
Genome Mining and Comparative Genome Analysis Revealed Niche-Specific Genome Expansion in Antibacterial Bacillus pumilus Strain SF-4

Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling, Isolation, and Antibacterial Activity
2.2. Genomic DNA Extraction and Strain Identification
2.3. Whole-Genome Sequence, Assembly, and Annotation
2.4. Genome Mining
2.5. Identification of Putative Horizontal Gene Transfer (HGT)
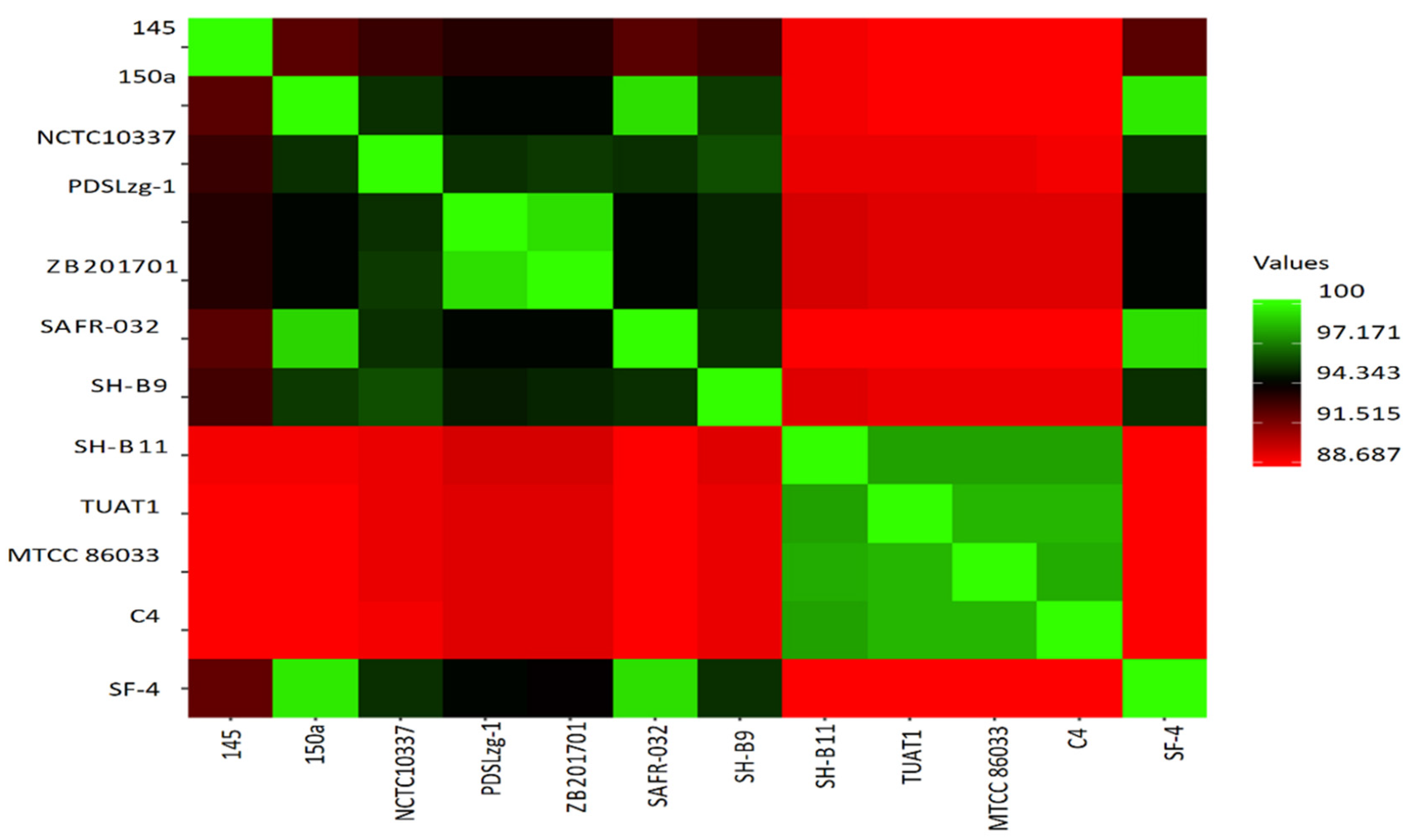
2.6. Comparative Genome Analysis
3. Results and Discussion
3.1. B. pumilus SF-4 Isolation and Antibacterial Activities
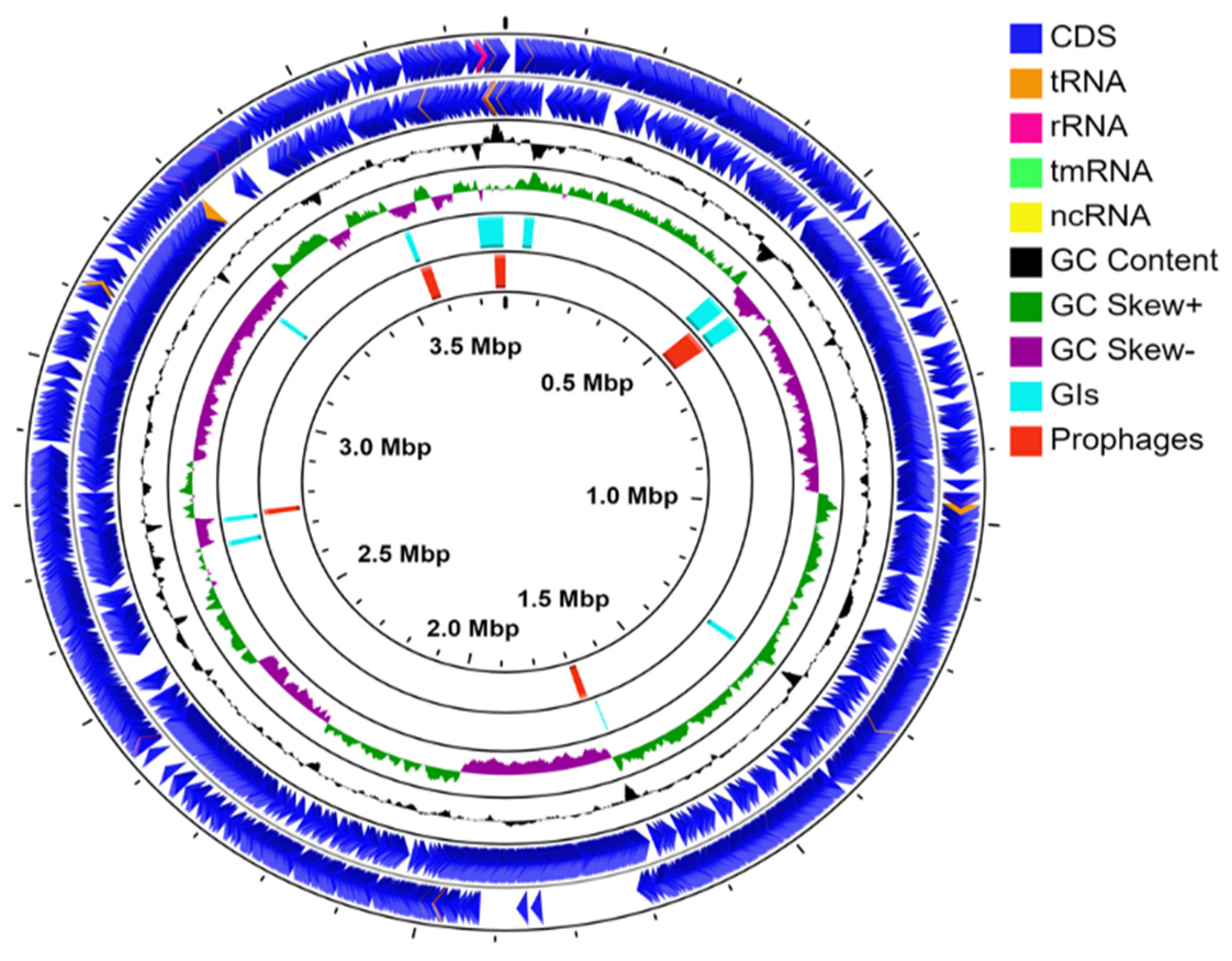
3.2. Genomic Features of B. pumilus SF-4
3.3. Genomic Islands (GIs) and Prophages
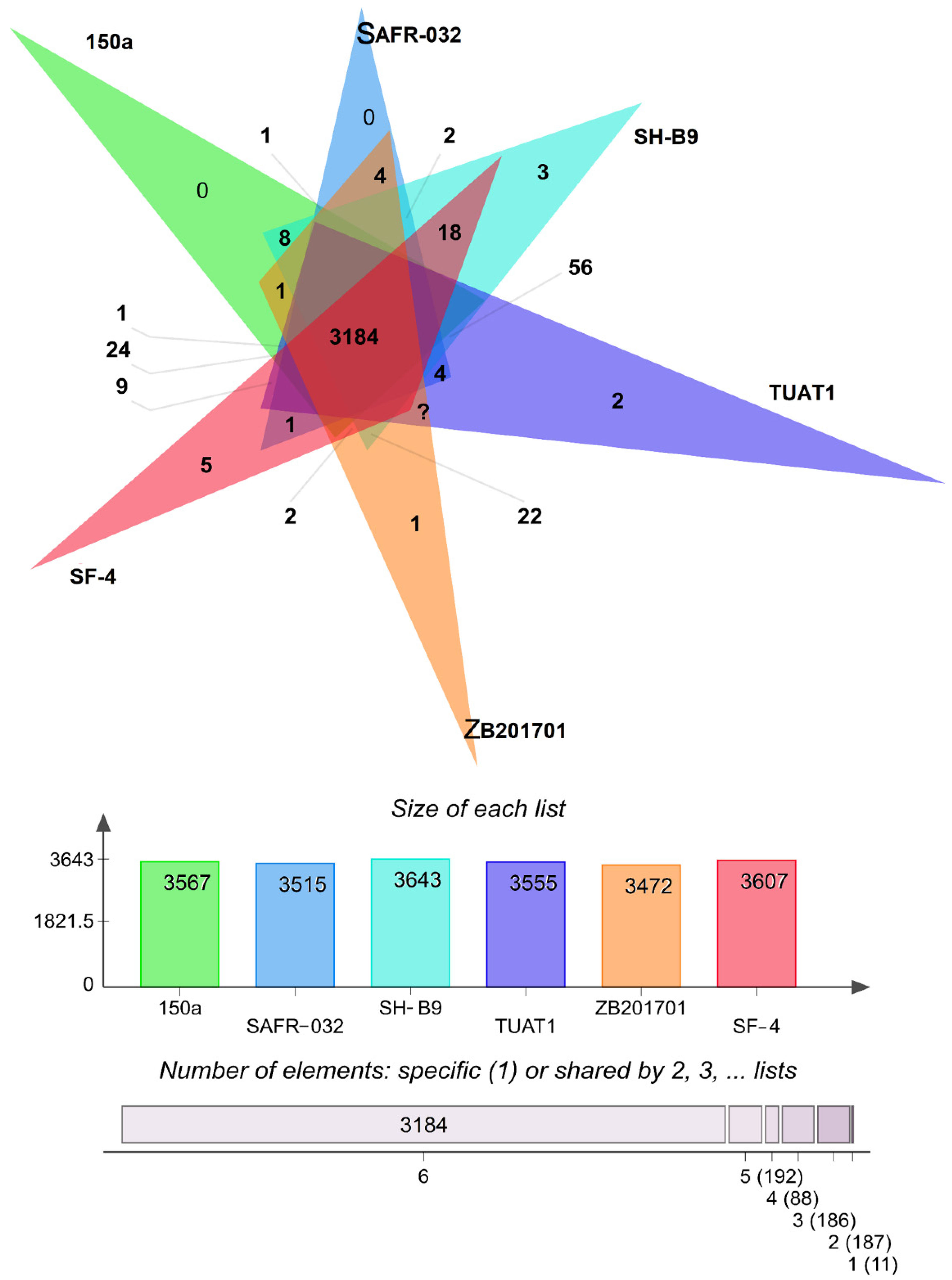
3.4. Pan-Core Genome Analysis
3.5. Unique Gene Pool in B. pumilus Strains
3.6. Comparison of BGCs in B. pumilus Strains
3.7. Comparative Genome Analysis
3.8. Plant Growth-Promoting Traits
3.8.1. Phosphate Solubilization
3.8.2. Iron Acquisition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forni, C.; Duca, D.; Glick, B.R. Mechanisms of plant response to salt and drought stress and their alteration by rhizobacteria. Plant. Soil 2017, 410, 335–356. [Google Scholar] [CrossRef]
- Han, Q.-Q.; Lü, X.-P.; Bai, J.-P.; Qiao, Y.; Paré, P.W.; Wang, S.-M.; Zhang, J.-L.; Wu, Y.-N.; Pang, X.-P.; Xu, W.-B.; et al. Beneficial soil bacterium Bacillus subtilis (GB03) augments salt tolerance of white clover. Front. Plant. Sci. 2014, 5, 525. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, M.; Wani, S.P. Rhizobacterial-plant interactions: Strategies ensuring plant growth promotion under drought and salinity stress. Agric. Ecosyst. Environ. 2016, 231, 68–78. [Google Scholar] [CrossRef]
