
Molecular and Metabolic Subtypes in Sporadic and Inherited Clear Cell Renal Cell Carcinoma


 , ,
, ,  and
and 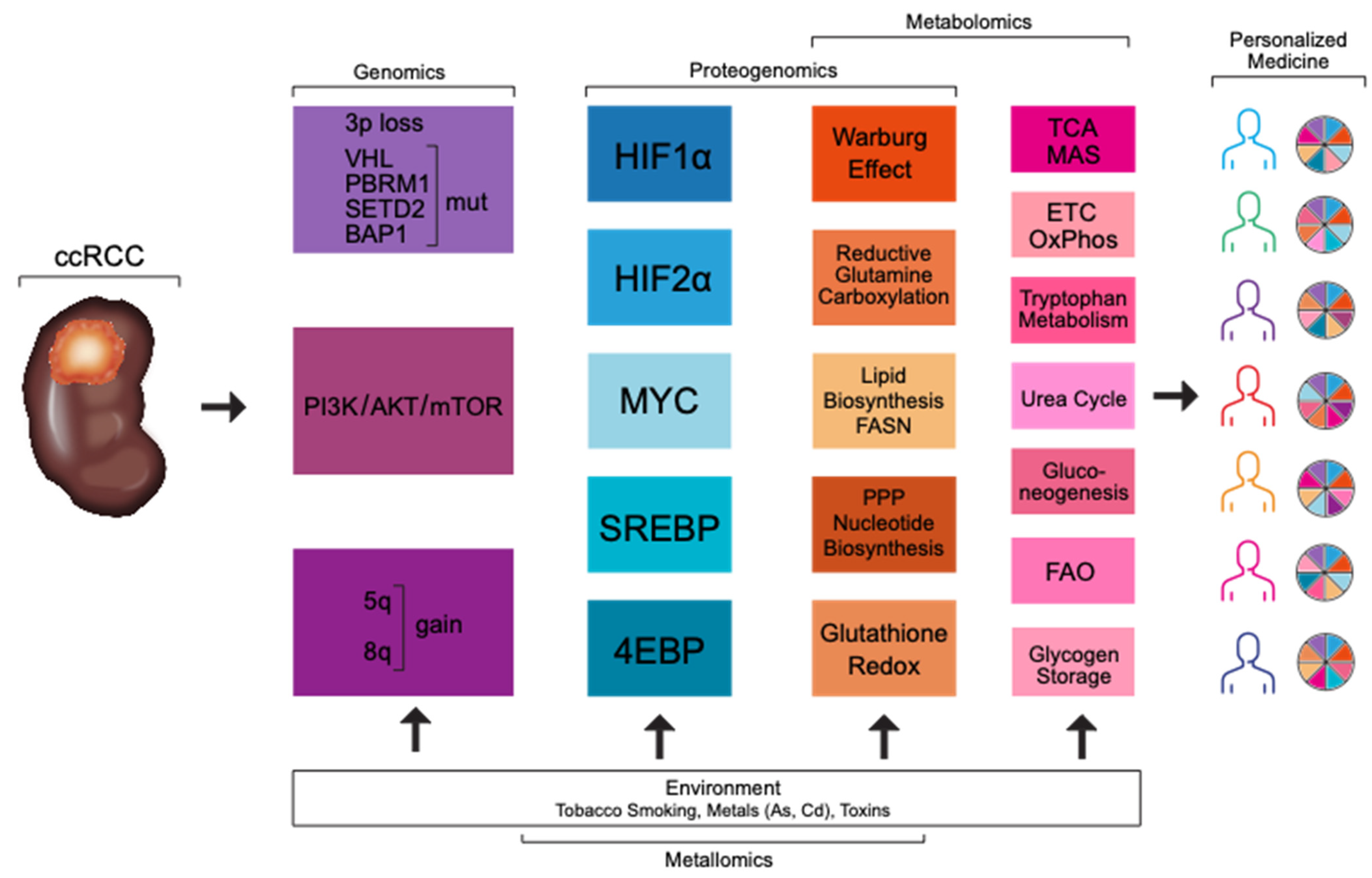{kind=link}
Abstract
:1. Introduction
2. Genomic Subtypes in Sporadic ccRCC
3. Transcriptomics
4. Proteomics
5. Metabolomics
6. Metallomics
7. ccRCC in VHL Disease
8. Moving Forwards
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610.e511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623.e617. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef]
- Espana-Agusti, J.; Warren, A.; Chew, S.K.; Adams, D.J.; Matakidou, A. Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat. Commun. 2017, 8, 2026. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pham, C.G.; Dong, Y.; Wang, P.I.; Osmangeyoglu, H.U.; Xie, Y.; Aras, O.; Han, S.; Oyama, T.; Takeda, S.; et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep. 2017, 18, 2893–2906. [Google Scholar] [CrossRef]
- Pawlowski, R.; Muhl, S.M.; Sulser, T.; Krek, W.; Moch, H.; Schraml, P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int. J. Cancer 2013, 132, E11–E17. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Kapur, P.; Pena-Llopis, S.; Christie, A.; Zhrebker, L.; Pavia-Jimenez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.C.; Park, I.Y.; Terzo, E.A.; Tripathi, D.N.; Mason, F.M.; Fahey, C.C.; Karki, M.; Shuster, C.B.; Sohn, B.H.; Chowdhury, P.; et al. SETD2 Haploinsufficiency for Microtubule Methylation Is an Early Driver of Genomic Instability in Renal Cell Carcinoma. Cancer Res. 2018, 78, 3135–3146. [Google Scholar] [CrossRef] [Green Version]
- Kanu, N.; Gronroos, E.; Martinez, P.; Burrell, R.A.; Yi Goh, X.; Bartkova, J.; Maya-Mendoza, A.; Mistrik, M.; Rowan, A.J.; Patel, H.; et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 2015, 34, 5699–5708. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, Y.; Yoshikawa, G.; Takasago, M.; Shimizu, S.; Tsutsumi, K. Extremely Delayed Multiple Brain Metastases from Renal Cell Carcinoma: Remission Achieved with Total Surgical Removal: Case Report and Literature Review. World Neurosurg. 2016, 92, 583.e513–583.e517. [Google Scholar] [CrossRef]
- Ishikawa, J.; Umezu, K.; Yamashita, H.; Maeda, S. Solitary brain metastasis from renal cell carcinoma 14 years after nephrectomy: A case report. Hinyokika Kiyo 1990, 36, 1439–1441. [Google Scholar]
- Kim, H.L.; Mayerson, E.; Lara, P.N.; Messing, E.; Tangen, C.; Shuch, B.M.; Vaishampayan, U. Considerations for the Next Clinical Trial Evaluating the Role of Cytoreductive Nephrectomy for Metastatic Renal Cell Carcinoma. Eur. Urol. Focus 2019, 5, 927–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejean, A.; Thezenas, S.; Chevreau, C.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H.; Guy, L.; Gravis, G.; et al. Cytoreductive nephrectomy (CN) in metastatic renal cancer (mRCC): Update on Carmena trial with focus on intermediate IMDC-risk population. J. Clin. Oncol. 2019, 37, 4508. [Google Scholar] [CrossRef]
