function of the mutant. The possibility to provide such treatment seems to be far away and will require substantial efforts. However, given the severity of claudin-related rare disease, it is worth the effort.
Author Contributions
C.P.-B. and P.H. have been equally involved in the drafting and the editing of the manuscript. Both approved the final version. C.P.-B. and P.H. agree to be personally accountable for the author’s own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved and documented in the literature. All authors have read and agreed to the published version of the manuscript.
Funding
C.P.-B. is supported by the Fondation pour la Recherche Médicale (FRM FDT201904007918). This work was supported by grants from Agence Nationale de la Recherche (ANR-12-BSV1-0031-01 and ANR-17-CE14-0032 to P.H.).
Conflicts of Interest
C.P.-B. and P.H. have no conflict of interest to declare regarding the content of the present review.
Abbreviations
| CLDN | human claudin protein |
| CLDN | human claudin gene/mRNA |
| Cldn | rodent claudin protein |
| Cldn | rodent claudin gene/mRNA)) |
References
- Pei, L.; Solis, G.; Nguyen, M.T.; Kamat, N.; Magenheimer, L.; Zhuo, M.; Li, J.; Curry, J.; McDonough, A.A.; Fields, T.A.; et al. Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J. Clin. Investig. 2016, 126, 2509–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, S. Physiological roles of claudins in kidney tubule paracellular transport. Am. J. Physiol. Ren. Physiol. 2017, 312, F9–F24. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.W. Barrier function of epithelia. Am. J. Physiol. 1981, 241, G275–G288. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. The molecular physiology of tight junction pores. Physiology 2004, 19, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Reuss, L. Tight junction permeability to ions and water. In Tight Junctions, 2nd ed.; Cereijido, M., Anderson, J.M., Eds.; CRC Press: Boca Raton, FL, USA, 2001; pp. 61–88. [Google Scholar]
- Knipp, G.T.; Ho, N.F.; Barsuhn, C.L.; Borchardt, R.T. Paracellular diffusion in Caco-2 cell monolayers: Effect of perturbation on the transport of hydrophilic compounds that vary in charge and size. J. Pharm. Sci. 1997, 86, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Rowland, M.; Warhurst, G. Functional modeling of tight junctions in intestinal cell monolayers using polyethylene glycol oligomers. Am. J. Physiol. Cell Physiol. 2001, 281, C388–C397. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, R.; Sugano, K.; Takata, N.; Tachibana, T.; Higashida, A.; Nabuchi, Y.; Aso, Y. Correction of permeability with pore radius of tight junctions in Caco-2 monolayers improves the prediction of the dose fraction of hydrophilic drugs absorbed by humans. Pharm. Res. 2004, 21, 749–755. [Google Scholar] [CrossRef]
- Guo, P.; Weinstein, A.M.; Weinbaum, S. A dual-pathway ultrastructural model for the tight junction of rat proximal tubule epithelium. Am. J. Physiol. Ren. Physiol. 2003, 285, F241–F257. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.S.; Cheng, M.H.; Angelow, S.; Gunzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular basis for cation selectivity in claudin-2-based paracellular pores: Identification of an electrostatic interaction site. J. Gen. Physiol. 2009, 133, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Volkov, A.G.; Paula, S.; Deamer, D.W. Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers. Bioelectrochem. Bioenerg. 1997, 42, 153–160. [Google Scholar] [CrossRef]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Furuse, M.; Fujimoto, K.; Tsukita, S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. USA 1999, 96, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Nishizawa, T.; Tani, K.; Yamazaki, Y.; Tamura, A.; Ishitani, R.; Dohmae, N.; Tsukita, S.; Nureki, O.; Fujiyoshi, Y. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014, 344, 304–307. [Google Scholar] [CrossRef]
- Gong, Y.; Yu, M.; Yang, J.; Gonzales, E.; Perez, R.; Hou, M.; Tripathi, P.; Hering-Smith, K.S.; Hamm, L.L.; Hou, J. The Cap1-claudin-4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2014, 111, E3766–E3774. [Google Scholar] [CrossRef] [Green Version]
- Piontek, J.; Winkler, L.; Wolburg, H.; Muller, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: Determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Tani, K.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J. Mol. Biol. 2015, 427, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberini, G.; Benfenati, F.; Maragliano, L. A refined model of claudin-15 tight junction paracellular architecture by molecular dynamics simulations. PLoS ONE 2017, 12, e0184190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Sasaki, H.; Furuse, M.; Ozaki, H.; Kita, T.; Tsukita, S. Junctional adhesion molecule (JAM) binds to PAR-3: A possible mechanism for the recruitment of PAR-3 to tight junctions. J. Cell Biol. 2001, 154, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Velazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999, 285, 103–106. [Google Scholar] [CrossRef]
- Kirk, A.; Campbell, S.; Bass, P.; Mason, J.; Collins, J. Differential expression of claudin tight junction proteins in the human cortical nephron. Nephrol. Dial. Transplant. 2010, 25, 2107–2119. [Google Scholar] [CrossRef] [Green Version]
- Hadj-Rabia, S.; Brideau, G.; Al-Sarraj, Y.; Maroun, R.C.; Figueres, M.L.; Leclerc-Mercier, S.; Olinger, E.; Baron, S.; Chaussain, C.; Nochy, D.; et al. Multiplex epithelium dysfunction due to CLDN10 mutation: The HELIX syndrome. Genet. Med. 2018, 20, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.M.; Gunzel, D.; Conrad, M.P.; Rosenthal, R.; Fromm, A.; Amasheh, S.; Schulzke, J.D.; Fromm, M. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol. Life Sci. 2012, 69, 2765–2778. [Google Scholar] [CrossRef]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.S. Claudins and the kidney. J. Am. Soc. Nephrol. 2015, 26, 11–19. [Google Scholar] [CrossRef] [Green Version]
- HGMD Professional. 2019. Available online: http://www.hgmd.org/ (accessed on 12 December 2019).
- Van Itallie, C.M.; Rogan, S.; Yu, A.; Vidal, L.S.; Holmes, J.; Anderson, J.M. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am. J. Physiol. Ren. Physiol. 2006, 291, F1288–F1299. [Google Scholar] [CrossRef] [PubMed]
- Bongers, E.; Shelton, L.M.; Milatz, S.; Verkaart, S.; Bech, A.P.; Schoots, J.; Cornelissen, E.A.M.; Bleich, M.; Hoenderop, J.G.J.; Wetzels, J.F.M.; et al. A Novel Hypokalemic-Alkalotic Salt-Losing Tubulopathy in Patients with CLDN10 Mutations. J. Am. Soc. Nephrol. 2017, 28, 3118–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, J.; Piontek, J.; Milatz, S.; Tariq, M.; Jameel, M.; Breiderhoff, T.; Schuster, J.; Fatima, A.; Asif, M.; Sher, M.; et al. Altered paracellular cation permeability due to a rare CLDN10B variant causes anhidrosis and kidney damage. PLoS Genet. 2017, 13, e1006897. [Google Scholar] [CrossRef] [PubMed]
- Meyers, N.; Nelson-Williams, C.; Malaga-Dieguez, L.; Kaufmann, H.; Loring, E.; Knight, J.; Lifton, R.P.; Trachtman, H. Hypokalemia Associated With a Claudin 10 Mutation: A Case Report. Am. J. Kidney Dis. 2019, 73, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Gunzel, D.; Stuiver, M.; Kausalya, P.J.; Haisch, L.; Krug, S.M.; Rosenthal, R.; Meij, I.C.; Hunziker, W.; Fromm, M.; Muller, D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci. 2009, 122, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Breiderhoff, T.; Himmerkus, N.; Stuiver, M.; Mutig, K.; Will, C.; Meij, I.C.; Bachmann, S.; Bleich, M.; Willnow, T.E.; Muller, D. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14241–14246. [Google Scholar] [CrossRef] [Green Version]
- Milatz, S.; Piontek, J.; Hempel, C.; Meoli, L.; Grohe, C.; Fromm, A.; Lee, I.M.; El-Athman, R.; Gunzel, D. Tight junction strand formation by claudin-10 isoforms and claudin-10a/-10b chimeras. Ann. N Y Acad. Sci. 2017, 1405, 102–115. [Google Scholar] [CrossRef]
- Gunzel, D.; Yu, A.S. Function and regulation of claudins in the thick ascending limb of Henle. Pflug. Arch. 2009, 458, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Angelow, S.; Schneeberger, E.E.; Yu, A.S. Claudin-8 expression in renal epithelial cells augments the paracellular barrier by replacing endogenous claudin-2. J. Membr. Biol. 2007, 215, 147–159. [Google Scholar] [CrossRef]
- Breiderhoff, T.; Himmerkus, N.; Drewell, H.; Plain, A.; Gunzel, D.; Mutig, K.; Willnow, T.E.; Muller, D.; Bleich, M. Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int. 2018, 93, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, S.; Rossignol, P.; Grelac, F.; Chalumeau, C.; Klein, C.; Laghmani, K.; Chambrey, R.; Bruneval, P.; Duong, J.P.; Poggioli, J.; et al. Differentiated thick ascending limb (TAL) cultured cells derived from SV40 transgenic mice express functional apical NHE2 isoform: Effect of nitric oxide. Pflug. Arch. 2003, 446, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Schneider, L.; Peters, M.; Misselwitz, J.; Ronnefarth, G.; Boswald, M.; Bonzel, K.E.; Seeman, T.; Sulakova, T.; Kuwertz-Broking, E.; et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol. 2001, 12, 1872–1881. [Google Scholar] [PubMed]
- Hou, J.; Paul, D.L.; Goodenough, D.A. Paracellin-1 and the modulation of ion selectivity of tight junctions. J. Cell Sci. 2005, 118, 5109–5118. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Renigunta, A.; Konrad, M.; Gomes, A.S.; Schneeberger, E.E.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J. Clin. Investig. 2008, 118, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Gunzel, D.; Amasheh, S.; Pfaffenbach, S.; Richter, J.F.; Kausalya, P.J.; Hunziker, W.; Fromm, M. Claudin-16 affects transcellular Cl-secretion in MDCK cells. J. Physiol. 2009, 587, 3777–3793. [Google Scholar] [CrossRef]
- Konrad, M.; Hou, J.; Weber, S.; Dotsch, J.; Kari, J.A.; Seeman, T.; Kuwertz-Broking, E.; Peco-Antic, A.; Tasic, V.; Dittrich, K.; et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol. 2008, 19, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Godron, A.; Harambat, J.; Boccio, V.; Mensire, A.; May, A.; Rigothier, C.; Couzi, L.; Barrou, B.; Godin, M.; Chauveau, D.; et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin. J. Am. Soc. Nephrol. 2012, 7, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Claverie-Martin, F.; Garcia-Nieto, V.; Loris, C.; Ariceta, G.; Nadal, I.; Espinosa, L.; Fernandez-Maseda, A.; Anton-Gamero, M.; Avila, A.; Madrid, A.; et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS ONE 2013, 8, e53151. [Google Scholar] [CrossRef]