- Numan, M.; Bashir, S.; Khan, Y.; Mumtaz, R.; Shinwari, Z.K.; Khan, A.L.; Khan, A.; Al-Harrasi, A. Plant growth promoting bacteria as an alternative strategy for salt tolerance in plants: A review. Microbiol. Res. 2018, 209, 21–32. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-Growth-Promoting Rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.W.; Islam, A.; Opdahl, L.; Davenport, J.R.; Sullivan, T.S. Comparative Genomics, Siderophore Production, and Iron Scavenging Potential of Root Zone Soil Bacteria Isolated from ‘Concord’ Grape Vineyards. Microb. Ecol. 2019, 78, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Fu, X.; Li, Y.; Wang, Q. Isolation and characterization of Bacillus amyloliquefaciens PG12 for the biological control of apple ring rot. Postharvest Biol. Technol. 2016, 115, 113–121. [Google Scholar] [CrossRef]
- Gioia, J.; Yerrapragada, S.; Qin, X.; Jiang, H.; Igboeli, O.C.; Muzny, D.; Dugan-Rocha, S.; Ding, Y.; Hawes, A.; Liu, W.; et al. Paradoxical DNA Repair and Peroxide Resistance Gene Conservation in Bacillus pumilus SAFR-032. PLoS ONE 2007, 2, e928. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Mañero, F.J.; Ramos-Solano, B.; Probanza, A.; Mehouachi, J.; Tadeo, F.R.; Talon, M. The plant-growth-promoting rhizobacteria Bacillus pumilus and Bacillus licheniformis produce high amounts of physiologically active gibberellins. Physiol. Plant. 2001, 111, 206–211. [Google Scholar] [CrossRef]