- Vaishampayan, U.; George, J.; Vigneau, F. Predictors of Cytoreductive Nephrectomy for Metastatic Kidney Cancer in SEER and Metropolitan Detroit Databases. J. Kidney Cancer VHL 2019, 6, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Ghidini, M.; Petrelli, F.; Ghidini, A.; Tomasello, G.; Hahne, J.C.; Passalacqua, R.; Barni, S. Clinical development of mTor inhibitors for renal cancer. Expert Opin. Investig. Drugs 2017, 26, 1229–1237. [Google Scholar] [CrossRef]
- González-Larriba, J.L.; Maroto, P.; Durán, I.; Lambea, J.; Flores, L.; Castellano, D. The role of mTOR inhibition as second-line therapy in metastatic renal carcinoma: Clinical evidence and current challenges. Expert Rev. Anticancer Ther. 2017, 17, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bedke, J.; Stühler, V.; Stenzl, A.; Brehmer, B. Immunotherapy for kidney cancer: Status quo and the future. Curr. Opin. Urol. 2018, 28, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Considine, B.; Hurwitz, M.E. Current Status and Future Directions of Immunotherapy in Renal Cell Carcinoma. Curr. Oncol. Rep. 2019, 21, 34. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Linehan, W.M. Gender Specific Mutation Incidence and Survival Associations in Clear Cell Renal Cell Carcinoma (CCRCC). PLoS ONE 2015, 10, e0140257. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e512. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007, 39, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Perez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; Lopez-Larrubia, P.; et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Mijn, J.C.; Fu, L.; Khani, F.; Zhang, T.; Molina, A.M.; Barbieri, C.E.; Chen, Q.; Gross, S.S.; Gudas, L.J.; Nanus, D.M. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple HIF1α-Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2020, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Gatto, F.; Nookaew, I.; Nielsen, J. Chromosome 3p loss of heterozygosity is associated with a unique metabolic network in clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, E866–E875. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Ochocki, J.D.; Khare, S.; Hess, M.; Ackerman, D.; Qiu, B.; Daisak, J.I.; Worth, A.J.; Lin, N.; Lee, P.; Xie, H.; et al. Arginase 2 Suppresses Renal Carcinoma Progression via Biosynthetic Cofactor Pyridoxal Phosphate Depletion and Increased Polyamine Toxicity. Cell Metab. 2018, 27, 1263–1280.e1266. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interpheron alpha or both fo advanced renal-cell carinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Brunet, J.P.; Di Napoli, A.; Mertz, K.D.; Seeley, A.; Pires, M.M.; Linhart, D.; Worrell, R.A.; Moch, H.; Rubin, M.A.; et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69, 4674–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Klatte, T.; Kroeger, N.; Rampersaud, E.N.; Birkhäuser, F.D.; Logan, J.E.; Sonn, G.; Riss, J.; Rao, P.N.; Kabbinavar, F.F.; Belldegrun, A.S.; et al. Gain of chromosome 8q is associated with metastases and poor survival of patients with clear cell renal cell carcinoma. Cancer 2012, 118, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-α effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grampp, S.; Platt, J.L.; Lauer, V.; Salama, R.; Kranz, F.; Neumann, V.K.; Wach, S.; Stöhr, C.; Hartmann, A.; Eckardt, K.U.; et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 2016, 7, 13183. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinié, V.; Sibony, M.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroto, P.; Esteban, E.; Parra, E.F.; Mendez-Vidal, M.J.; Domenech, M.; Pérez-Valderrama, B.; Calderero, V.; Pérez-Gracia, J.L.; Grande, E.; Algaba, F. HIF pathway and c-Myc as biomarkers for response to sunitinib in metastatic clear-cell renal cell carcinoma. OncoTargets Ther. 2017, 10, 4635–4643. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.T.; Smith, A.M.; Kardos, J.; Wobker, S.E.; Wilson, H.L.; Krishnan, B.; Saito, R.; Lee, H.J.; Zhang, J.; Eaton, S.C.; et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun. 2017, 8, 15770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Gallo, C.A.; Khatri, S.; Barger, J.F.; Yepiskoposyan, H.; Plas, D.R. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 2361–2365. [Google Scholar] [CrossRef] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.A.; Brannon, A.R.; Parker, J.S.; Fisher, J.C.; Sen, O.; Kattan, M.W.; Hakimi, A.A.; Hsieh, J.J.; Choueiri, T.K.; Tamboli, P.; et al. ClearCode34: A prognostic risk predictor for localized clear cell renal cell carcinoma. Eur. Urol. 2014, 66, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, L.; Yu, Z.; Chai, K.; Chen, J. Identification of a Glucose Metabolism-related Signature for prediction of Clinical Prognosis in Clear Cell Renal Cell Carcinoma. J. Cancer 2020, 11, 4996–5006. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e804. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor microenvironment dynamics in clear cell renal cell carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, L.; Bonetti, L.; Brenner, D. Metabolic modulation of immunity: A new concept in cancer immunotherapy. Cell Rep. 2020, 32, 107848. [Google Scholar] [CrossRef]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.M.; Chang, H.Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e931. [Google Scholar] [CrossRef] [Green Version]
- Neely, B.A.; Wilkins, C.E.; Marlow, L.A.; Malyarenko, D.; Kim, Y.; Ignatchenko, A.; Sasinowska, H.; Sasinowski, M.; Nyalwidhe, J.O.; Kislinger, T.; et al. Proteotranscriptomic Analysis Reveals Stage Specific Changes in the Molecular Landscape of Clear-Cell Renal Cell Carcinoma. PLoS ONE 2016, 11, e0154074. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Masui, O.; Desouza, L.V.; Krakovska, O.; Metias, S.; Romaschin, A.D.; Honey, R.J.; Stewart, R.; Pace, K.; Lee, J.; et al. Quantitative proteomic analysis reveals potential diagnostic markers and pathways involved in pathogenesis of renal cell carcinoma. Oncotarget 2014, 5, 506–518. [Google Scholar] [CrossRef]
