
The Genes of Freedom: Genome-Wide Insights into Marronage, Admixture and Ethnogenesis in the Gulf of Guinea

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Samples
2.2. Expanded Exome Sequencing, Variant Calling and Quality Control
2.3. Population Structure Analyses
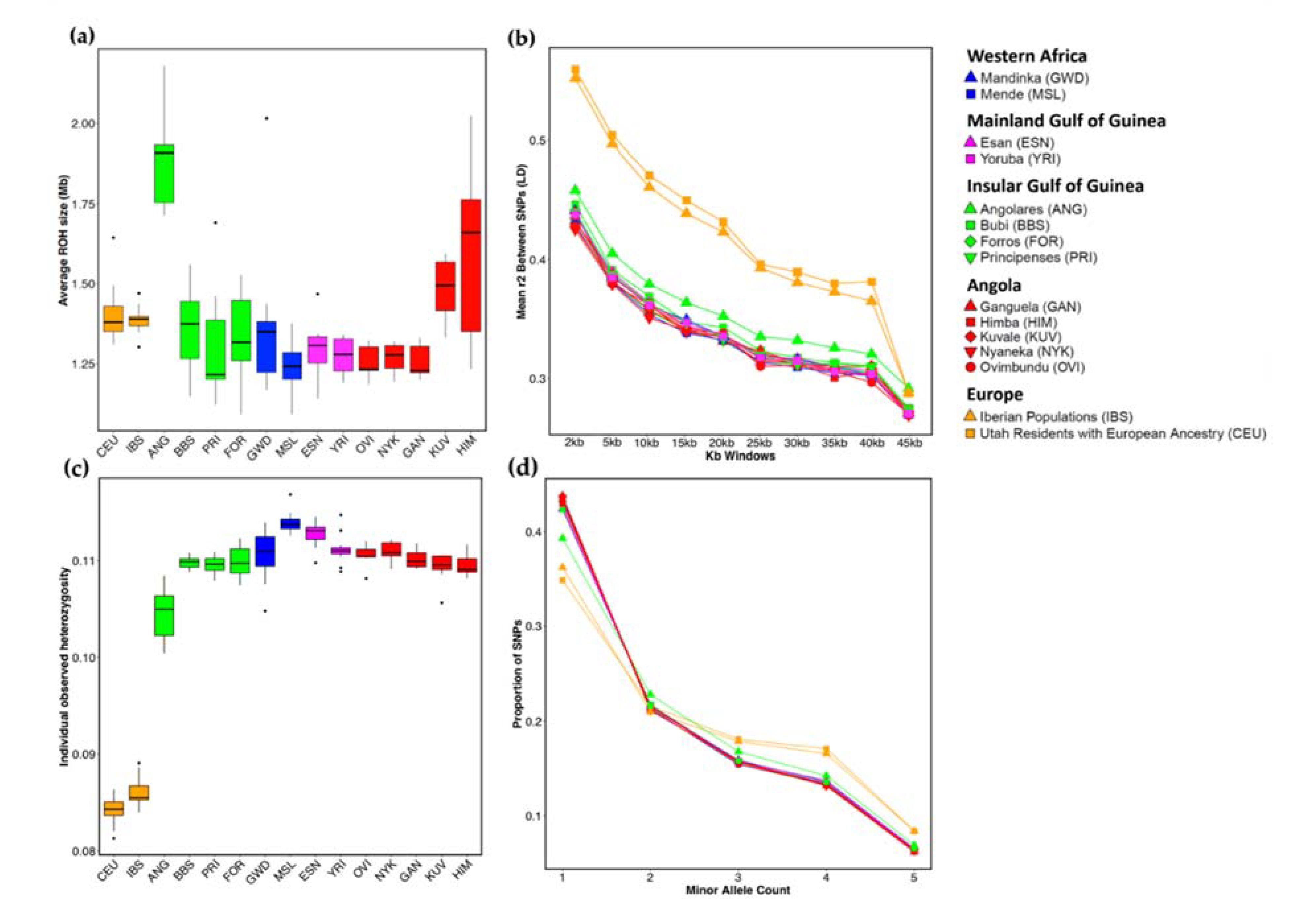
2.4. Genetic Diversity
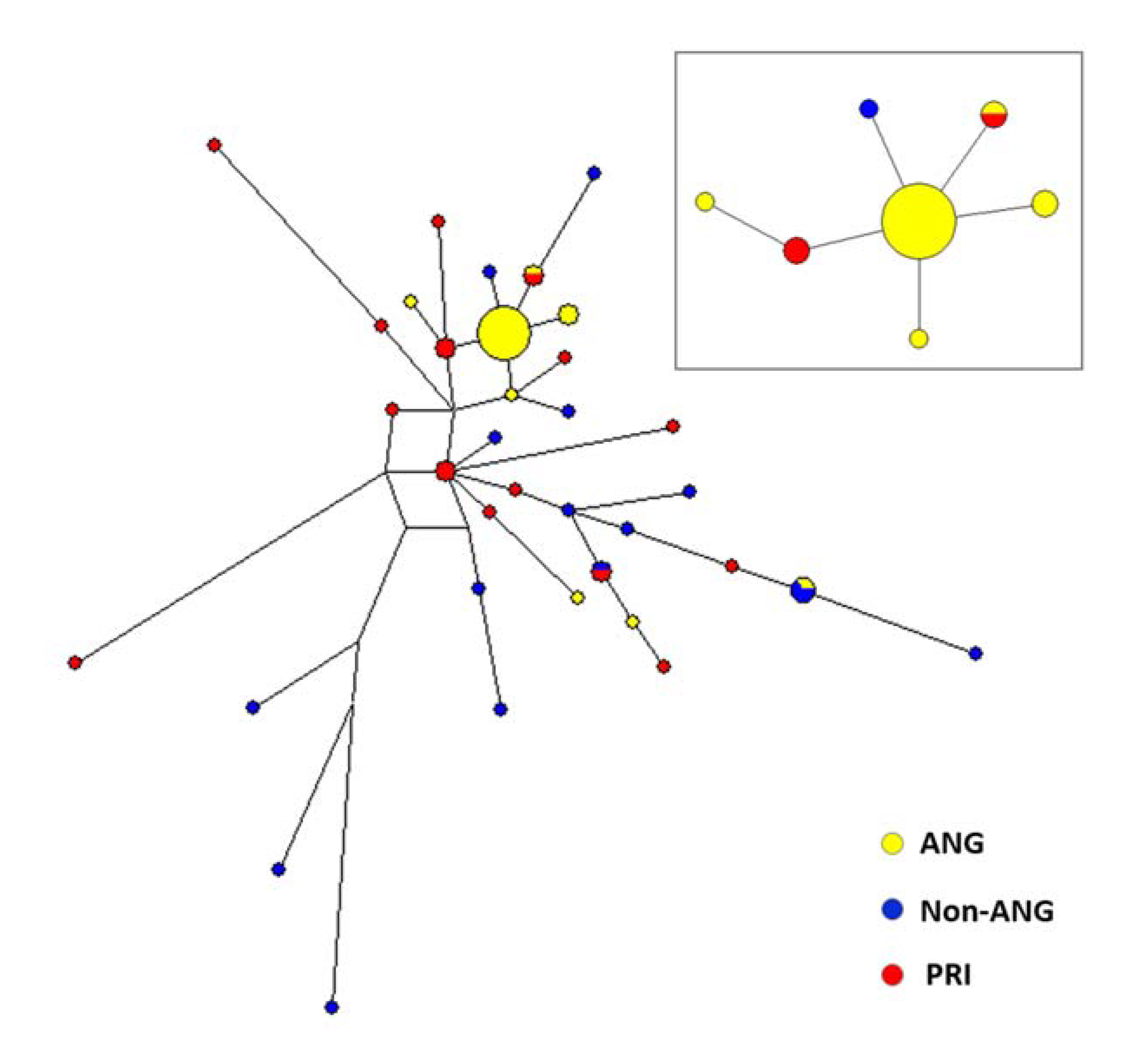
2.5. Mitochondrial DNA and Y-Chromosome Variation
3. Results
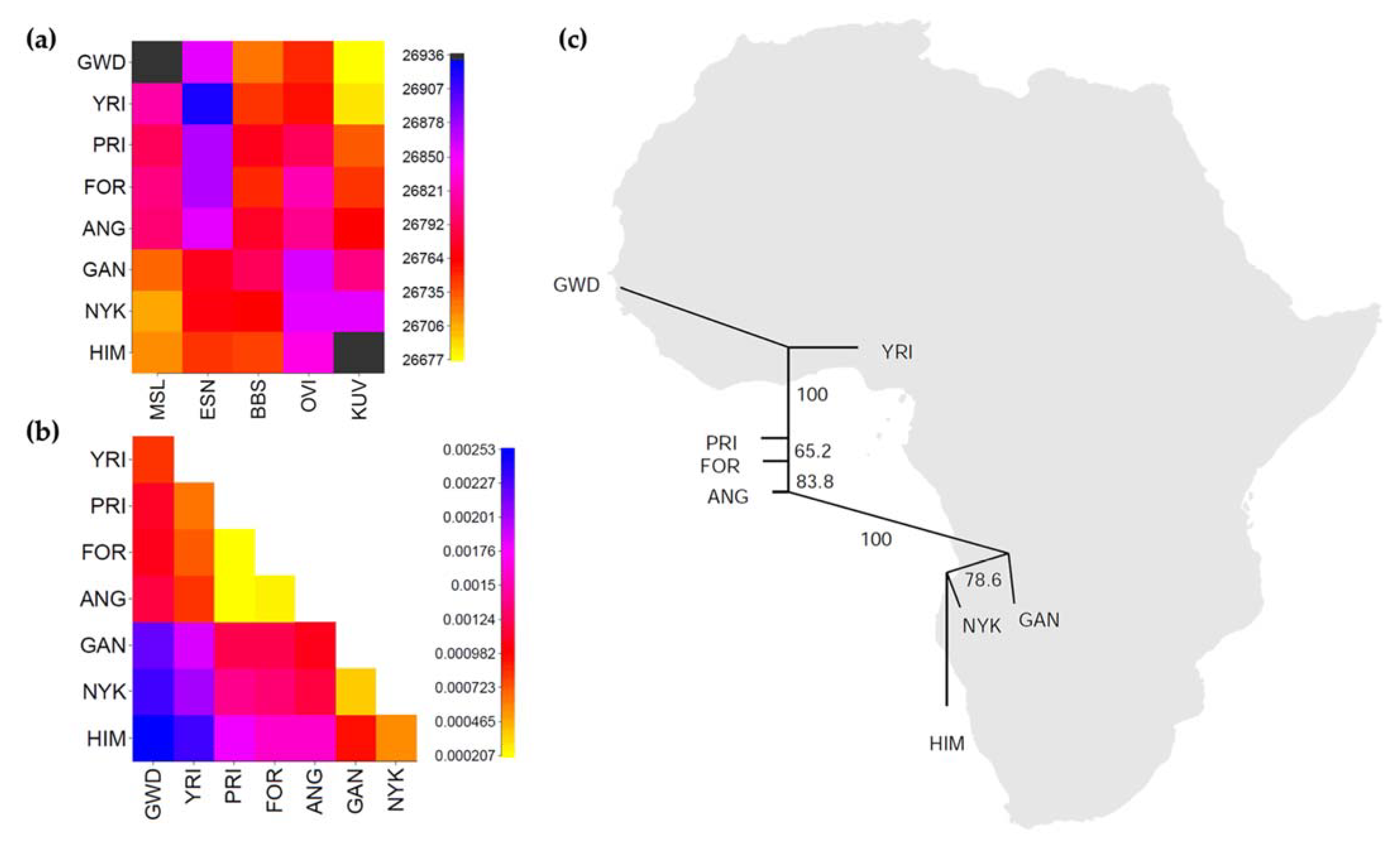
3.1. Genetic Structure
3.2. Genetic Diversity
3.3. Reanalyzing Previously Generated Uniparental Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Curtin, P.D. The Rise and Fall of the Plantation Complex; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Gelabert, P.; Ferrando-Bernal, M.; de-Dios, T.; Mattorre, B.; Campoy, E.; Gorostiza, A.; Patin, E.; González-Martín, A.; Lalueza-Fox, C. Genome-Wide Data from the Bubi of Bioko Island Clarifies the Atlantic Fringe of the Bantu Dispersal. BMC Genom. 2019, 20, 179. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, A.M. Mulheres, Sexualidade e Casamento Em São Tomé e Príncipe (Séculos XV-XVIII); Edições Cosmos: Lisbon, Portugal, 1999. [Google Scholar]
- Caldeira, A.M. Do Refúgio Nos Picos da Ilha de São Tomé à Absorção Colonial: A Questão Dos Angolares [From the Refuge on the Peaks of São Tomé Island to Colonial Absorption: The Issue of the Angolares]. Biblos 2018, 4, 123–147. [Google Scholar] [CrossRef]
- Tenreiro, F. A Ilha de São Tomé; Memórias da Junta de Investigaçãoes Científicas do Ultramar: Lisbon, Portugal, 1961. [Google Scholar]