- Hill, J.E.; Baiano, J.C.F.; Barnes, A. Isolation of a novel strain of Bacillus pumilusfrom penaeid shrimp that is inhibitory against marine pathogens. J. Fish. Dis. 2009, 32, 1007–1016. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. Feeding on plastic. Science 2016, 351, 1154–1155. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Sano, N.; Yamada, T.; Ishii, K.; Kojima, K.; Djedidi, S.; Ramírez, M.D.A.; Yuan, K.; Kanekatsu, M.; Ohkama-Ohtsu, N.; et al. Complete Genome Sequence of Plant Growth-Promoting Bacillus pumilus TUAT1. Microbiol. Resour. Announc. 2019, 8, e00076-19. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, I.; Fazalur-Rahman; Ihsanullah; Fazlul-Haq. Degradation of Communal Natural Resources and Their Impacts on Mountain Women: A Case Study of Karak District Pakistan. Pak. J. Soc. Sci. 2012, 32, 157–169. [Google Scholar]
- Hockett, K.L.; Baltrus, D.A. Use of the Soft-agar Overlay Technique to Screen for Bacterially Produced Inhibitory Compounds. J. Vis. Exp. 2017, 55064, e55064. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, I.J.; Otto, T.D.; Berriman, M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11, R41. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; di Cuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Hsiao, W.W.L.; Brinkman, F. Detecting genomic islands using bioinformatics approaches. Nat. Rev. Genet. 2010, 8, 373–382. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabelsi, I.; Oves, D.; Manteca, A.; Genilloud, O.; Altalhi, A.; Nour, M. Antimicrobial Activities of Some Actinomycetes Isolated from Different Rhizospheric Soils in Tunisia. Curr. Microbiol. 2016, 73, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Rakhisi, Z.; Ahmady, A.Z. Isolation and Identification of Bacillus Species from Soil and Evaluation of Their Antibacterial Properties. Avicenna J. Clin. Microbiol. Infect. 2015, 2, 10–13. [Google Scholar] [CrossRef]