- Sikora, P.; Zaniew, M.; Haisch, L.; Pulcer, B.; Szczepanska, M.; Moczulska, A.; Rogowska-Kalisz, A.; Bienias, B.; Tkaczyk, M.; Ostalska-Nowicka, D.; et al. Retrospective cohort study of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis due to CLDN16 mutations. Nephrol. Dial. Transplant. 2015, 30, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Alparslan, C.; Oncel, E.P.; Akbay, S.; Alaygut, D.; Mutlubas, F.; Tatli, M.; Konrad, M.; Yavascan, O.; Kasap-Demir, B. A novel homozygous W99G mutation in CLDN-16 gene causing familial hypomagnesemic hypercalciuric nephrocalcinosis in Turkish siblings. Turk. J. Pediatrics 2018, 60, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Jeunemaitre, X.; Coudol, P.; Dechaux, M.; Froissart, M.; May, A.; Demontis, R.; Fournier, A.; Paillard, M.; Houillier, P. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int. 2001, 59, 2206–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, A.; Abood, S.A.; Simon, J.; Dastoor, H.; Pearce, S.H.; Sayer, J.A. A novel CLDN16 mutation in a large family with familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. BMC Res. Notes 2013, 6, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zhao, X.; Paiardini, A.; Lang, Y.; Bottillo, I.; Shao, L. Familial hypomagnesaemia, Hypercalciuria and Nephrocalcinosis associated with a novel mutation of the highly conserved leucine residue 116 of Claudin 16 in a Chinese patient with a delayed diagnosis: A case report. BMC Nephrol. 2018, 19, 181. [Google Scholar] [CrossRef]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, M.E.; Hunziker, W.; Teo, A.S.; Hillmer, A.M.; Mutchinick, O.M. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Variable phenotypic expression in three affected sisters from Mexican ancestry. Ren. Fail. 2015, 37, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Hampson, G.; Konrad, M.A.; Scoble, J. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC): Compound heterozygous mutation in the claudin 16 (CLDN16) gene. BMC Nephrol. 2008, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Hanssen, O.; Castermans, E.; Bovy, C.; Weekers, L.; Erpicum, P.; Dubois, B.; Bours, V.; Krzesinski, J.M.; Jouret, F. Two novel mutations of the CLDN16 gene cause familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. Clin. Kidney J. 2014, 7, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Choi, H.J.; Cho, H.Y.; Lee, J.H.; Ha, I.S.; Cheong, H.I.; Choi, Y. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis associated with CLDN16 mutations. Pediatrics Nephrol. 2005, 20, 1490–1493. [Google Scholar] [CrossRef]
- Lv, F.; Xu, X.J.; Wang, J.Y.; Liu, Y.; Jiang, Y.; Wang, O.; Xia, W.B.; Xing, X.P.; Li, M. A novel mutation in CLDN16 results in rare familial hypomagnesaemia with hypercalciuria and nephrocalcinosis in a Chinese family. Clin. Chim. Acta 2016, 457, 69–74. [Google Scholar] [CrossRef]
- Perdomo-Ramirez, A.; Aguirre, M.; Davitaia, T.; Ariceta, G.; Ramos-Trujillo, E.; RenalTube, G.; Claverie-Martin, F. Characterization of two novel mutations in the claudin-16 and claudin-19 genes that cause familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Gene 2019, 689, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Poussou, R.; Cochat, P.; Le Pottier, N.; Roncelin, I.; Liutkus, A.; Blanchard, A.; Jeunemaitre, X. Report of a family with two different hereditary diseases leading to early nephrocalcinosis. Pediatrics Nephrol. 2008, 23, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Vianna, J.G.P.; Simor, T.G.; Senna, P.; De Bortoli, M.R.; Costalonga, E.F.; Seguro, A.C.; Luchi, W.M. Atypical presentation of familial hypomagnesemia with hypercalciuria and nephrocalcinosis in a patient with a new claudin-16 gene mutation. Clin. Nephrol. Case Stud. 2019, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Hoffmann, K.; Jeck, N.; Saar, K.; Boeswald, M.; Kuwertz-Broeking, E.; Meij, I.I.; Knoers, N.V.; Cochat, P.; Sulakova, T.; et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene. Eur. J. Hum. Genet. 2000, 8, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Faguer, S.; Chauveau, D.; Cintas, P.; Tack, I.; Cointault, O.; Rostaing, L.; Vargas-Poussou, R.; Ribes, D. Renal, ocular, and neuromuscular involvements in patients with CLDN19 mutations. Clin. J. Am. Soc. Nephrol. 2011, 6, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Al-Shibli, A.; Konrad, M.; Altay, W.; Al Masri, O.; Al-Gazali, L.; Al Attrach, I. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC): Report of three cases with a novel mutation in CLDN19 gene. Saudi J. Kidney Dis. Transplant. 2013, 24, 338–344. [Google Scholar] [CrossRef]
- Peng, S.; Rao, V.S.; Adelman, R.A.; Rizzolo, L.J. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1392–1403. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Xu, T.; Peng, S.; Singh, D.; Ghiassi-Nejad, M.; Adelman, R.A.; Rizzolo, L.J. Disease-associated mutations of claudin-19 disrupt retinal neurogenesis and visual function. Commun. Biol. 2019, 2, 113. [Google Scholar] [CrossRef]
- Bardet, C.; Courson, F.; Wu, Y.; Khaddam, M.; Salmon, B.; Ribes, S.; Thumfart, J.; Yamaguti, P.M.; Rochefort, G.Y.; Figueres, M.L.; et al. Claudin-16 Deficiency Impairs Tight Junction Function in Ameloblasts, Leading to Abnormal Enamel Formation. J. Bone Miner. Res. 2016, 31, 498–513. [Google Scholar] [CrossRef] [Green Version]
- Yamaguti, P.M.; Neves, F.A.; Hotton, D.; Bardet, C.; de La Dure-Molla, M.; Castro, L.C.; Scher, M.D.; Barbosa, M.E.; Ditsch, C.; Fricain, J.C.; et al. Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations. J. Med. Genet. 2017, 54, 26–37. [Google Scholar] [CrossRef]
- Miyamoto, T.; Morita, K.; Takemoto, D.; Takeuchi, K.; Kitano, Y.; Miyakawa, T.; Nakayama, K.; Okamura, Y.; Sasaki, H.; Miyachi, Y.; et al. Tight junctions in Schwann cells of peripheral myelinated axons: A lesson from claudin-19-deficient mice. J. Cell Biol. 2005, 169, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, L.; Khosravi, M.; Dumitriu, S.; Klootwijk, E.; Kleta, R.; Yaqoob, M.M.; Walsh, S.B. A novel claudin-16 mutation, severe bone disease, and nephrocalcinosis. Lancet 2014, 383, 98. [Google Scholar] [CrossRef]
- Naeem, M.; Hussain, S.; Akhtar, N. Mutation in the tight-junction gene claudin 19 (CLDN19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (FHHNC) and severe ocular disease. Am. J. Nephrol. 2011, 34, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Pang, Q.; Xing, X.; Wang, X.; Li, Y.; Li, J.; Wu, X.; Li, M.; Wang, O.; Jiang, Y.; et al. First report of a novel missense CLDN19 mutations causing familial hypomagnesemia with hypercalciuria and nephrocalcinosis in a Chinese family. Calcif. Tissue Int. 2015, 96, 265–273. [Google Scholar] [CrossRef]
- Sanjad, S.A.; Hariri, A.; Habbal, Z.M.; Lifton, R.P. A novel PCLN-1 gene mutation in familial hypomagnesemia with hypercalciuria and atypical phenotype. Pediatrics Nephrol. 2007, 22, 503–508. [Google Scholar] [CrossRef]
- Seeley, H.H.; Loomba-Albrecht, L.A.; Nagel, M.; Butani, L.; Bremer, A.A. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis in three siblings having the same genetic lesion but different clinical presentations. World J. Pediatrics 2012, 8, 177–180. [Google Scholar] [CrossRef]
- Zimmermann, B.; Plank, C.; Konrad, M.; Stohr, W.; Gravou-Apostolatou, C.; Rascher, W.; Dotsch, J. Hydrochlorothiazide in CLDN16 mutation. Nephrol. Dial. Transplant. 2006, 21, 2127–2132. [Google Scholar] [CrossRef] [Green Version]
- Alexander, R.T.; Dimke, H. Effect of diuretics on renal tubular transport of calcium and magnesium. Am. J. Physiol. Ren. Physiol. 2017, 312, F998–F1015. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.; Kausalya, P.J.; Bockenhauer, D.; Thumfart, J.; Meij, I.C.; Dillon, M.J.; van’t Hoff, W.; Hunziker, W. Unusual clinical presentation and possible rescue of a novel claudin-16 mutation. J. Clin. Endocrinol. Metab. 2006, 91, 3076–3079. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.; Kausalya, P.J.; Meij, I.C.; Hunziker, W. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Blocking endocytosis restores surface expression of a novel Claudin-16 mutant that lacks the entire C-terminal cytosolic tail. Hum. Mol. Genet. 2006, 15, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Marunaka, K.; Fujii, N.; Kimura, T.; Furuta, T.; Hasegawa, H.; Matsunaga, T.; Endo, S.; Ikari, A. Rescue of tight junctional localization of a claudin-16 mutant D97S by antimalarial medicine primaquine in Madin-Darby canine kidney cells. Sci. Rep. 2019, 9, 9647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillano, D.; Bertoli-Avella, A.M.; Kumar Kandaswamy, K.; Weiss, M.E.; Koster, J.; Marais, A.; Paknia, O.; Schroder, R.; Garcia-Aznar, J.M.; Werber, M.; et al. Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 2017, 25, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guran, T.; Akcay, T.; Bereket, A.; Atay, Z.; Turan, S.; Haisch, L.; Konrad, M.; Schlingmann, K.P. Clinical and molecular characterization of Turkish patients with familial hypomagnesaemia: Novel mutations in TRPM6 and CLDN16 genes. Nephrol. Dial. Transplant. 2012, 27, 667–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdomo-Ramirez, A.; de Armas-Ortiz, M.; Ramos-Trujillo, E.; Suarez-Artiles, L.; Claverie-Martin, F. Exonic CLDN16 mutations associated with familial hypomagnesemia with hypercalciuria and nephrocalcinosis can induce deleterious mRNA alterations. BMC Med. Genet. 2019, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016, 89, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Staiger, K.; Staiger, H.; Haas, C.; Thamer, C.; Risler, T.; Machicao, F.; Haring, H.U. Hypomagnesemia and nephrocalcinosis in a patient with two heterozygous mutations in the CLDN16 gene. J. Nephrol. 2007, 20, 107–110. [Google Scholar] [PubMed]
- Tajima, T.; Nakae, J.; Fujieda, K. Two heterozygous mutations of CLDN16 in a Japanese patient with FHHNC. Pediatrics Nephrol. 2003, 18, 1280–1282. [Google Scholar] [CrossRef]
- Yavarna, T.; Al-Dewik, N.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; Lakhani, S.; AlMulla, M.; Nawaz, Z.; et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 2015, 134, 967–980. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Tumer, L.; Okur, I.; Hasanoglu, A. A novel mutation of the claudin 16 gene in familial hypomagnesemia with hypercalciuria and nephrocalcinosis mimicking rickets. Genet. Couns. 2011, 22, 187–192. [Google Scholar]
- Margabandhu, S.; Doshi, M. Familial Hypomagnesemia, Hypercalciuria and Nephrocalcinosis with Novel Mutation. Indian J. Nephrol. 2019, 29, 57–61. [Google Scholar] [CrossRef]
- Kutluturk, F.; Temel, B.; Uslu, B.; Aral, F.; Azezli, A.; Orhan, Y.; Konrad, M.; Ozbey, N. An unusual patient with hypercalciuria, recurrent nephrolithiasis, hypomagnesemia and G227R mutation of Paracellin-1. An unusual patient with hypercalciuria and hypomagnesemia unresponsive to thiazide diuretics. Horm. Res. 2006, 66, 175–181. [Google Scholar] [PubMed]
- Daga, A.; Majmundar, A.J.; Braun, D.A.; Gee, H.Y.; Lawson, J.A.; Shril, S.; Jobst-Schwan, T.; Vivante, A.; Schapiro, D.; Tan, W.; et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018, 93, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkmen, M.; Kasap, B.; Soylu, A.; Bober, E.; Konrad, M.; Kavukcu, S. Paracellin-1 gene mutation with multiple congenital abnormalities. Pediatrics Nephrol. 2006, 21, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Kausalya, P.J.; Claverie-Martin, F.; Meij, I.C.; Eggert, P.; Garcia-Nieto, V.; Hunziker, W. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am. J. Hum. Genet. 2003, 73, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Mao, J.; Ye, Q.; Zhu, X.; Zhang, Y.; Ye, Y.; Fu, H.; Shen, H.; Lu, Z.; Xia, Y.; et al. Clinical features and genetic findings in Chinese children with distal renal tubular acidosis. Int. J. Clin. Exp. Pathol. 2018, 11, 3523–3532. [Google Scholar]
- Yamaguti, P.M.; dos Santos, P.A.; Leal, B.S.; Santana, V.B.; Mazzeu, J.F.; Acevedo, A.C.; Neves Fde, A. Identification of the first large deletion in the CLDN16 gene in a patient with FHHNC and late-onset of chronic kidney disease: Case report. BMC Nephrol. 2015, 16, 92. [Google Scholar] [CrossRef] [Green Version]
- Uniprot. Available online: https://www.uniprot.org (accessed on 20 December 2019).
- Claverie-Martin, F.; Vargas-Poussou, R.; Muller, D.; Garcia-Nieto, V. Clinical utility gene card for: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis with/without severe ocular involvement. Eur. J. Hum. Genet. 2015, 23. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.R.; Machado Gde, A.; dos Santos, M.M.; Lopes Pde, F.; de Matos, J.P.; Neves, A.C.; Lugon, J.R. Five years results after intrafamilial kidney post-transplant in a case of familial hypomagnesemia due to a claudin-19 mutation. J. Bras. Nefrol. 2014, 36, 401–405. [Google Scholar] [CrossRef]
- Haisch, L.; Almeida, J.R.; Abreu da Silva, P.R.; Schlingmann, K.P.; Konrad, M. The role of tight junctions in paracellular ion transport in the renal tubule: Lessons learned from a rare inherited tubular disorder. Am. J. Kidney Dis. 2011, 57, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Martin-Nunez, E.; Cordoba-Lanus, E.; Gonzalez-Acosta, H.; Oliet, A.; Izquierdo, E.; Claverie-Martin, F. Haplotype analysis of CLDN19 single nucleotide polymorphisms in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. World J. Pediatrics 2015, 11, 272–275. [Google Scholar] [CrossRef]
- Khan, A.O.; Patel, N.; Ghazi, N.G.; Alzahrani, S.S.; Arold, S.T.; Alkuraya, F.S. Familial non-syndromic macular pseudocoloboma secondary to homozygous CLDN19 mutation. Ophthalmic Genet. 2018, 39, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, Z.; Karabas, L.; Konrad, M. Hypomagnesemia-hypercalciuria-nephrocalcinosis and ocular findings: A new claudin-19 mutation. Turk. J. Pediatrics 2012, 54, 168–170. [Google Scholar]
- Sharma, S.; Place, E.; Lord, K.; Leroy, B.P.; Falk, M.J.; Pradhan, M. Claudin 19-based familial hypomagnesemia with hypercalciuria and nephrocalcinosis in a sibling pair. Clin. Nephrol. 2016, 85, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Dimke, H.; Schnermann, J. Axial and cellular heterogeneity in electrolyte transport pathways along the thick ascending limb. Acta Physiol. 2018, 223, e13057. [Google Scholar] [CrossRef] [PubMed]
- Greger, R. Chloride reabsorption in the rabbit cortical thick ascending limb of the loop of Henle. A sodium dependent process. Pflug. Arch. 1981, 390, 38–43. [Google Scholar] [CrossRef]
- Greger, R.; Schlatter, E. Properties of the basolateral membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. A model for secondary active chloride transport. Pflug. Arch. 1983, 396, 325–334. [Google Scholar] [CrossRef]
- Greger, R.; Schlatter, E. Properties of the lumen membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflug. Arch. 1983, 396, 315–324. [Google Scholar] [CrossRef]
- Carney, S.L.; Wong, N.L.; Quamme, G.A.; Dirks, J.H. Effect of magnesium deficiency on renal magnesium and calcium transport in the rat. J. Clin. Investig. 1980, 65, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Le Grimellec, C.; Roinel, N.; Morel, F. Simultaneous Mg, Ca, P,K,Na and Cl analysis in rat tubular fluid. I. During perfusion of either inulin or ferrocyanide. Pflug. Arch. 1973, 340, 181–196. [Google Scholar] [CrossRef]
- Costanzo, L.S.; Windhager, E.E. Calcium and sodium transport by the distal convoluted tubule of the rat. Am. J. Physiol. 1978, 235, F492–F506. [Google Scholar] [CrossRef]
- Edwards, B.R.; Baer, P.G.; Sutton, R.A.; Dirks, J.H. Micropuncture study of diuretic effects on sodium and calcium reabsorption in the dog nephron. J. Clin. Investig. 1973, 52, 2418–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpe, K.O.; Lowitz, H.D.; Ochwadt, B. Fluid reabsorption in Henle’sloop and urinary excretion of sodium and water in normal rats and rats with chronic hypertension. J. Clin. Investig. 1970, 49, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10, 2569–2576. [Google Scholar] [PubMed]
- Vallon, V.; Osswald, H.; Blantz, R.C.; Thomson, S. Potential role of luminal potassium in tubuloglomerular feedback. J. Am. Soc. Nephrol. 1997, 8, 1831–1837. [Google Scholar]
- Schnermann, J.; Briggs, J.; Schubert, G. In situ studies of the distal convoluted tubule in the rat. I. Evidence for NaCl secretion. Am. J. Physiol. 1982, 243, F160–F166. [Google Scholar] [CrossRef]
- Luke, R.G.; Wright, F.S.; Fowler, N.; Kashgarian, M.; Giebisch, G.H. Effects of potassium depletion on renal tubular chloride transport in the rat. Kidney Int. 1978, 14, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.; Castrop, H.; Laghmani, K.; Vallon, V.; Layton, A.T. Effects of NKCC2 isoform regulation on NaCl transport in thick ascending limb and macula densa: A modeling study. Am. J. Physiol. Ren. Physiol. 2014, 307, F137–F146. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A. Regulation of calcium reabsorption along the rat nephron: A modeling study. Am. J. Physiol. Ren. Physiol. 2015, 308, F553–F566. [Google Scholar] [CrossRef]
- Layton, A.T.; Vallon, V.; Edwards, A. A computational model for simulating solute transport and oxygen consumption along the nephrons. Am. J. Physiol. Ren. Physiol. 2016, 311, F1378–F1390. [Google Scholar] [CrossRef]
- Tournus, M.; Seguin, N.; Perthame, B.; Thomas, S.R.; Edwards, A. A model of calcium transport along the rat nephron. Am. J. Physiol. Ren. Physiol. 2013, 305, F979–F994. [Google Scholar] [CrossRef] [Green Version]
- Nieves-Gonzalez, A.; Clausen, C.; Layton, A.T.; Layton, H.E.; Moore, L.C. Transport efficiency and workload distribution in a mathematical model of the thick ascending limb. Am. J. Physiol. Ren. Physiol. 2013, 304, F653–F664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, A.M. A mathematical model of rat ascending Henle limb. III. Tubular function. Am. J. Physiol Ren. Physiol 2010, 298, F543–F556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, A.M. A mathematical model of rat proximal tubule and loop of Henle. Am. J. Physiol. Ren. Physiol. 2015, 308, F1076–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milatz, S.; Himmerkus, N.; Wulfmeyer, V.C.; Drewell, H.; Mutig, K.; Hou, J.; Breiderhoff, T.; Muller, D.; Fromm, M.; Bleich, M.; et al. Mosaic expression of claudins in thick ascending limbs of Henle results in spatial separation of paracellular Na+ and Mg2+ transport. Proc. Natl. Acad. Sci. USA 2017, 114, E219–E227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plain, A.; Wulfmeyer, V.C.; Milatz, S.; Klietz, A.; Hou, J.; Bleich, M.; Himmerkus, N. Corticomedullary difference in the effects of dietary Ca2+ on tight junction properties in thick ascending limbs of Henle’s loop. Pflug. Arch. 2016, 468, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Hirai, N.; Shiroma, M.; Harada, H.; Sakai, H.; Hayashi, H.; Suzuki, Y.; Degawa, M.; Takagi, K. Association of paracellin-1 with ZO-1 augments the reabsorption of divalent cations in renal epithelial cells. J. Biol. Chem. 2004, 279, 54826–54832. [Google Scholar] [CrossRef] [Green Version]
- Ikari, A.; Matsumoto, S.; Harada, H.; Takagi, K.; Hayashi, H.; Suzuki, Y.; Degawa, M.; Miwa, M. Phosphorylation of paracellin-1 at Ser217 by protein kinase A is essential for localization in tight junctions. J. Cell Sci. 2006, 119, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Kausalya, P.J.; Amasheh, S.; Gunzel, D.; Wurps, H.; Muller, D.; Fromm, M.; Hunziker, W. Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J. Clin. Investig. 2006, 116, 878–891. [Google Scholar] [CrossRef]
- Ikari, A.; Kinjo, K.; Atomi, K.; Sasaki, Y.; Yamazaki, Y.; Sugatani, J. Extracellular Mg(2+) regulates the tight junctional localization of claudin-16 mediated by ERK-dependent phosphorylation. Biochim. Biophys. Acta 2010, 1798, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Tang, V.W.; Goodenough, D.A. Paracellular ion channel at the tight junction. Biophys. J. 2003, 84, 1660–1673. [Google Scholar] [CrossRef] [Green Version]
- Angelow, S.; El-Husseini, R.; Kanzawa, S.A.; Yu, A.S. Renal localization and function of the tight junction protein, claudin-19. Am. J. Physiol. Ren. Physiol. 2007, 293, F166–F177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Renigunta, V.; Zhou, Y.; Sunq, A.; Wang, J.; Yang, J.; Renigunta, A.; Baker, L.A.; Hou, J. Biochemical and biophysical analyses of tight junction permeability made of claudin-16 and claudin-19 dimerization. Mol. Biol. Cell 2015, 26, 4333–4346. [Google Scholar] [CrossRef] [PubMed]
- Will, C.; Breiderhoff, T.; Thumfart, J.; Stuiver, M.; Kopplin, K.; Sommer, K.; Gunzel, D.; Querfeld, U.; Meij, I.C.; Shan, Q.; et al. Targeted deletion of murine Cldn16 identifies extra- and intrarenal compensatory mechanisms of Ca2+ and Mg2+ wasting. Am. J. Physiol. Ren. Physiol. 2010, 298, F1152–F1161. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Renigunta, A.; Gomes, A.S.; Hou, M.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc. Natl. Acad. Sci. USA 2009, 106, 15350–15355. [Google Scholar] [CrossRef] [Green Version]