- Reigle, J.; Secic, D.; Biesiada, J.; Wetzel, C.; Shamsaei, B.; Chu, J.; Zang, Y.; Zhang, X.; Talbot, N.J.; Bischoff, M.E.; et al. Tobacco smoking induces metabolic reprogramming of renal cell carcinoma. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Steuer, R. Review: On the analysis and interpretation of correlations in metabolomic data. Brief. Bioinform. 2006, 7, 151–158. [Google Scholar] [CrossRef]
- Rosato, A.; Tenori, L.; Cascante, M.; De Atauri Carulla, P.R.; Martins Dos Santos, V.A.P.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Wettersten, H.I.; Hakimi, A.A.; Morin, D.; Bianchi, C.; Johnstone, M.E.; Donohoe, D.R.; Trott, J.F.; Aboud, O.A.; Stirdivant, S.; Neri, B.; et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. 2015, 75, 2541–2552. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Tannir, N.M.; Mier, J.W.; DeMichele, A.; Telli, M.L.; Fan, A.C.; Munster, P.N.; Carvajal, R.D.; Orford, K.W.; Bennett, M.K.; et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS), alone and in combination with everolimus (E) in patients (pts) with renal cell cancer (RCC). J. Clin. Oncol. 2016, 34, 4568. [Google Scholar] [CrossRef]
- Tannir, N.M.; Fan, A.C.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Meric-Bernstam, F.; DeMichele, A.; Voss, M.H.; et al. Phase 1 study of glutaminase (GLS) inhibitor CB-839 combined with either everolimus (E) or cabozantinib (Cabo) in patients (pts) with clear cell (cc) and papillary (pap) metastatic renal cell cancer (mRCC). J. Clin. Oncol. 2018, 36, 603. [Google Scholar] [CrossRef]
- Gulati, S.; Vaishampayan, U. Current State of Systemic Therapies for Advanced Renal Cell Carcinoma. Curr. Oncol. Rep. 2020, 22, 26. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Minton, D.R.; Fu, L.; Mongan, N.P.; Shevchuk, M.M.; Nanus, D.M.; Gudas, L.J. Role of NADH Dehydrogenase (Ubiquinone) 1 α Subcomplex 4-Like 2 in Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 2018, 28, 793–800.e792. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Furberg, H.; Zabor, E.C.; Jacobsen, A.; Schultz, N.; Ciriello, G.; Mikklineni, N.; Fiegoli, B.; Kim, P.H.; Voss, M.H.; et al. An epidemiologic and genomic investigation into the obesity paradox in renal cell carcinoma. J. Natl. Cancer Inst. 2013, 105, 1862–1870. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Hakimi, A.A.; Xie, W.; McKay, R.R.; Simantov, R.; Lin, X.; Lee, J.L.; Rini, B.I.; Srinivas, S.; Bjarnason, G.A.; et al. Body Mass Index and Metastatic Renal Cell Carcinoma: Clinical and Biological Correlations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020, 21, 283–293. [Google Scholar] [CrossRef]
- Singh, S.; Karthikeyan, C.; Moorthy, N. Recent Advances in the Development of Fatty Acid Synthase Inhibitors as Anticancer Agents. Mini Rev. Med. Chem. 2020, 20, 1820–1837. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. 2017, 35, 461.e415–461.e427. [Google Scholar] [CrossRef]
- Kim, K.; Taylor, S.L.; Ganti, S.; Guo, L.; Osier, M.V.; Weiss, R.H. Urine metabolomic analysis identifies potential biomarkers and pathogenic pathways in kidney cancer. Omics 2011, 15, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bullock, K.; Gurjao, C.; Braun, D.; Shukla, S.A.; Bossé, D.; Lalani, A.A.; Gopal, S.; Jin, C.; Horak, C.; et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 2019, 10, 4346. [Google Scholar] [CrossRef] [Green Version]
- Lara, P.; Bauer, T.M.; Hamid, O.; Smith, D.C.; Gajewski, T.; Gangadhar, T.C.; Somer, B.G.; Schmidt, E.V.; Zhao, Y.; Gowda, H.; et al. Epacadostat plus pembrolizumab in patients with advanced RCC: Preliminary phase I/II results from ECHO-202/KEYNOTE-037. J. Clin. Oncol. 2017, 35, 4515. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol. 2018, 36, 108. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.H.M.; Theiner, S.; Kornauth, C.; Meier-Menches, S.M.; Heffeter, P.; Berger, W.; Koellensperger, G.; Keppler, B.K. Bioimaging of isosteric osmium and ruthenium anticancer agents by LA-ICP-MS. Metallomics 2018, 10, 388–396. [Google Scholar] [CrossRef]
- Espina, M.; Corte-Rodríguez, M.; Aguado, L.; Montes-Bayón, M.; Sierra, M.I.; Martínez-Camblor, P.; Blanco-González, E.; Sierra, L.M. Cisplatin resistance in cell models: Evaluation of metallomic and biological predictive biomarkers to address early therapy failure. Metallomics 2017, 9, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Wolters, D.A.; Stefanopoulou, M.; Dyson, P.J.; Groessl, M. Combination of metallomics and proteomics to study the effects of the metallodrug RAPTA-T on human cancer cells. Metallomics 2012, 4, 1185–1196. [Google Scholar] [CrossRef]
- Bartnicka, J.J.; Blower, P.J. Insights into Trace Metal Metabolism in Health and Disease from PET: “PET Metallomics”. J. Nucl. Med. 2018, 59, 1355–1359. [Google Scholar] [CrossRef]
- Hueting, R.; Kersemans, V.; Tredwell, M.; Cornelissen, B.; Christlieb, M.; Gee, A.D.; Passchier, J.; Smart, S.C.; Gouverneur, V.; Muschel, R.J.; et al. A dual radiolabelling approach for tracking metal complexes: Investigating the speciation of copper bis(thiosemicarbazonates) in vitro and in vivo. Metallomics 2015, 7, 795–804. [Google Scholar] [CrossRef]
- Markolovic, S.; Wilkins, S.E.; Schofield, C.J. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20712–20722. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Zhang, X.; Vertino, P.M.; Cheng, X. The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations. J. Biol. Chem. 2015, 290, 20723–20733. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [Green Version]
- Mou, Y.; Wu, J.; Zhang, Y.; Abdihamid, O.; Duan, C.; Li, B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer 2021, 21, 18. [Google Scholar] [CrossRef]
- Mou, Y.; Zhang, Y.; Wu, J.; Hu, B.; Zhang, C.; Duan, C.; Li, B. The Landscape of Iron Metabolism-Related and Methylated Genes in the Prognosis Prediction of Clear Cell Renal Cell Carcinoma. Front. Oncol. 2020, 10, 788. [Google Scholar] [CrossRef]
- Rehwald, C.; Schnetz, M.; Urbschat, A.; Mertens, C.; Meier, J.K.; Bauer, R.; Baer, P.; Winslow, S.; Roos, F.C.; Zwicker, K.; et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br. J. Cancer 2020, 122, 421–433. [Google Scholar] [CrossRef]