- Klein, H.S. The Atlantic Slave Trade; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Hagemeijer, T. From Creoles to Portuguese. In The Portuguese Language Continuum in Africa and Brazil; López, L.Á., Gonçalves, P., de Avelar, J.O., Eds.; John Benjamins: Amsterdam, The Netherlands, 2018; pp. 169–184. [Google Scholar] [CrossRef]
- Seibert, G. Castaways, Autochthons, or Maroons? The Debate on the Angolares of São Tomé Island. In Creole Societies in the Portuguese Colonial Empire; Havik, P.J., Newitt, M., Eds.; Bristol University Press: Bristol, UK, 2007; pp. 105–126. [Google Scholar]
- Maurer, P.; Angolar. Languages Portuguese-Based, Spanish-Based and French-Based Languages. In The Survey of Pidgin and Creole; Michaelis, S.M., Maurer, P., Haspelmath, M., Huber, M., Eds.; Oxford University Press: Oxford, UK, 2013; Volume 2, pp. 59–71. [Google Scholar]
- Hagemeijer, T.; Zamora, A. Fa d’Ambô: From Past to Present. Int. J. Sociol. Lang. 2016, 193–209. [Google Scholar] [CrossRef] [Green Version]
- Hagemeijer, T. The Gulf of Guinea Creoles: Genetic and Typological Relations. J. Pidgin Creole Lang. 2011, 26, 111–154. [Google Scholar] [CrossRef]
- Hagemeijer, T.; Rocha, J. Creole Languages and Genes: The Case of São Tomé and Príncipe. Faits Lang. 2019, 49, 167–182. [Google Scholar] [CrossRef]
- Ryder, A. Benin and the Europeans 1485–1897; Longman: London, UK, 1969. [Google Scholar]
- Lorenzino, G. The Angolar Creole Portuguese of São Tomé: Its Grammar and Sociolinguistic History. Ph.D. Thesis, City University of New York, New York, NY, USA, 1998. [Google Scholar]