- Ramachandran, R.; Chalasani, A.G.; Lal, R.; Roy, U. A Broad-Spectrum Antimicrobial Activity of Bacillus subtilis RLID 12.1. Sci. World J. 2014, 2014, 968487. [Google Scholar] [CrossRef] [Green Version]
- Shafi, J.; Tian, H.; Ji, M. Bacillus species as versatile weapons for plant pathogens: A review. Biotechnol. Biotechnol. Equip. 2017, 31, 446–459. [Google Scholar] [CrossRef] [Green Version]
- Cook, R.J.; Weller, D.M.; El-Banna, A.Y.; Vakoch, D.; Zhang, H. Yield Responses of Direct-Seeded Wheat to Rhizobacteria and Fungicide Seed Treatments. Plant. Dis. 2002, 86, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Genet. 2009, 8, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, X.; Liang, Y.; Guo, X.; Xiao, Y.; Ma, L.; Miao, B.; Liu, H.; Peng, D.; Huang, W.; et al. Adaptive Evolution of Extreme Acidophile Sulfobacillus thermosulfidooxidans Potentially Driven by Horizontal Gene Transfer and Gene Loss. Appl. Environ. Microbiol. 2017, 83, e03098-16. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, G.W.; Franklin, M.; Dilworth, M.J. Effect of sulfur supply on sulfate uptake, and alkaline sulfatase activity in free-living and symbiotic bradyrhizobia. Arch. Microbiol. 1987, 149, 163–167. [Google Scholar] [CrossRef]
- Woods, L.C.; Gorrell, R.J.; Taylor, F.; Connallon, T.; Kwok, T.; McDonald, M.J. Horizontal gene transfer potentiates adaptation by reducing selective constraints on the spread of genetic variation. Proc. Natl. Acad. Sci. USA 2020, 117, 26868–26875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, D.; Kendall, J.R.A.; Borriss, R.; Druzhinina, I.S.; