
LTBP3 Frameshift Variant in British Shorthair Cats with Complex Skeletal Dysplasia

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Clinical Examinations
2.3. Necropsy and Histopathological Examination
2.4. Immunohistochemistry
2.5. DNA Isolation
2.6. Whole Genome Sequencing of an Affected Kitten
2.7. Variant Calling
2.8. Gene Analysis
2.9. Targeted Genotyping and Sanger Sequencing
3. Results
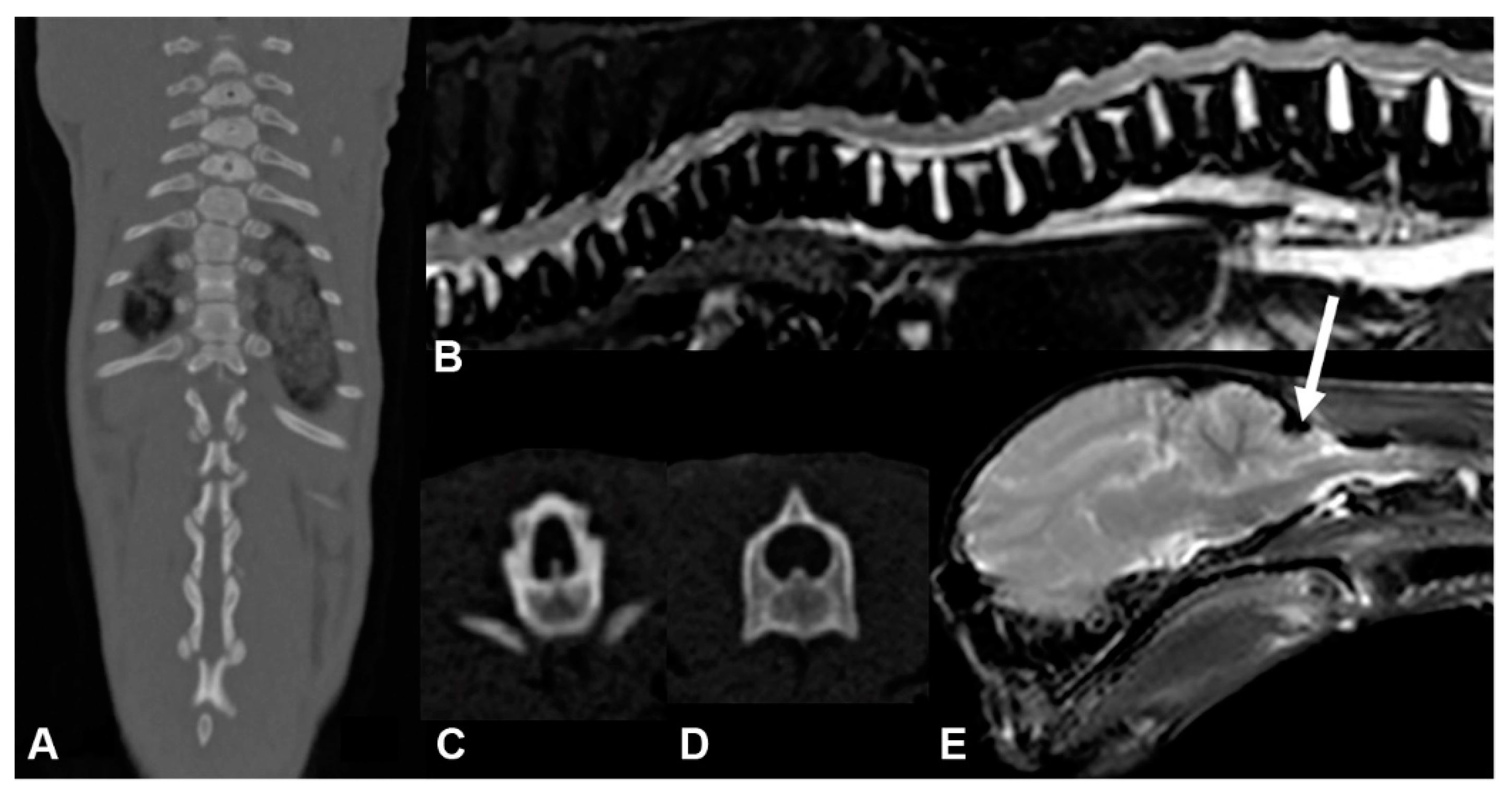
3.1. Phenotype Description
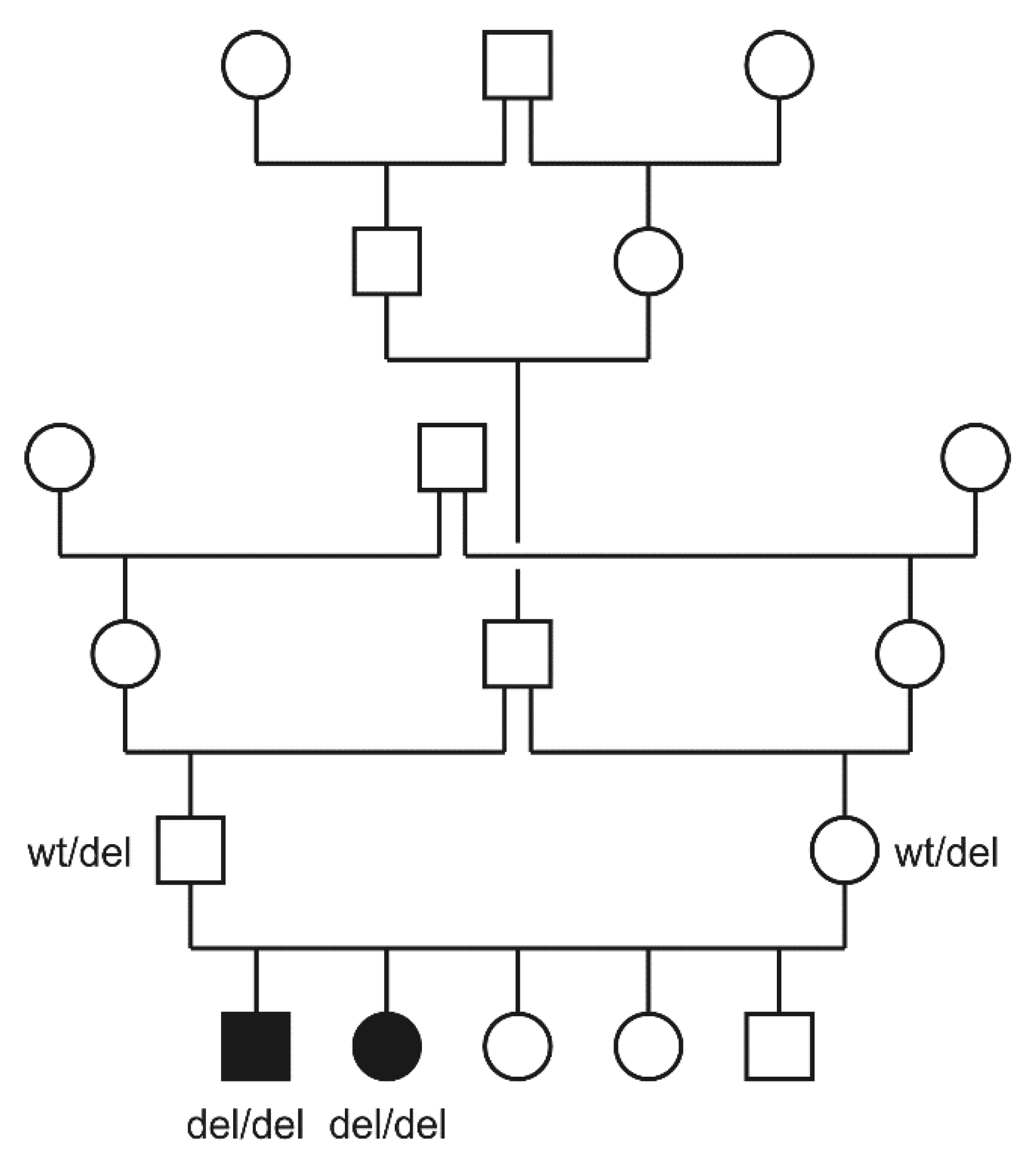
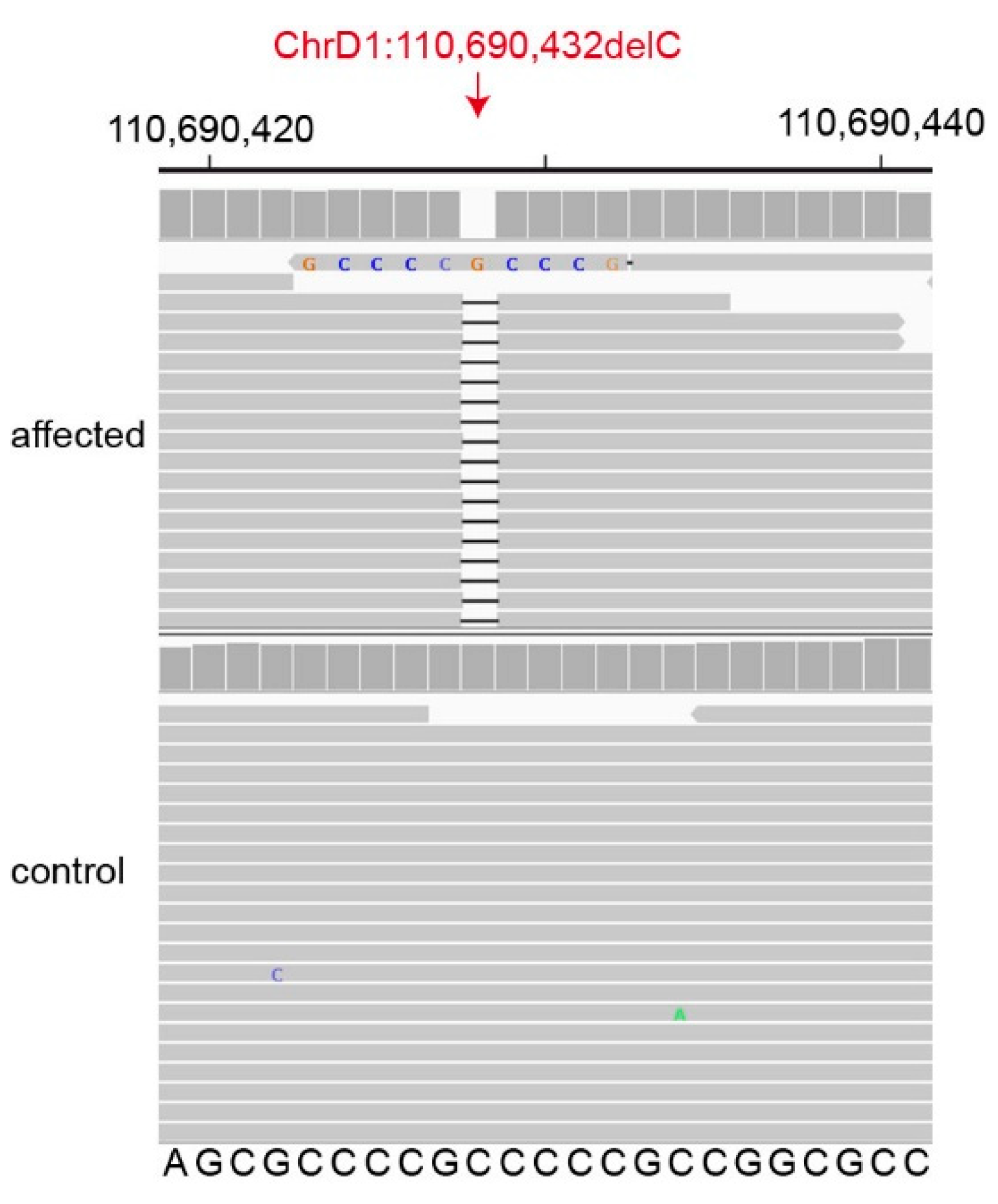
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krakow, D. Skeletal dysplasias. Clin. Perinatol. 2015, 42, 301–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Haase, B.; Mazrier, H.; Wade, C.M. Digging for known genetic mutations underlying inherited bone and cartilage characteristics and disorders in the dog and cat. Vet. Comp. Orthop. Traumatol. 2016, 29, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Bannasch, D.L.; Baes, C.F.; Leeb, T. Genetic variants affecting skeletal morphology in domestic dogs. Trends Genet. 2020, 36, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Westworth, D.R.; Sturges, B.K. Congenital spinal malformations in small animals. Vet. Clin. N. Am. Small Anim Pract. 2010, 40, 951–981. [Google Scholar] [CrossRef]
- Takanosu, M.; Takanosu, T.; Suzuki, H.; Suzuki, K. Incomplete dominant osteochondrodysplasia in heterozygous Scottish Fold cats. J. Small Anim. Pract. 2008, 49, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, B.; Alamri, S.; Darby, W.G.; Adhikari, B.; Lattimer, J.C.; Malik, R.; Wade, C.M.; Lyons, L.A.; Cheng, J.; Bateman, J.F.; et al. A dominant TRPV4 variant underlies osteochondrodysplasia in Scottish fold cats. Osteoarthr. Cartil. 2016, 24, 1441–1450. [Google Scholar] [CrossRef] [Green Version]
- Buckley, R.M.; Davis, B.W.; Brashear, W.A.; Farias, F.H.G.; Kuroki, K.; Graves, T.; Hillier, L.W.; Kremitzki, M.; Li, G.; Middleton, R.P.; et al. A new domestic cat genome assembly based on long sequence reads empowers feline genomic medicine and identifies a novel gene for dwarfism. PLoS Genet. 2020, 16, e1008926. [Google Scholar] [CrossRef] [PubMed]
- Struck, A.-K.; Braun, M.; Detering, K.A.; Dziallas, P.; Neßler, J.; Fehr, M.; Metzger, J.; Distl, O. A structural UGDH variant associated with standard Munchkin cats. BMC Genet. 2020, 21, 67. [Google Scholar] [CrossRef] [PubMed]
- Lyons, L.A.; Erdman, C.A.; Grahn, R.A.; Hamilton, M.J.; Carter, M.J.; Helps, C.R.; Alhaddad, H.; Gandolfi, B. Aristaless-like homeobox protein 1 (ALX1) variant associated with craniofacial structure and frontonasal dysplasia in Burmese cats. Dev. Biol. 2016, 409, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lettice, L.A.; Hill, A.E.; Devenney, P.S.; Hill, R.E. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 2008, 17, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casal, M.L.; Engiles, J.B.; Pipan, M.Z.; Berkowitz, A.; Porat-Mosenco, Y.; Mai, W.; Wurzburg, K.; Xu, M.Q.; Allen, R.; O’Donnell, P.A.; et al. Identification of the identical human mutation in ACVR1 in 2 cats with fibrodysplasia ossificans progressiva. Vet. Pathol. 2019, 56, 614–618. [Google Scholar] [CrossRef]
- Buckingham, K.J.; McMillin, M.J.; Brassil, M.M.; Shively, K.M.; Magnaye, K.M.; Cortes, A.; Weinmann, A.S.; Lyons, L.A.; Bamshad, M.J. Multiple mutant T alleles cause haploinsufficiency of Brachyury and short tails in Manx cats. Mamm Genome 2013, 24, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Lyons, L.A.; Creighton, E.K.; Alhaddad, H.; Beale, H.C.; Grahn, R.A.; Rah, H.; Maggs, D.J.; Helps, C.R.; Gandolfi, B. Whole genome sequencing in cats, identifies new models for blindness in AIPL1 and somite segmentation in HES7. BMC Genom. 2016, 17, 265. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sun, X.; Hu, X.-S.; Zhuang, Y.; Liu, Y.-C.; Meng, H.; Miao, L.; Yu, H.; Luo, S.-J. Whole Genome Sequencing Identifies a Missense Mutation in HES7 Associated with Short Tails in Asian Domestic Cats. Sci. Rep. 2016, 6, 31583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Rusbridge, C.; Knowler, S.P. Hereditary aspects of occipital bone hypoplasia and syringomyelia (Chiari type I malformation) in cavalier King Charles spaniels. Vet. Rec. 2003, 153, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Minato, S.; Baroni, M. Chiari-like malformation in two cats. J. Small Anim Pract. 2018, 59, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Rusbridge, C. New considerations about Chiari-like malformation, syringomyelia and their management. Practice 2020, 42, 252–267. [Google Scholar] [CrossRef]
- Driver, C.J.; Volk, H.A.; Rusbridge, C.; Van Ham, L.M. An update on the pathogenesis of syringomyelia secondary to Chiari-like malformations in dogs. Vet. J. 2013, 198, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Roe, K.A.M.; Syme, H.M.; Brooks, H.W. Congenital large intestinal hypoganglionosis in a domestic shorthair kitten. J. Feline Med. Surg. 2010, 12, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Szylberg, L.; Marszałek, A. Diagnosis of Hirschsprung’s disease with particular emphasis on histopathology. A systematic review of current literature. Prz. Gastroenterol. 2014, 9, 264–269. [Google Scholar] [CrossRef]
- Amiel, J.; Lyonnet, S. Hirschsprung disease, associated syndromes, and genetics: A review. J. Med. Genet. 2001, 38, 729–739. [Google Scholar] [CrossRef]
- Jaroy, E.G.; Acosta-Jimenez, L.; Hotta, R.; Goldstein, A.M.; Emblem, R.; Klungland, A.; Ougland, R. “Too much guts and not enough brains”: (epi)genetic mechanisms and future therapies of Hirschsprung disease—A review. Clin. Epigenetics 2019, 11, 1–11. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Verlepe, J.; Androschuk, A.; Laurent, S.B.; Rochesfort, D.; Spiegelman, D.; et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Forman, O.P.; De Risio, L.; Mellersh, C.S. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS ONE 2013, 8, e64627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.B.; Rifkin, W.J.; Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018, 71–72, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Koli, K.; Saharinen, J.; Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-β. Microsc. Res. Tech. 2001, 52, 354–362. [Google Scholar] [CrossRef]
- Chen, Y.; Dabovic, B.; Annes, J.P.; Rifkin, D.B. Latent TGF-β binding protein-3 (LTBP-3) requires binding to TGF-β for secretion. FEBS Lett. 2002, 517, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Zilberberg, L.; Todorovic, V.; Dabovic, B.; Horiguchi, M.; Couroussé, T.; Sakai, L.Y.; Rifkin, D.B. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: Role of fibrillins and fibronectin. J. Cell. Physiol. 2012, 227, 3828–3836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koli, K.; Ryynänen, M.J.; Keski-Oja, J. Latent TGF-β binding proteins (LTBPs)-1 and-3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone 2008, 43, 679–688. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-β family signaling in connective tissue and skeletal diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a022269. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Windpassinger, C.; Vitcu, I.; Orlic, M.; Rafiq, M.A.; Khalid, M.; Malik, M.N.; Ayub, M.; Alman, B.; Vincent, J.B. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-β binding protein 3. Am. J. Hum. Genet. 2009, 84, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huckert, M.; Stoetzel, C.; Morkmued, S.; Laugel-Haushalter, V.; Geoffroy, V.; Muller, J.; Clauss, F.; Prasad, M.K.; Obry, F.; Raymond, J.L.; et al. Mutations in the latent TGF-β binding protein 3 (LTBP3) gene cause brachyolmia with amelogenesis imperfecta. Hum. Mol. Genet. 2015, 24, 3038–3049. [Google Scholar] [CrossRef] [Green Version]
- Dugan, S.L.; Temme, R.T.; Olson, R.A.; Mikhailov, A.; Law, R.; Mahmood, H.; Noor, A.; Vincent, J.B. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. Part A 2015, 167, 1396–1399. [Google Scholar] [CrossRef]
- Kaur, R.; Siddiqui, I.; Mathur, V.; Jana, M.; Kabra, M.; Gupta, N. Bi-allelic loss-of-function novel variants in LTBP3 -related skeletal dysplasia: Report of first patient from India. Am. J. Med. Genet. Part A 2020, 182, 1944–1946. [Google Scholar] [CrossRef]
- Stanley, S.; Balic, Z.; Hubmacher, D. Acromelic dysplasias: How rare musculoskeletal disorders reveal biological functions of extracellular matrix proteins. Ann. N. Y. Acad. Sci. 2021, 1490, 57–76. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Le Goff, C.; Leo, P.J.; Kenna, T.J.; Keith, P.; Harris, J.E.; Steer, R.; Bole-Feysot, C.; Nitschke, P.; Kielty, C.; et al. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J. Med. Genet. 2016, 53, 457–464. [Google Scholar] [CrossRef]
- Marzin, P.; Thierry, B.; Dancasius, A.; Cavau, A.; Michot, C.; Rondeau, S.; Baujat, G.; Phan, G.; Bonnière, M.; Le Bourgeois, M.; et al. Geleophysic and acromicric dysplasias: Natural history, genotype-phenotype correlations, and management guidelines from 38 cases. Genet. Med. 2021, 23, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Dabovic, B.; Chen, Y.; Colarossi, C.; Zambuto, L.; Obata, H.; Rifkin, D.B. Bone defects in latent TGF-β binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-β presentation. J. Endocrinol. 2002, 175, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Dabovic, B.; Chen, Y.; Colarossi, C.; Obata, H.; Zambuto, L.; Perle, M.A.; Rifkin, D.B. Bone abnormalities in latent TGF-β binding protein (Ltbp)-3–null mice indicate a role for Ltbp-3 in modulating TGF-β bioavailability. J. Cell Biol. 2002, 156, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabovic, B.; Levasseur, R.; Zambuto, L.; Chen, Y.; Karsenty, G.; Rifkin, D.B. Osteopetrosis-like phenotype in latent TGF-β binding protein 3 deficient mice. Bone 2005, 37, 25–31. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Variants |
|---|---|
| Variants in whole genome | 5,759,180 |
| Private variants (absent from 62 control genomes) | 7398 |
| Protein-changing private variants | 55 |
| Phenotype | CAPN1:c.1295G>A | LTBP3:c.158delG |
|---|---|---|
| Cases (n = 2) | A/A | del/del |
| Non-affected parents (n = 2) | G/A | wt/del |
| Non-affected British Shorthair cats (n = 6) | G/G | wt/wt |
| Random-bred cats and cats from other breeds (n = 90) | G/G | wt/wt |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudd Garces, G.; Knebel, A.; Hülskötter, K.; Jagannathan, V.; Störk, T.; Hewicker-Trautwein, M.; Leeb, T.; Volk, H.A. LTBP3 Frameshift Variant in British Shorthair Cats with Complex Skeletal Dysplasia. Genes 2021, 12, 1923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121923
Rudd Garces G, Knebel A, Hülskötter K, Jagannathan V, Störk T, Hewicker-Trautwein M, Leeb T, Volk HA. LTBP3 Frameshift Variant in British Shorthair Cats with Complex Skeletal Dysplasia. Genes. 2021; 12(12):1923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121923
Chicago/Turabian StyleRudd Garces, Gabriela, Anna Knebel, Kirsten Hülskötter, Vidhya Jagannathan, Theresa Störk, Marion Hewicker-Trautwein, Tosso Leeb, and Holger A. Volk. 2021. "LTBP3 Frameshift Variant in British Shorthair Cats with Complex Skeletal Dysplasia" Genes 12, no. 12: 1923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121923