- Himmerkus, N.; Shan, Q.; Goerke, B.; Hou, J.; Goodenough, D.A.; Bleich, M. Salt and acid-base metabolism in claudin-16 knockdown mice: Impact for the pathophysiology of FHHNC patients. Am. J. Physiol. Ren. Physiol. 2008, 295, F1641–F1647. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Shan, Q.; Wang, T.; Gomes, A.S.; Yan, Q.; Paul, D.L.; Bleich, M.; Goodenough, D.A. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J. Biol. Chem. 2007, 282, 17114–17122. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, E.R.; Burton, Q.L.; Naz, S.; Riazuddin, S.; Smith, T.N.; Ploplis, B.; Belyantseva, I.; Ben-Yosef, T.; Liburd, N.A.; Morell, R.J.; et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 2001, 104, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Bashir, Z.E.; Latief, N.; Belyantseva, I.A.; Iqbal, F.; Riazuddin, S.A.; Khan, S.N.; Friedman, T.B.; Riazuddin, S.; Riazuddin, S. Phenotypic variability of CLDN14 mutations causing DFNB29 hearing loss in the Pakistani population. J. Hum. Genet. 2013, 58, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yosef, T.; Belyantseva, I.A.; Saunders, T.L.; Hughes, E.D.; Kawamoto, K.; Van Itallie, C.M.; Beyer, L.A.; Halsey, K.; Gardner, D.J.; Wilcox, E.R.; et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum. Mol. Genet. 2003, 12, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Duran, D.; Zeng, X.; Jin, S.C.; Choi, J.; Nelson-Williams, C.; Yatsula, B.; Gaillard, J.; Furey, C.G.; Lu, Q.; Timberlake, A.T.; et al. Mutations in Chromatin Modifier and Ephrin Signaling Genes in Vein of Galen Malformation. Neuron 2019, 101, 429–443 e424. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Renigunta, V.; Himmerkus, N.; Zhang, J.; Renigunta, A.; Bleich, M.; Hou, J. Claudin-14 regulates renal Ca(+)(+) transport in response to CaSR signalling via a novel microRNA pathway. EMBO J. 2012, 31, 1999–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimke, H.; Desai, P.; Borovac, J.; Lau, A.; Pan, W.; Alexander, R.T. Activation of the Ca2+-sensing receptor increases renal claudin-14 expression and urinary Ca2+ excretion. Am. J. Physiol. Ren. Physiol. 2013, 304, F761–F769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, T.; Olinger, E.; Harris, S.E.; Traglia, M.; Ulivi, S.; Lenarduzzi, S.; Belge, H.; Youhanna, S.; Tokonami, N.; Bonny, O.; et al. Common variants in CLDN14 are associated with differential excretion of magnesium over calcium in urine. Pflugers Arch. 2017, 469, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Hou, J. Claudin-14 underlies Ca(+)(+)-sensing receptor-mediated Ca(+)(+) metabolism via NFAT-microRNA-based mechanisms. J. Am. Soc. Nephrol. 2014, 25, 745–760. [Google Scholar] [CrossRef] [Green Version]
- Thorleifsson, G.; Holm, H.; Edvardsson, V.; Walters, G.B.; Styrkarsdottir, U.; Gudbjartsson, D.F.; Sulem, P.; Halldorsson, B.V.; de Vegt, F.; d’Ancona, F.C.; et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat. Genet. 2009, 41, 926–930. [Google Scholar] [CrossRef]
- Guha, M.; Bankura, B.; Ghosh, S.; Pattanayak, A.K.; Ghosh, S.; Pal, D.K.; Puri, A.; Kundu, A.K.; Das, M. Polymorphisms in CaSR and CLDN14 Genes Associated with Increased Risk of Kidney Stone Disease in Patients from the Eastern Part of India. PLoS ONE 2015, 10, e0130790. [Google Scholar] [CrossRef] [Green Version]
- Oddsson, A.; Sulem, P.; Helgason, H.; Edvardsson, V.O.; Thorleifsson, G.; Sveinbjornsson, G.; Haraldsdottir, E.; Eyjolfsson, G.I.; Sigurdardottir, O.; Olafsson, I.; et al. Common and rare variants associated with kidney stones and biochemical traits. Nat. Commun. 2015, 6, 7975. [Google Scholar] [CrossRef] [Green Version]
- Ure, M.E.; Heydari, E.; Pan, W.; Ramesh, A.; Rehman, S.; Morgan, C.; Pinsk, M.; Erickson, R.; Herrmann, J.M.; Dimke, H.; et al. A variant in a cis-regulatory element enhances claudin-14 expression and is associated with pediatric-onset hypercalciuria and kidney stones. Hum. Mutat. 2017, 38, 649–657. [Google Scholar] [CrossRef]
- Toka, H.R.; Genovese, G.; Mount, D.B.; Pollak, M.R.; Curhan, G.C. Frequency of rare allelic variation in candidate genes among individuals with low and high urinary calcium excretion. PLoS ONE 2013, 8, e71885. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, T.; Simonini, M.; Lanzani, C.; Citterio, L.; Salvi, E.; Barlassina, C.; Spotti, D.; Cusi, D.; Manunta, P.; Vezzoli, G. Claudin-14 Gene Polymorphisms and Urine Calcium Excretion. Clin. J. Am. Soc. Nephrol. 2018, 13, 1542–1549. [Google Scholar] [CrossRef] [Green Version]
- Loupy, A.; Ramakrishnan, S.K.; Wootla, B.; Chambrey, R.; de la Faille, R.; Bourgeois, S.; Bruneval, P.; Mandet, C.; Christensen, E.I.; Faure, H.; et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J. Clin. Investig. 2012, 122, 3355–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Pattern of expression of claudin (CLDN) proteins along the human renal tubule and collecting duct. The figure is a summary of data published in References [26,27,28,29] (the expression pattern in rodent kidney has recently been summarized in several reviews [2,30,31]). Question mark means that direct evidence of the expression of the considered claudin in human is not available but that indirect evidence suggests that it should be expressed there. Brackets mean that the expression is low. The numbers above the various segments are: 1, proximal convoluted and straight tubule; 2, descending thin limb; 3, ascending thin limb; 4, distal straight tubule (thick ascending limb); 5, distal convoluted tubule; 6, cortical and outer medullary collecting duct. Colors illustrate the fact that composition and/or volume of tubular fluid changes along the renal tubule. Readers interested in mRNA expression pattern can visit https://hpcwebapps.cit.nih.gov/ESBL/Database/NephronRNAseq/All_transcripts.html.
Figure 1.
Pattern of expression of claudin (CLDN) proteins along the human renal tubule and collecting duct. The figure is a summary of data published in References [26,27,28,29] (the expression pattern in rodent kidney has recently been summarized in several reviews [2,30,31]). Question mark means that direct evidence of the expression of the considered claudin in human is not available but that indirect evidence suggests that it should be expressed there. Brackets mean that the expression is low. The numbers above the various segments are: 1, proximal convoluted and straight tubule; 2, descending thin limb; 3, ascending thin limb; 4, distal straight tubule (thick ascending limb); 5, distal convoluted tubule; 6, cortical and outer medullary collecting duct. Colors illustrate the fact that composition and/or volume of tubular fluid changes along the renal tubule. Readers interested in mRNA expression pattern can visit https://hpcwebapps.cit.nih.gov/ESBL/Database/NephronRNAseq/All_transcripts.html.
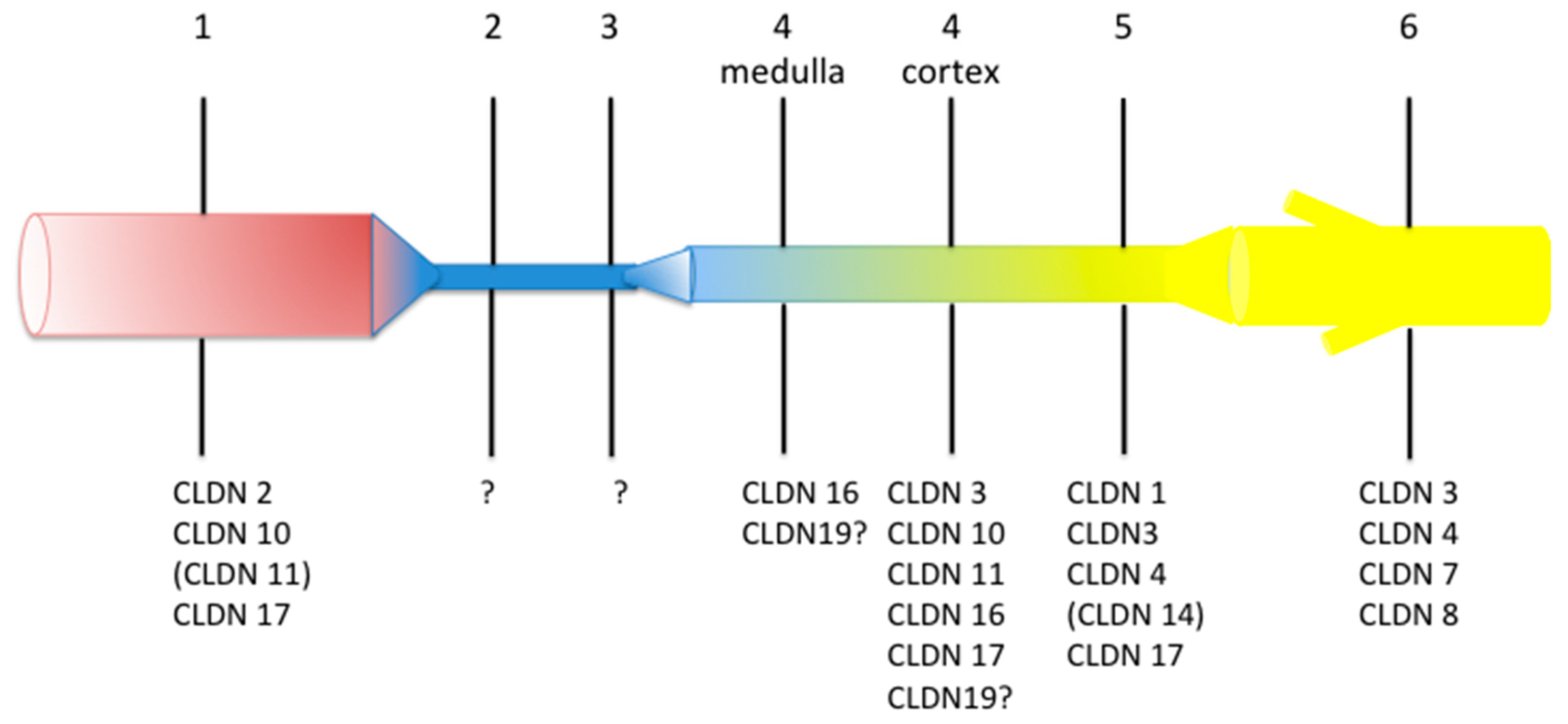
Figure 2.
Model of ion transport in the inner stripe of outer medulla (IS-) (A) and in the cortical thick ascending limb of the loop of Henle (C-TAL) (B) and estimated ion concentration (C). The original works used to build the model have been published in References [108,109,110]. NaCl is reabsorbed via the apical cotransporter NKCC2. Most of the potassium that enters the cell recycles back to the lumen via the potassium channel ROMK, thereby hyperpolarizing the apical membrane, while most of the chloride leaves the cell across the basolateral chloride channel CLCKB, resulting in a depolarization of the membrane. Sodium exits the cell via the Na+,K+-ATPase at the basolateral membrane. The difference in voltage of the two membranes accounts for the lumen positive transepithelial potential difference, the driving force for the paracellular diffusion of divalent cations in the C-TAL and of sodium in the IS-TAL. Claudin (Cldn)16 and Cldn19 may confer a paracellular permeability and selectivity to cations. Cldn14 may interact with Cldn16 and inhibit the Cldn16/Cldn19 complex. It is suggested that Cldn10b drives paracellular NaCl back flux in cortical TAL, adding to the lumen-positive voltage as the paracellular pathway is more permeable to sodium than to chloride. High Ca2+ diet and allosteric agonists of calcium-sensing receptor (CaSR) may trigger the expression of Cldn14 via the inhibition of the transcription of two microRNAs miR-9 and miR-374 suppressing Cldn14 gene expression. Ø Direct measurement of luminal ion concentration in the TAL is not possible because the segment is inaccessible to micropuncture. Luminal concentrations of Na+, chloride (Cl−), Mg2+ and Ca2+ in the early distal convoluted tubule, the segment just downstream the late cortical thick ascending limb (CTAL) have been measured during micropuncture experiments in rodents [111,112,113,114,115,116,117,118,119]. In most studies, concentrations are expressed as a ratio between tubular fluid and plasma ultrafilterable ion concentration. Plasma NaCl is freely filtered, whereas around 60% of Ca and 80% of Mg are ultrafilterable. Luminal ion concentrations in the early medullary thick ascending limb (MTAL) can be estimated based on a Ca2+ and Mg2+ reabsorption equaling 20–25% and 60–70% of filtered load, respectively and the lack of substantial water reabsorption in the TAL. ∞ Values of ion concentrations in the cortical interstitial fluid are similar to those in plasma, due to the dense capillary network and high blood flow in the cortex. Those concentrations have been reported in mathematical models except for magnesium (*). Estimates of the luminal and interstitial concentrations of Na+, Cl− and Ca2+ in the early medullary thick ascending limbs have been published in mathematical models [120,121,122,123,124,125,126].