- Maurer, P. L’apport Lexical Bantou En Angolar. Afr. Arbeitspap. 1992, 29, 163–174. [Google Scholar]
- Ferraz, L.I. The Creole of São Tomé; Witwatersrand University Press: Johannesburg, South Africa, 1979. [Google Scholar]
- Freyre, G. Aventura e Rotina: Sugestões de Uma Viagem à Procura Das Constantes Portuguesas de Caráter e Ação; Livraria, J.O., Ed.; Topbooks: Rio de Janeiro, Brazil, 1953. [Google Scholar]
- Crawford, M.H. The Anthropological Genetics of the Black Caribs (Garifuna) of Central America and the Caribbean. Yearb. Phys. Anthropol. 1983, 26, 161–192. [Google Scholar] [CrossRef]
- Salas, A.; Richards, M.; Lareu, M.V.; Sobrino, B.; Silva, S.; Matamoros, M.; Macaulay, V.; Carracedo, A. Shipwrecks and Founder Effects: Divergent Demographic Histories Reflected in Caribbean MtDNA. Am. J. Phys. Anthropol. 2005, 128, 855–860. [Google Scholar] [CrossRef]
- Vansina, J. Quilombos on São Tomé, or in Search of Original Sources. Hist. Afr. 1996, 23, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, L.I. A Linguistic Appraisal of Angolar. In Memoriam Antonio Jorge Dias; Instituto de Alta Cultura/Junta de Investigações Cientificas do Ultramar: Lisbon, Portugal, 1974; Volume 2, pp. 177–186. [Google Scholar]
- Coelho, M.; Coia, C.A.V.; Luiselli, D.; Useli, A.; Hagemeijer, T.; Amorim, A.; Destro-Bisol, G.; Rocha, J. Human Microevolution and the Atlantic Slave Trade: A Case Study from Säo Tomé. Curr. Anthropol. 2008, 49, 134–143. [Google Scholar] [CrossRef]
- Coelho, M.; Sequeira, F.; Luiselli, D.; Beleza, S.; Rocha, J. On the Edge of Bantu Expansions: MtDNA, y Chromosome and Lactase Persistence Genetic Variation in Southwestern Angola. BMC Evol. Biol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, S.; Fehn, A.-M.; Aço, T.; Lages, F.; Gayà-Vidal, M.; Pakendorf, B.; Stoneking, M.; Rocha, J. Matriclans Shape Populations: Insights from the Angolan Namib Desert into the Maternal Genetic History of Southern Africa. Am. J. Phys. Anthropol. 2018, 165, 518–535. [Google Scholar] [CrossRef] [PubMed]
- Tomás, G.; Seco, L.; Seixas, S.; Faustino, P.; Lavinha, J.; Rocha, J. The Peopling of São Tomé (Gulf of Guinea): Origins of Slave Settlers and Admixture with the Portuguese. Hum. Biol. 2002, 74, 397–411. [Google Scholar] [CrossRef]
- 1000 Genomes Project, Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; [Software]; 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 27 May 2021).
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33); [Software]; 2011. Available online: https://github.com/najoshi/sickle (accessed on 27 May 2021).
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arxiv 2013, arXiv:1303.3997v2 [q-bio.GN]. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinforma. 2013, 43 (Suppl. 43). [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust Relationship Inference in Genome-Wide Association Studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs Explain a Large Proportion of the Heritability for Human Height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of Population Structure Using Dense Haplotype Data. PLoS Genet. 2012, 8, e1002453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies by Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Leslie, S.; Winney, B.; Hellenthal, G.; Davison, D.; Boumertit, A.; Day, T.; Hutnik, K.; Royrvik, E.C.; Cunliffe, B.; Lawson, D.J.; et al. The Fine-Scale Genetic Structure of the British Population. Nature 2015. [Google Scholar] [CrossRef] [Green Version]
- Van Dorp, L.; Balding, D.; Myers, S.; Pagani, L.; Tyler-Smith, C.; Bekele, E.; Tarekegn, A.; Thomas, M.G.; Bradman, N.; Hellenthal, G. Evidence for a Common Origin of Blacksmiths and Cultivators in the Ethiopian Ari within the Last 4500 Years: Lessons for Clustering-Based Inference. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 254–267. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, A.A.; Liu, K.Z.; Liu-Fang, G.; Nakka, P.; Ramachandran, S. Pong: Fast Analysis and Visualization of Latent Clusters in Population Genetic Data. Bioinformatics 2016, 32, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of Levels of Gene Flow from DNA Sequence Data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef]
- Keinan, A.; Mullikin, J.C.; Patterson, N.; Reich, D. Measurement of the Human Allele Frequency Spectrum Demonstrates Greater Genetic Drift in East Asians than in Europeans. Nat. Genet. 2007, 39, 1251–1255. [Google Scholar] [CrossRef] [Green Version]
- Kolde, R. Pheatmap: Pretty Heatmaps; 2013. [Software]. Available online: http://CRAN.R-project.org/package=pheatmap (accessed on 27 May 2021).
- Bernstein, F. Die Geographische Verteilung der Blutgruppen Und Ihre Anthropologische Bedeutung. In Comitato Italiano per lo Studio dei Problemi della Populazione; Instituto Poligrafico dello Stato: Rome, Italy, 1931; pp. 227–243. [Google Scholar]
- Verdu, P.; Jewett, E.M.; Pemberton, T.J.; Rosenberg, N.A.; Baptista, M. Parallel Trajectories of Genetic and Linguistic Admixture in a Genetically Admixed Creole Population. Curr. Biol. 2017, 27, 2529–2535.e3. [Google Scholar] [CrossRef]
- Bryc, K.; Auton, A.; Nelson, M.R.; Oksenberg, J.R.; Hauser, S.L.; Williams, S.; Froment, A.; Bodo, J.M.; Wambebe, C.; Tishkoff, S.A.; et al. Genome-Wide Patterns of Population Structure and Admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. USA 2010, 107, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Kloss-Brandstätter, A.; Pacher, D.; Schönherr, S.; Weissensteiner, H.; Binna, R.; Specht, G.; Kronenberg, F. HaploGrep: A Fast and Reliable Algorithm for Automatic Classification of Mitochondrial DNA Haplogroups. Hum. Mutat. 2011, 32, 25–32. [Google Scholar] [CrossRef]
- Athey, T.W. Haplogroup Prediction from Y-STR Values Using a Bayesian-Allele- Frequency Approach. J Genetic Genealogy 2006, 2, 34–39. [Google Scholar]
- Bandelt, H.J.; Forster, P.; Sykes, B.C.; Richards, M.B. Mitochondrial Portraits of Human Populations Using Median Networks. Genetics 1995, 141, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (Version 3.0): An Integrated Software Package for Population Genetics Data Analysis. Evol. Bioinforma. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Forster, P.; Harding, R.; Torroni, A.; Bandelt, H.J. Origin and Evolution of Native American MtDNA Variation: A Reappraisal. Am. J. Hum. Genet. 1996, 59, 935–945. [Google Scholar]
- Macaulay, V.; Soares, P.; Richards, M.B. Rectifying Long-Standing Misconceptions about the ρ Statistic for Molecular Dating. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Gusmão, L.; Sánchez-Diz, P.; Calafell, F.; Martín, P.; Alonso, C.A.; Álvarez-Fernández, F.; Alves, C.; Borjas-Fajardo, L.; Bozzo, W.R.; Bravo, M.L.; et al. Mutation Rates at Y Chromosome Specific Microsatellites. Hum. Mutat. 2005, 26, 520–528. [Google Scholar] [CrossRef]
- Balaresque, P.; Poulet, N.; Cussat-Blanc, S.; Gerard, P.; Quintana-Murci, L.; Heyer, E.; Jobling, M.A. Y-Chromosome Descent Clusters and Male Differential Reproductive Success: Young Lineage Expansions Dominate Asian Pastoral Nomadic Populations. Eur. J. Hum. Genet. 2015, 23, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, J.N. Cross-Cultural Estimation of the Human Generation Interval for Use in Genetics-Based Population Divergence Studies. Am. J. Phys. Anthropol. 2005, 128, 415–423. [Google Scholar] [CrossRef]
- Patin, E.; Lopez, M.; Grollemund, R.; Verdu, P.; Harmant, C.; Quach, H.; Laval, G.; Perry, G.H.; Barreiro, L.B.; Froment, A.; et al. Dispersals and Genetic Adaptation of Bantu-Speaking Populations in Africa and North America. Science 2017, 356, 543–546. [Google Scholar] [CrossRef] [Green Version]
- Semo, A.; Gayà-Vidal, M.; Fortes-Lima, C.; Alard, B.; Oliveira, S.; Almeida, J.; Prista, A.; Damasceno, A.; Fehn, A.-M.; Schlebusch, C.; et al. Along the Indian Ocean Coast: Genomic Variation in Mozambique Provides New Insights into the Bantu Expansion. Mol. Biol. Evol. 2020, 37. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Akey, J.M.; Jakobsson, M.; Pritchard, J.K.; Tishkoff, S.; Willerslev, E. Tracing the Peopling of the World through Genomics. Nature 2017, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of Homozygosity: Windows into Population History and Trait Architecture. Nat. Rev. Genet. 2018, 220–234. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; Mckeigue, P.M.; Wilson, J.F. Genomic Runs of Homozygosity Record Population History and Consanguinity. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.D. Double Descent and Its Correlates among the Herero of Ngamiland. Am. Anthropol. 1956, 58, 109–139. [Google Scholar] [CrossRef]
- Price, R. Maroon Societies. Rebel Slave Communities in the Americas; The John Hopkins University Press: Baltimore, MD, USA; London, UK, 1996. [Google Scholar]
- Seibert, G. Comrades, Clients, and Cousins. Colonialism, Socialism and Democratization in São Tomé and Príncipe; CNWS Publication: Leiden, The Netherlands, 1999. [Google Scholar]
- Zerjal, T.; Xue, Y.; Bertorelle, G.; Wells, R.S.; Bao, W.; Zhu, S.; Qamar, R.; Ayub, Q.; Mohyuddin, A.; Fu, S.; et al. The Genetic Legacy of the Mongols. Am. J. Hum. Genet. 2003, 72, 717–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lansing, J.S.; Watkins, J.C.; Hallmark, B.; Cox, M.P.; Karafet, T.M.; Sudoyo, H.; Hammer, M.F. Male Dominance Rarely Skews the Frequency Distribution of Y Chromosome Haplotypes in Human Populations. Proc. Natl. Acad. Sci. USA 2008, 105, 11645–11650. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P.W. Hopi Indians, “Cultural” Selection, and Albinism. Am. J. Phys. Anthropol. 2003, 121, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Neel, J.V. Lessons from a “Primitive” People. Science 1970, 170, 815–822. [Google Scholar] [CrossRef]
- MacCluer, J.W.; Neel, J.V.; Chagnon, N.A. Demographic Structure of a Primitive Population: A Simulation. Am. J. Phys. Anthropol. 1971, 35, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Heyer, E.; Sibert, A.; Austerlitz, F. Cultural Transmission of Fitness: Genes Take the Fast Lane. Trends Genet. 2005, 234–239. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.; Fehn, A.-M.; Ferreira, M.; Machado, T.; Hagemeijer, T.; Rocha, J.; Gayà-Vidal, M. The Genes of Freedom: Genome-Wide Insights into Marronage, Admixture and Ethnogenesis in the Gulf of Guinea. Genes 2021, 12, 833. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060833
Almeida J, Fehn A-M, Ferreira M, Machado T, Hagemeijer T, Rocha J, Gayà-Vidal M. The Genes of Freedom: Genome-Wide Insights into Marronage, Admixture and Ethnogenesis in the Gulf of Guinea. Genes. 2021; 12(6):833. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060833
Chicago/Turabian StyleAlmeida, João, Anne-Maria Fehn, Margarida Ferreira, Teresa Machado, Tjerk Hagemeijer, Jorge Rocha, and Magdalena Gayà-Vidal. 2021. "The Genes of Freedom: Genome-Wide Insights into Marronage, Admixture and Ethnogenesis in the Gulf of Guinea" Genes 12, no. 6: 833. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060833