- Wu, G.; Wang, Q.; Xu, Y.; Li, Q.; Cheng, L. A new survival model based on ferroptosis-related genes for prognostic prediction in clear cell renal cell carcinoma. Aging 2020, 12, 14933–14948. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, L.; Bai, Q.; Long, Q.; Wang, J.; Xi, W.; Xu, J.; Guo, J. Prognostic value of copper transporter 1 expression in patients with clear cell renal cell carcinoma. Oncol. Lett. 2017, 14, 5791–5800. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Liu, L.; Long, Q.; Bai, Q.; Wang, J.; Xu, J.; Guo, J. Decreased expression of CTR2 predicts poor prognosis of patients with clear cell renal cell carcinoma. Urol. Oncol. 2016, 34, 5.e1–5.e9. [Google Scholar] [CrossRef]
- Adam, T.; Becker, T.M.; Chua, W.; Bray, V.; Roberts, T.L. The Multiple Potential Biomarkers for Predicting Immunotherapy Response-Finding the Needle in the Haystack. Cancers 2021, 13, 277. [Google Scholar] [CrossRef]
- Mikhail, M.I.; Singh, A.K. Von Hippel Lindau Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, Finland, 2020. [Google Scholar]
- Ashouri, K.; Mohseni, S.; Tourtelot, J.; Sharma, P.; Spiess, P.E. Implications of Von Hippel-Lindau Syndrome and Renal Cell Carcinoma. J. Kidney Cancer VHL 2015, 2, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.; Horswell, S.; Rowan, A.; Salm, M.P.; de Bruin, E.C.; Gulati, S.; McGranahan, N.; Stares, M.; Gerlinger, M.; Varela, I.; et al. Development of synchronous VHL syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol. 2014, 15, 433. [Google Scholar] [CrossRef] [PubMed]
- Fei, S.S.; Mitchell, A.D.; Heskett, M.B.; Vocke, C.D.; Ricketts, C.J.; Peto, M.; Wang, N.J.; Sönmez, K.; Linehan, W.M.; Spellman, P.T. Patient-specific factors influence somatic variation patterns in von Hippel-Lindau disease renal tumours. Nat. Commun. 2016, 7, 11588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann. Oncol. 2011, 22, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: A single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. J. Clin. Oncol. 2020, 38, 5003. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czyzyk-Krzeska, M.F.; Landero Figueroa, J.A.; Gulati, S.; Cunningham, J.T.; Meller, J.; ShamsaeI, B.; Vemuri, B.; Plas, D.R. Molecular and Metabolic Subtypes in Sporadic and Inherited Clear Cell Renal Cell Carcinoma. Genes 2021, 12, 388. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030388
Czyzyk-Krzeska MF, Landero Figueroa JA, Gulati S, Cunningham JT, Meller J, ShamsaeI B, Vemuri B, Plas DR. Molecular and Metabolic Subtypes in Sporadic and Inherited Clear Cell Renal Cell Carcinoma. Genes. 2021; 12(3):388. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030388
Chicago/Turabian StyleCzyzyk-Krzeska, Maria F., Julio A. Landero Figueroa, Shuchi Gulati, John T. Cunningham, Jarek Meller, Behrouz ShamsaeI, Bhargav Vemuri, and David R. Plas. 2021. "Molecular and Metabolic Subtypes in Sporadic and Inherited Clear Cell Renal Cell Carcinoma" Genes 12, no. 3: 388. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030388