Figure 2.
Model of ion transport in the inner stripe of outer medulla (IS-) (A) and in the cortical thick ascending limb of the loop of Henle (C-TAL) (B) and estimated ion concentration (C). The original works used to build the model have been published in References [108,109,110]. NaCl is reabsorbed via the apical cotransporter NKCC2. Most of the potassium that enters the cell recycles back to the lumen via the potassium channel ROMK, thereby hyperpolarizing the apical membrane, while most of the chloride leaves the cell across the basolateral chloride channel CLCKB, resulting in a depolarization of the membrane. Sodium exits the cell via the Na+,K+-ATPase at the basolateral membrane. The difference in voltage of the two membranes accounts for the lumen positive transepithelial potential difference, the driving force for the paracellular diffusion of divalent cations in the C-TAL and of sodium in the IS-TAL. Claudin (Cldn)16 and Cldn19 may confer a paracellular permeability and selectivity to cations. Cldn14 may interact with Cldn16 and inhibit the Cldn16/Cldn19 complex. It is suggested that Cldn10b drives paracellular NaCl back flux in cortical TAL, adding to the lumen-positive voltage as the paracellular pathway is more permeable to sodium than to chloride. High Ca2+ diet and allosteric agonists of calcium-sensing receptor (CaSR) may trigger the expression of Cldn14 via the inhibition of the transcription of two microRNAs miR-9 and miR-374 suppressing Cldn14 gene expression. Ø Direct measurement of luminal ion concentration in the TAL is not possible because the segment is inaccessible to micropuncture. Luminal concentrations of Na+, chloride (Cl−), Mg2+ and Ca2+ in the early distal convoluted tubule, the segment just downstream the late cortical thick ascending limb (CTAL) have been measured during micropuncture experiments in rodents [111,112,113,114,115,116,117,118,119]. In most studies, concentrations are expressed as a ratio between tubular fluid and plasma ultrafilterable ion concentration. Plasma NaCl is freely filtered, whereas around 60% of Ca and 80% of Mg are ultrafilterable. Luminal ion concentrations in the early medullary thick ascending limb (MTAL) can be estimated based on a Ca2+ and Mg2+ reabsorption equaling 20–25% and 60–70% of filtered load, respectively and the lack of substantial water reabsorption in the TAL. ∞ Values of ion concentrations in the cortical interstitial fluid are similar to those in plasma, due to the dense capillary network and high blood flow in the cortex. Those concentrations have been reported in mathematical models except for magnesium (*). Estimates of the luminal and interstitial concentrations of Na+, Cl− and Ca2+ in the early medullary thick ascending limbs have been published in mathematical models [120,121,122,123,124,125,126].
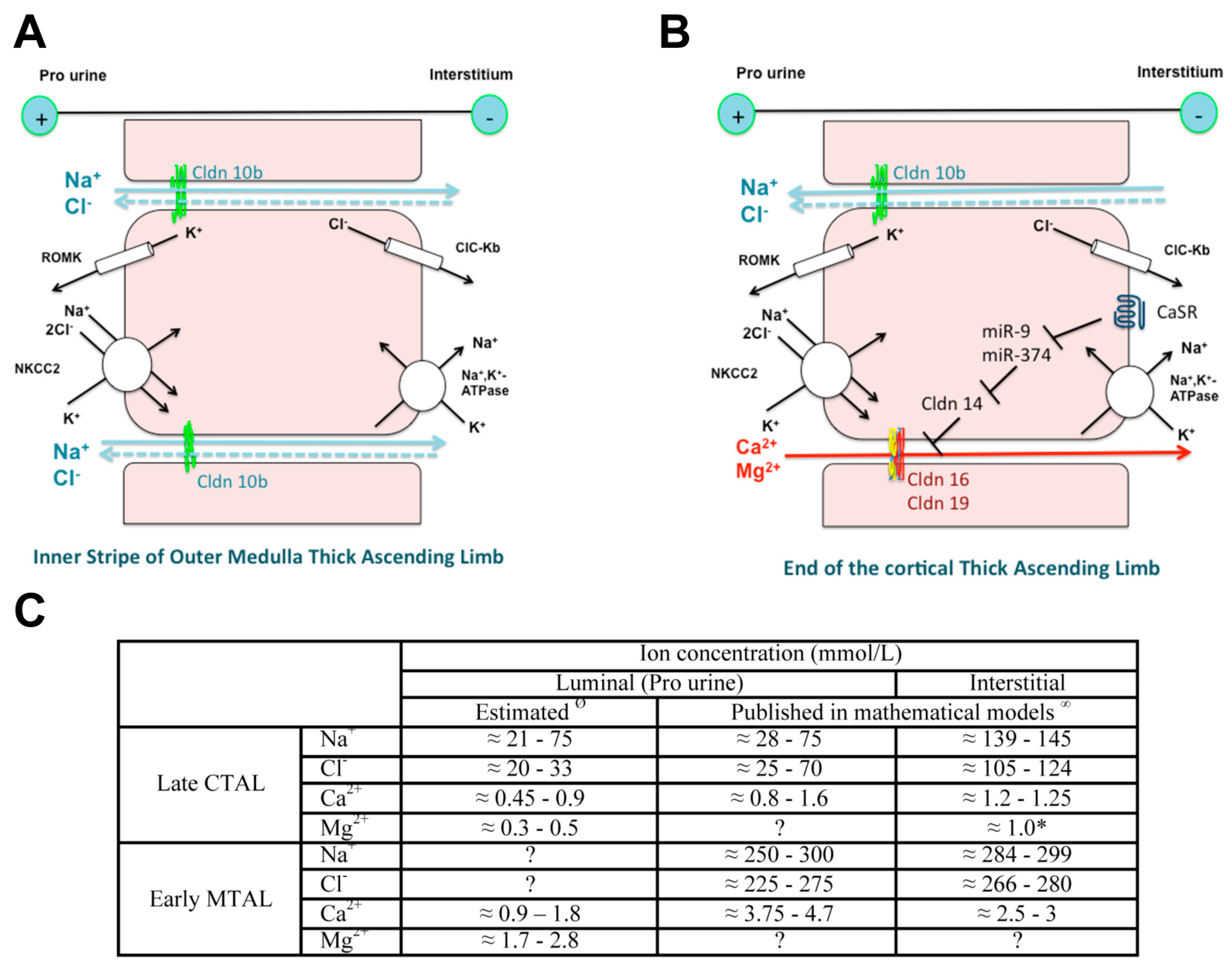
Figure 3.
Expression of claudin 10, claudin 16 and claudin 19 in the murine cortical thick ascending limb in immunofluorescence. (A) Pattern of expression of claudin 10 and claudin 16 in the mouse C-TAL. Either claudin 10 (Cldn 10, in green) or claudin 16 (Cldn 16, in red) are expressed at tight junction. (B) Pattern of expression of claudin 16 and claudin 19 in the mouse C-TAL. Claudin 16 (Cldn 16, in red) and claudin 19 (Cldn 19, in green) are colocalized at tight junction. Bar = 20 µm.
Figure 3.
Expression of claudin 10, claudin 16 and claudin 19 in the murine cortical thick ascending limb in immunofluorescence. (A) Pattern of expression of claudin 10 and claudin 16 in the mouse C-TAL. Either claudin 10 (Cldn 10, in green) or claudin 16 (Cldn 16, in red) are expressed at tight junction. (B) Pattern of expression of claudin 16 and claudin 19 in the mouse C-TAL. Claudin 16 (Cldn 16, in red) and claudin 19 (Cldn 19, in green) are colocalized at tight junction. Bar = 20 µm.
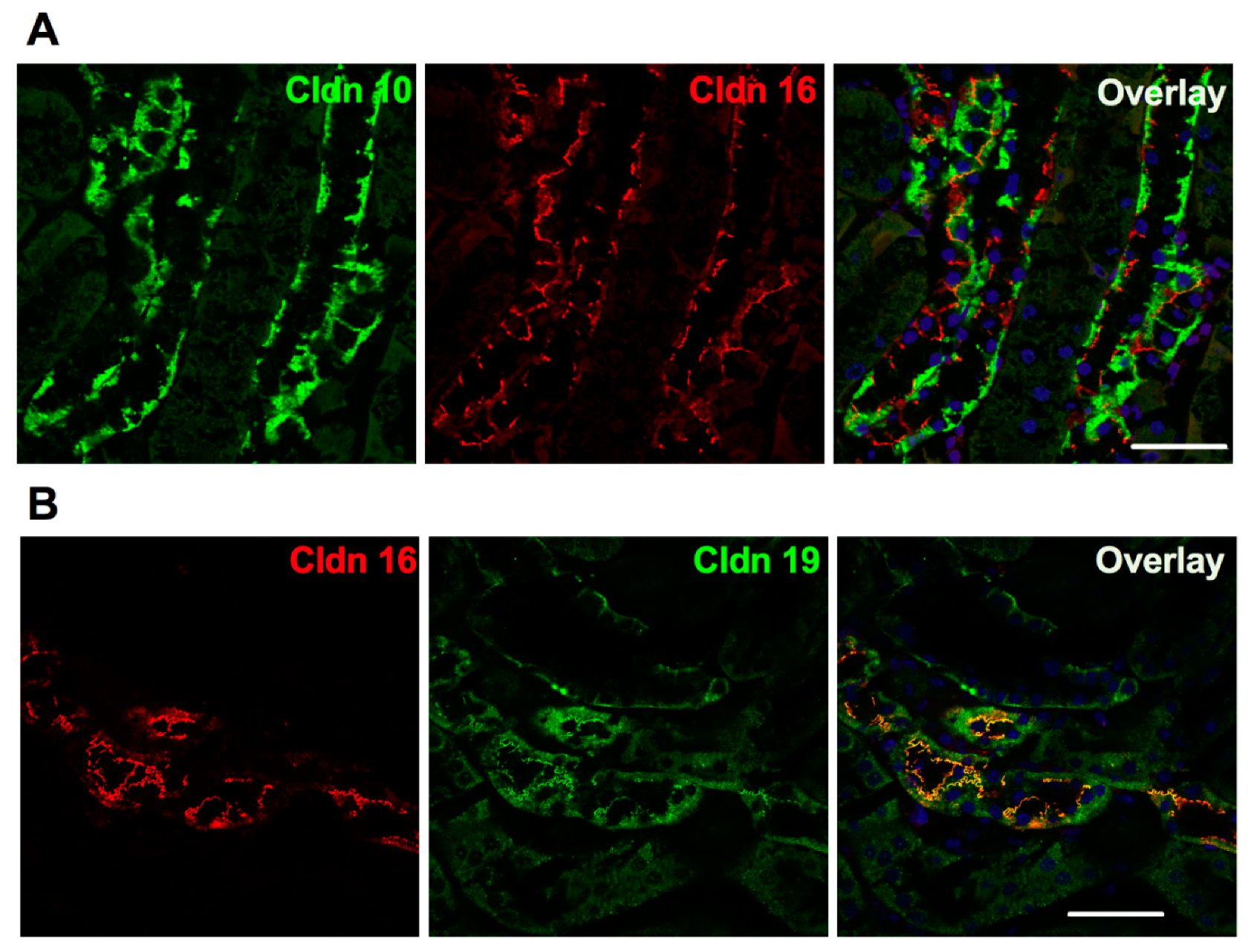

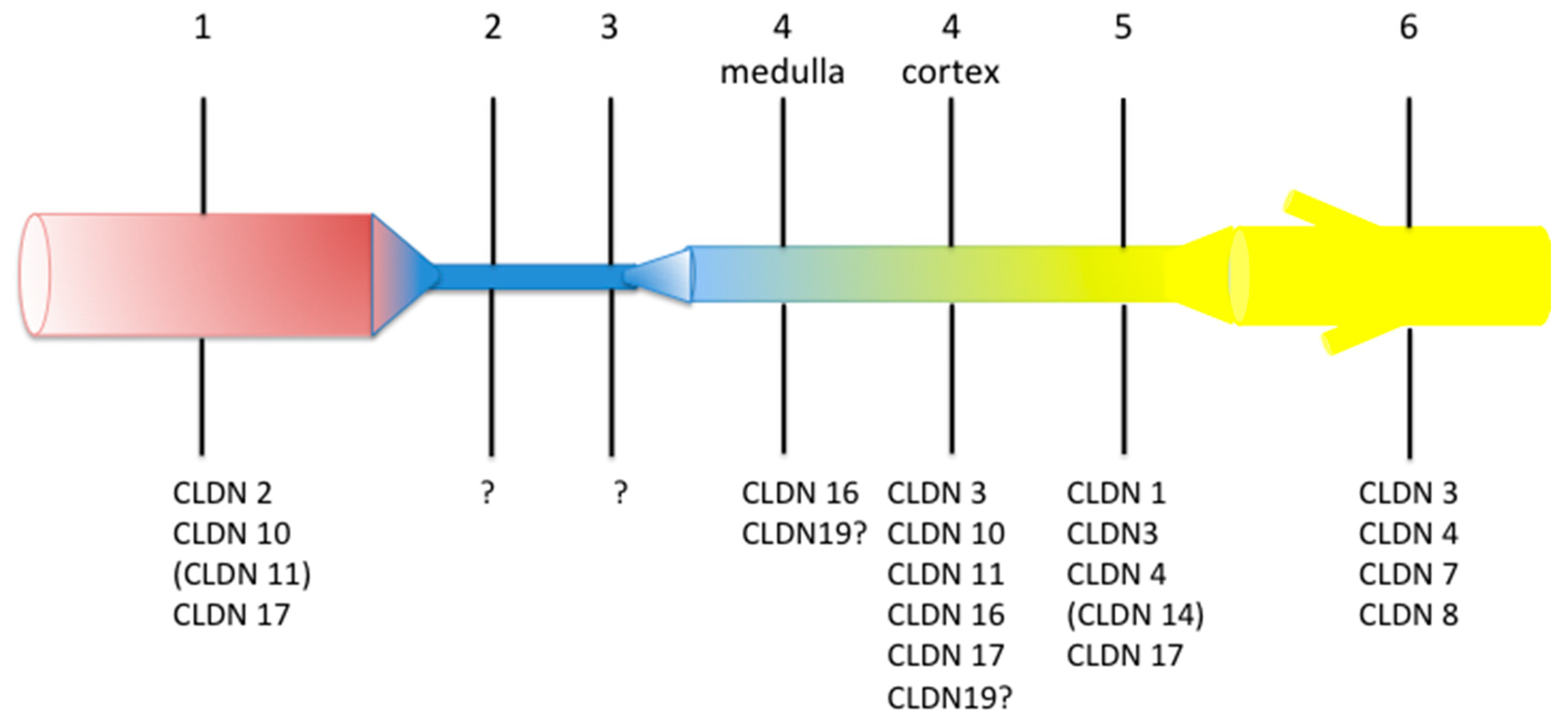{kind=link}
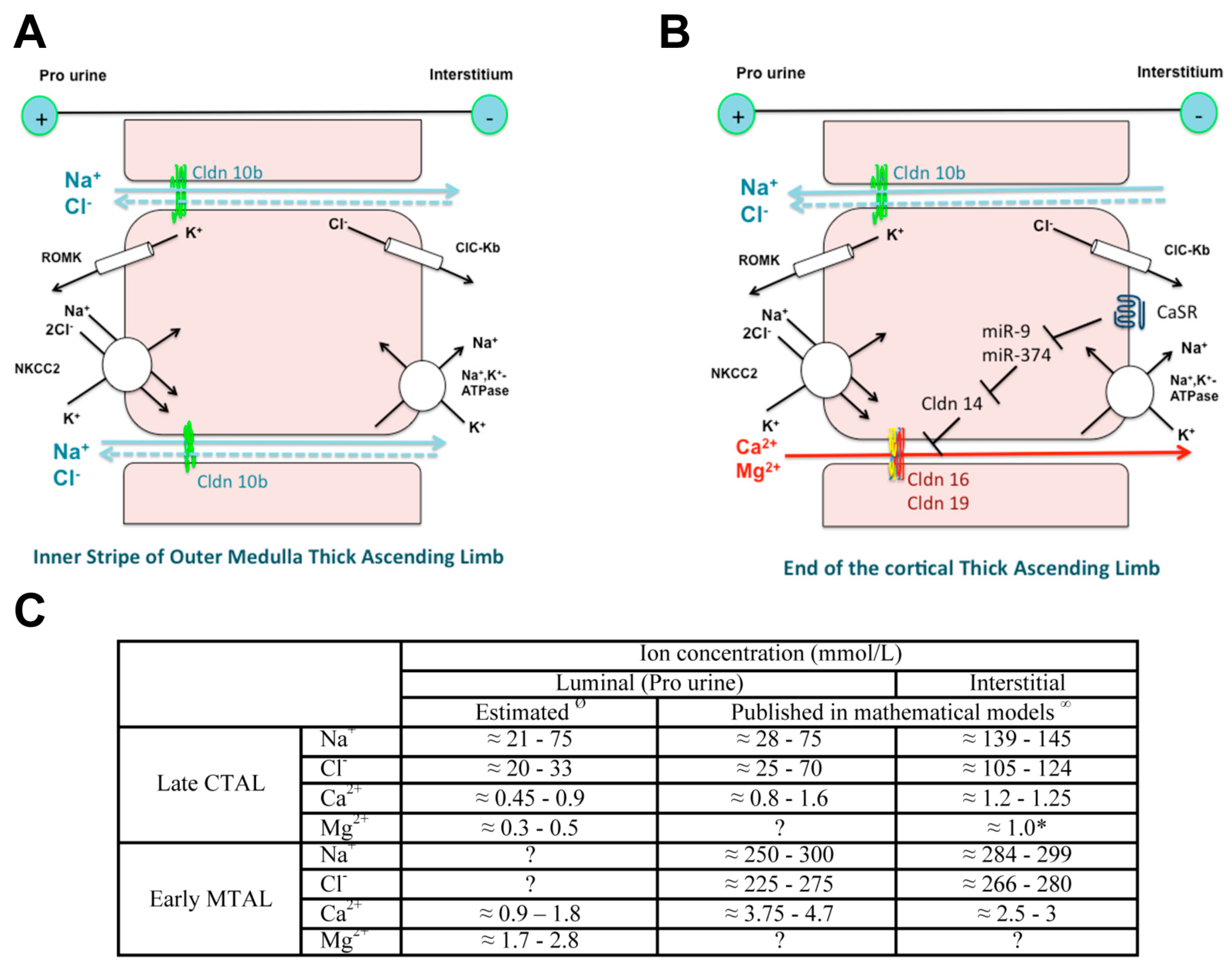{kind=link}
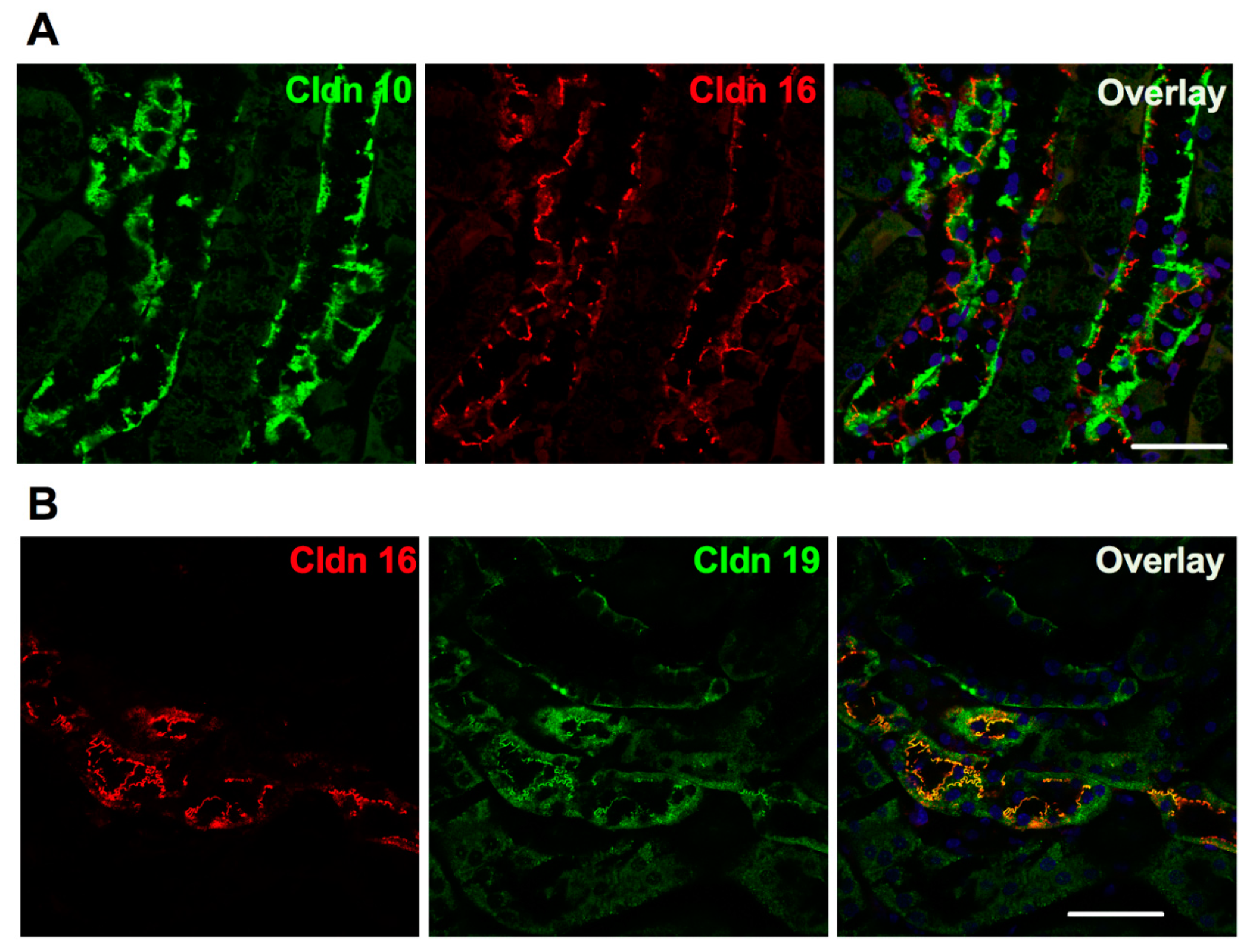{kind=link}
Table 1.
Similarities and differences among phenotype in patients with HELIX syndrome.
| Reference | Bongers [34] | Klar [35] | Hadj-Rabia [28] | Meyers [36] | Overall (%) |
|---|---|---|---|---|---|
| Area of Origin | Europe | Pakistan | North Africa, Pakistan | South America | |
| Consanguinity | No | Yes | Yes | Yes | |
| Hypohidrosis | N.D. | 13/13 | 6/6 | 1/1 | 20/20 (100%) |
| Electrolyte imbalance | 2/2 | 6/7 | 6/6 | 1/1 | 15/16 (94%) |
| Hypolacrimia | N.D. | 13/13 | 6/6 | 1/1 | 20/20 (100%) |
| Ichthyosis | N.D. | N.D. | 6/6 | 0/1 | 6/7 (86%) |
| Xerostomia | N.D. | 13/13 | 6/6 | 1/1 | 20/20 (100%) |
| Plasma abnormalities | |||||
| Hypokalemia | 2/2 | 0/7 | 3/6 | 1/1 | 6/16 (38%) |
| Hypermagnesemia | 1/2 | 6/7 | 6/6 | 1/1 | 14/16 (88%) |
| eGFR < 60 mL/min/1.73 m2 | 1/2 | 0/3 | 1/6 | 1/1 | 3/12 (25%) |
| Secondary hyperaldosteronism | N.D. | N.D. | 6/6 | Hyperaldosteronism without hyperreninism | |
| Nephrolithiasis | 0/2 | 4/13 | 0/6 | 0/1 | 4/22 (18%) |
N.D.: not determined.
Table 2.
CLDN10 disease-causing variants [32].
Table 2.
CLDN10 disease-causing variants [32].
| Missense/Nonsense Mutations | ||||||
|---|---|---|---|---|---|---|
| Ref. | Nucleotide Change | Amino Acid Change | Protein Change | Variant Class | Exon | Domain |
| [28] | c.2T>C | Met1Thr | p.M1? | DM | 1b | Helical |
| [35] | c.144C>G | Asn48Lys | p.N48K | DM | 1b | ECS1 |
| [34] | c.217G>A | Asp73Asn | p.D73N | DM? | 1b | ECS1 |
| [36] | c.238A>G | Arg80Gly | p.R80G | DM | 2 | ECS1 |
| [28] | c.386C>T | Ser131Leu | p.S131L | DM | 3 | Helical |
| [34] | c.446C>G | Pro149Arg | p.P149R | DM? | 3 | ECS2 |
| Splicing Mutations | ||||||
| [34] | c.465–1G>A | p.E157_T192del | DM? | 4 | Helical | |
Variant class is described according the Human Gene Mutation Database [37] DM: Disease-causing mutations; DM?: probable/possible pathological mutation; ECS1: first extracellular segment; ECS2: second extracellular segment.
Table 3.
Function of claudin 10b according to heterologous expression studies in cell lines.
| Claudin | Cell Line | Transfection | TER | PNa/PCl | PNa | PCl | PMg/PCl | PCa/PCl | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Mouse Cldn10b | MDCK II | Stable | NS | NS | NS | NS | [33] | ||
| Mouse Cldn10b | LLC-PK1 | Stable | ↘ | NS | ↗ | NS | [33] | ||
| Mouse Cldn10b | MDCK-C7 | Stable | ↘ | ↗ | ↗ | ↗ | [38] | ||
| Human CLDN10b | MDCK-C7 | Stable | ↘ | ↗ | ↗ | ↗ | [38] | ||
| Human CLDN10b | MDCK-C7 | Stable | ↘ | [40] | |||||
| Mouse Cldn10a | MCDK II | Stable | NS | ↘ | ↘ | ↗ | [33] | ||
| Mouse Cldn10a | LLC-PK1 | Stable | ↘ | NS | NS | ↗ | [33] | ||
| Mouse Cldn10a | MDCK II | Stable | NS | ↘ | [38] | ||||
| Mouse Cldn10a | MDCK-C7 | Stable | NS | NS | NS | NS | [38] | ||
| Human CLDN10a | MDCK-C7 | Stable | ↘ | NS | NS | NS | [38] | ||
| Human CLDN10a | MDCK-C7 | Stable | NS | [40] |
In electrophysiological studies, relative epithelial permeabilities (e.g., PNa/PCl) are calculated using the Goldman-Hodgkin-Katz equation and the diffusion potential caused by the application of distinct solutions at the apical and basolateral compartment. Absolute permeabilities can be calculated if the transepithelial conductance is known using the Kimizuka-Koketsu equation [41]. The effects of claudin 10a have been included, for comparison with those of claudin 10b. TER: trans epithelial resistance; Px: permeability to ion X; NS: not significantly different from control.
Table 4.
CLDN16 disease-causing variants [32].
Table 4.
CLDN16 disease-causing variants [32].
| Missense/Nonsense Mutations | ||||||
|---|---|---|---|---|---|---|
| Ref. | Nucleotide Change | Amino Acid Change | Protein Change | Variant Class | Exon | Domain |
| [84] | c.114C>A | Cys38Term | p.C38 * | DM a | 1 | N term |
| [78] | c.211A>G | Met71Val | p.M71V | DM | 1 | N term |
| [26] | c.212T>G | Met71Arg | p.M71R | DM | 1 | N term |
| [49] | c.212T>C | Met71Thr | p.M71T | DM | 1 | N term |
| [50] | c.239G>A | Cys80Tyr | p.C80Y | DM | 1 | TM1 |
| [49] | c.263G>A | Gly88Glu | p.G88E | DM | 1 | TM1 |
| [85] | c.290A>G | Asp97Gly | p.D97G | DM | 1 | ECS1 |
| [53] | c.295T>G | Trp99Gly | p.W99G | DM | 1 | ECS1 |
| [49,52] | c.330C>G | Ser110Arg b | p.S110R | DM | 2 | ECS1 |
| [49,86] | c.341G>A | Arg114Gln | p.R114Q | DM | 2 | ECS1 |
| [60] | c.340C>T | Arg114Term | p.R114 * | DM | 2 | ECS1 |
| [56] | c.346C>G | Leu116Val | p.L116V | DM | 2 | ECS1 |
| [45,66] | c.350G>A | Trp117Term | p.W117 * | DM | 2 | ECS1 |
| [87] | c.354G>A | Trp118Term | p.W118 * | DM | 2 | ECS1 |
| [52,88] | c.358T>C | Cys120Arg | p.C120R | DM | 2 | ECS1 |
| [49] | c.385C>T | Arg129Cys | p.R129C | DM | 2 | ECS1 |
| [50,54,64,71,86] | c.416C>T | Ala139Val | p.A139V | DM | 2 | ECS1 |
| [45,49,66,86] | c.421C>G | His141Asp | p.H141D | DM | 2 | ECS1 |
| [45,46,47,52,66,86] | c.434T>C | Leu145Pro | p.L145P | DM | 3 | ECS1 |
| [86,89] | c.446G>A | Arg149Gln | p.R149Q | DM | 3 | ECS1 |
| [45,46,86] | c.446G>T | Arg149Leu | p.R149L | DM | 3 | ECS1 |
| [26,50] | c.445C>T | Arg149Term | p.R149 * | DM | 3 | ECS1 |
| [45,46,47,49,52,66,86,88] | c.453G>T | Leu151Phe | p.L151F | DM | 3 | ECS1or TM2? |
| [45,49,66,86] | c.452T>G | Leu151Trp | p.L151W | DM | 3 | ECS1or TM2? |
| [50,54,71,86] | c.485G>T | Gly162Val | p.G162V | DM | 3 | TM2 |
| [26,46] | c.500T>C | Leu167Pro | p.L167P | DM | 3 | TM2 |
| [90] | c.539C>T | Pro180Leu | p.P180L | DM | 3 | ICL |
| [50,87,91] | c.547A>G | Lys183Glu | p.K183E | DM | 3 | ICL |
| [26,46,47,86] | c.571G>A | Gly191Arg | p.G191R | DM | 3 | TM3 |
| [65] | c.592G>C | Gly198Arg | p.G198R | DM | 3 | TM3 |
| [45,86] | c.593G>C | Gly198Ala | p.G198A | DM | 4 | TM3 |
| [26,46,66,86] | c.593G>A | Gly198Asp | p.G198D | DM | 4 | TM3 |
| [63] | c.602G>A | Gly201Glu | p.G201E | DM | 4 | TM3 |
| [92] | c.620G>A | Trp207Term | p.W207 * | DM | 4 | ECS2 |
| [45,46,47,49] | c.625G>A | Ala209Thr | p.A209T | DM | 4 | ECS2 |
| [49,59,89] | c.646C>T | Arg216Cys | p.R216C | DM | 4 | ECS2 |
| [55] | c.647G>A | Arg216His | p.R216H | DM | 4 | ECS2 |
| [49,93] | c.679G>C | Gly227Arg | p.G227R | DM | 4 | ECS2 |
| [26,46,47,94] | c.695T>G | Phe232Cys | p.F232C | DM | 4 | ECS2 |
| [50] | c.697G>C | Gly233Arg | p.G233R | DM | 4 | ECS2 |
| [26,46] | c.698G>A | Gly233Asp | p.G233D | DM | 4 | ECS2 |
| [61] | c.697G>T | Gly233Cys | p.G233C | DM | 4 | ECS2 |
| [52] | c.702G>T | Trp234Cys | p.W234C | DM | 4 | ECS2 |
| [26] | c.704C>T | Ser235Phe | p.S235F | DM | 4 | ECS2 |
| [45,46] | c.703T>C | Ser235Pro | p.S235P | DM | 4 | ECS2 |
| [77] | c.704C>A | Ser235Tyr | p.S235Y | DM | 4 | ECS2 |
| [49,95] | c.710G>A | Trp237Term | p.W237 * | DM | 4 | ECS2 |
| [26,45,46,52,66,71,87] | c.715G>A | Gly239Arg | p.G239R | DM | 4 | ECL2 or TM4? |
| [52] | c.734G>A | Gly245Asp | p.G245D | DM | 4 | TM4 |
| [82] | c.823A>T c | Lys275Term | p.K275 * | DM | 5 | C term |
| [81] | c.831T>G d | Tyr277Term | p.Y277 * | DM | 5 | C term |
| [52] | c.864C>G | Tyr288Term | p.Y288 * | DM | 5 | C term |
| [96] | c.908C>G e | Thr303Arg | p.T303R | DM | 5 | C term |
| Splicing Mutations | ||||||
| Ref. | Nucleotide Change | Splicing Mutation | Variant Class | |||
| [45] | c.325-5T>G | IVS1 as T-G -5 | DM | |||
| [60] | c.427+5G>A | IVS2 ds G-A +5 | DM | |||
| [26] | c.593-2A>G | IVS3 as A-G -2 | DM | |||
| [49,59] | c.784+1G>T | IVS4 ds G-T +1 | DM | |||
| [45] | c.785-14T>G | IVS4 as T-G -14 | DM | |||
| Small Deletions | ||||||
| Nucleotide Change | Protein Change | Variant Class | Exon | Domain | ||
| [97] | c.166delG f | p.(Ala56Leufs*16) | DM? | 1 | N term | |
| [49] | c.235delG g | p.(Ala79fsX90) | 1 | TM1 | ||
| [45] | c.368delA | p.(Asn123Metfs*21) | DM | 2 | ECS1 | |
| [49] | c.408_410delCAT | p.(Ile137del) | DM | 2 | ECS1 | |
| [61] | c.800delG | p.(Arg267Lysfs*7) | DM | 5 | C term | |
| Small Insertions | ||||||
| Nucleotide Change | Protein Change | Variant Class | Exon | Domain | ||
| [74] | c.324+3_324+4insT | Not available | DM | intron 1 | ||
| [71] | c.545_548dupTTAA | p.(Lys183Asnfs*2) | DM | 3 | ICL | |
| Small Indels | ||||||
| Nucleotide Change | Protein Change | Variant Class | Exon | Domain | ||
| [45,66] | c.165_166delGGinsC | p.(Arg55Serfs*17) | R | 1 | N term | |
| [45,46] | c.646_647delCGinsAC | p.(Arg216Thr) | DM | 4 | ECS2 | |
| Gross Deletions | ||||||
| DNA Level | Description | Variant Class | Exon | Domain | ||
| [71,98] | g.DNA | Ex. 2-5 | DM | 2-5 | ||
| Complex Mutations | ||||||
| Description | Variant Class | Exon | Domain | |||
| [62] | c.574_589delins23bp | p.(A192Yfs∗ 25) | DM | 3 | TM3 | |
Variant class is described according the Human Gene Mutation Database [37] DM: Disease-causing mutations; DM?: probable/possible pathological mutation; «Retired records (R)», a variant that has been removed from HGMD if found to have been erroneously included ab initio or if the variant has been subject to retraction/correction in the literature resulting in the record becoming obsolete, merged or otherwise invalid. Domains are described according to authors and [99]. C Term, COOH terminus; TM, transmembrane domain; ECS1, first extracellular segment; ICL, intracellular loop; ECS2, second extracellular segment; N Term, NH2 terminus. a: the «significance» described by Trujillano was “likely pathogenic according to ACMG guidelines.” They categorized patients’ phenotypes according to the Human Phenotype Ontology nomenclature based on the clinical data and preceding workup provided by the referring physician. The phenotype described was: psychosis, seizures, muscle weakness, respiratory failure, reduced dihydropyrimidine dehydrogenase activity, decreased body weight, reduced consciousness/confusion, lower limb muscle weakness; b: reported as p.S110R 329AGC>AGG; c: reported as p.L203*, c.822A>T; d: reported as p.Y207*, c.620T>G; e: reported as p.T233R, c.697C>G; f: reported as c.164delG; g: reported as 236delG, p.A80fsX91,; * indicates that the predicted consequence is a termination codon.
Table 5.
CLDN19 disease-causing variants [32].
Table 5.
CLDN19 disease-causing variants [32].
| Missense/Nonsense Mutations | ||||||
|---|---|---|---|---|---|---|
| Ref. | Nucleotide Change | Amino Acid Change | Protein Change | Variant Class | Exon | Domain |
| [50] | c.54G>A | Trp18Term | p.W18 * | DM | 1 | TM1 |
| [47,50,51,57,58,67,70,72,101,102,103] | c.59G>A a,b | Gly20Asp | p.G20D | DM | 1 | TM1 |
| [50,101,102] | c.83C>T | Pro28Leu | p.P28L | DM | 1 | TM1 |
| [51] | c.122T>C | Ile41Thr | p.I41T | DM | 1 | ECS1 |
| [50,67] | c.130G>A | Val44Met | p.V44M | DM | 1 | ECS1 |
| [47,51,57] | c.169C>G | Gln57Glu | p.Q57E | DM | 1 | ECS1 |
| [50,72] | c.169C>T | Gln57Term | p.Q57 * | DM | 1 | ECS1 |
| [51] | c.223G>T | Gly75Cys | p.G75C | DM? | 1 | ECS1 |
| [51] | c.223G>A | Gly75Ser | p.G75S | DM? | 1 | ECS1 |
| [68,76] c | c.241C>T | Arg81Trp | p.R81W | DM | 2 | ECS1 or TM2? |
| [104] | c.263T>A | Val88Glu | p.V88E | DM | 2 | TM2 |
| [72] | c.269T>G | Leu90Arg | p.L90R | DM | 2 | TM2 |
| [47,57] | c.269T>C | Leu90Pro | p.L90P | DM | 2 | TM2 |
| [51] | c.364G>A | Gly122Arg | p.G122R | DM | 2 | TM3 |
| [63] | c.388G>T | Gly130Asp | p.G130C | DM | 2 | TM3 |
| [63,75] | c.389G>A | Gly130Asp | p.G130D | DM | 3 | TM3 |
| [105] | c.506G>A d | Trp169Term | p.W169 * | DM | 4 | TM4 |
| [94] | c.535G>A | Gly179Ser | p.G179S | DM | 4 | TM4 |
| [72] | c.599G>A | Arg200Gln | p.R200Q | DM? | 4 | C term |
| Small Deletions | ||||||
| Nucleotide Change | Protein Change | Variant Class | Exon | Domain | ||
| [106] | c.140_141delAT | p.(Tyr47 *) | DM | 1 | ECS1 | |
| [50] | c.403_406delACTG | p.(Thr135Leufs*9) | DM | 3 | TM3 | |
| Gross Deletions | ||||||
| DNA Level | Description | Variant Class | Exon | Domain | ||
| [50] | g.DNA | Ex. 1-4 | DM | 1-4 | ||
Variant class is described according the Human Gene Mutation Database [37]. DM: Disease-causing mutations; DM?: probable/possible pathological mutation. Domains are described according to authors and [99]. C Term, COOH terminus; TM, transmembrane domain; ECS1, first extracellular segment; ICL, intracellular loop; ECS2, second extracellular segment; N Term, NH2 terminus. a: reported as c.C>T in ref [58]; b: reported as c.69G>A in ref [72]; c: reported as p.Arg81Cys in ref [68]; d: reported as c.697G>A in ref [105]; * indicates that the predicted consequence is a termination codon..
Table 6.
Function of claudin 16 according to heterologous expression studies in cell lines.
| Claudin | Cell Line | Transfection | TER | PNa/PCl | PNa | PCl | PMg | PCa | Mg2+ Flux | Ca2+ Flux | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rat Cldn16 | MDCK | stable | ↗ | ↗ *∫◊ | [129] | ||||||
| Rat? Cldn16 | MDCK | stable | ↗ | ↘ | ↘ | NS | ↗ §ø | [130] | |||
| Rat Cldn16 | MDCK Tet-OFF | Inducible expression | ↗ | ↗ §* | [132] | ||||||
| Human ∆70 CLDN16 | LLC-PK1 | Stable? | ↘ | ↗ | ↗ | NS | ↗ † | [46] | |||
| Human ∆70 CLDN16 | MDCK II | Stable? | NS | NS | NS | [46] | |||||
| Full length Human CLDN16 | LLC-PK1 | Stable? | ↘ | ↗ | ↗ | NS | [47] | ||||
| Full length Human CLDN16 | MDCK II | Stable? | NS | NS | [47] | ||||||
| Short and long version of human CLDN16 | MDCK-C7 | Stable | *** | ↗ £ | NS ∫ | [48] | |||||
| Long version of human CLDN16 | MDCK-C7 | Stable | NS | ↗ ∞ | NS ∞ | [48] | |||||
| Full-length human CLDN16 | MDCK-C7 | stable | NS | ↗ ¥ | [131] |
Relative epithelial permeabilities (e.g., PNa/PCl) are calculated using the Goldman-Hodgkin-Katz equation and the diffusion potential caused by the application of distinct solutions at the apical and basolateral compartment. Absolute permeabilities can be calculated if the transepithelial conductance is known using Kimizuka-Koketsu equation [41]. Measuring ion flux can be performed using either radioactive isotopes or fluorescent compounds or non-radioactive ions. Flux measurement include trans- and paracellular transport [41]. TER: trans epithelial resistance; NS: not significantly different from control; Px: permeability to ion X; ∫: Transepithelial transport of 45Ca2+ measurement; §: The transepithelial transport of Mg2+ was measured using Xylidyl Blue-I.; †: The permeability of Mg2+ across monolayers was determined according to Tang and Goodenough [133]; £: Mg2+ flux was measured employing atomic absorption spectrometry; ∞: Mg2+ and Ca2+ permeabilities, calculated from dilution potential/bionic potential measurements; ¥: Measurement of unidirectional fluxes from the basolateral to the apical side was performed under short-circuit conditions with MgSO4. The atomic absorption of Mg2+ was measured in an oxidizing air-acetylene flame at 285.2 nm. PMg was calculated from resulting fluxes (PMg = flux/concentration); ∂Permeability PC = Flux/Substrate concentration in cis compartment. This was then corrected for the permeability of blank filters, PB, to obtain the true transepithelial permeability (PT), using the following equation PT = [(1/PC) & (1/PB)]−1; ***: Increased basolateral Mg2+ concentration induces a short circuit current (may activate a transcellular Cl− current); *: from apical to basal compartment, without affecting transport from basal to apical compartments; ø: from apical to basal compartment; ◊: The apical to basolateral flux was competitively inhibited by Mg2+.
Table 7.
Function of claudin 19 and of claudin 16 and claudin 19 co-expression according to heterologous expression studies in cell lines.
Table 7.
Function of claudin 19 and of claudin 16 and claudin 19 co-expression according to heterologous expression studies in cell lines.
| Claudin | Cell Line | Transfection | TER | PNa/PCl | PNa | PCl | PMg | PCa | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Human CLDN19 | LLC-PK1 | Stable? | ↗ | ↗ | NS | ↘ | † | [47] | |
| Human CLDN19 | MDCK II | Stable? | NS | NS | [47] | ||||
| Mouse Cldn19 | MDCK II Tet-Off cells | Stable inducible expression | ↗ | ↘ | NS | ↘ £∂ | ↘∫∂ | [134] | |
| Human CLDN19 + full length human CLDN16 | LLC-PK1 | Stable? | ↘ | ↗ | ↗ | ↘ | ↘ † | [47] |
Relative epithelial permeabilities (e.g., PNa/PCl) are calculated using the Goldman-Hodgkin-Katz equation and the diffusion potential caused by the application of distinct solutions at the apical and basolateral compartment. Absolute permeabilities can be calculated if the transepithelial conductance is known using Kimizuka-Koketsu equation [41]. TER: trans epithelial resistance; NS: not significantly different from control; Px: permeability to ion X; ∫: Transepithelial transport of 45Ca2+ measurement; †: The permeability of Mg2+ across monolayers was determined according to Tang and Goodenough [133]; £: Mg2+ flux was measured employing atomic absorption spectrometry; ∂: Permeability PC = Flux/Substrate concentration in cis compartment. This was then corrected for the permeability of blank filters, PB, to obtain the true transepithelial permeability (PT), using the following equation PT = [(1/PC) & (1/PB)]−1.
Table 8.
Function of claudin 14 according to heterologous expression studies in cell lines.
| Claudin | Cell Line | Transfection | TER | PNa/PCl | PNa | PCl | PCa | Ca2+ Flux | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Human CLDN14 | MDCK II Tet-Off cells | Stable inducible expression | ↗ | ↘ | ↘ | [142] | |||
| Mouse Cldn14 | OK | Stable | ↗ | ↘ | ↘ | NS | ↘∫ ø | [145] | |
| Mouse Cldn14 | MDCK II Tet-Off cells | Stable inducible expression | ↗ | ↘ | ↘ | NS | ↘∫ | [145] |
Relative epithelial permeabilities (e.g., PNa/PCl) are calculated using the Goldman-Hodgkin-Katz equation and the diffusion potential caused by the application of distinct solutions at the apical and basolateral compartment. Absolute permeabilities can be calculated if the transepithelial conductance is known using Kimizuka-Koketsu equation [41]. Flux measurement include trans- and paracellular transport [41]. TER: trans epithelial resistance; NS: not significantly different from control; Px: permeability to ion X; ∫: Transepithelial transport of 45Ca2+ measurement; ø: from apical to basal compartment.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Prot-Bertoye, C.; Houillier, P. Claudins in Renal Physiology and Pathology. Genes 2020, 11, 290. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030290
AMA Style
Prot-Bertoye C, Houillier P. Claudins in Renal Physiology and Pathology. Genes. 2020; 11(3):290. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030290
Chicago/Turabian StyleProt-Bertoye, Caroline, and Pascal Houillier. 2020. "Claudins in Renal Physiology and Pathology" Genes 11, no. 3: 290. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030290
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.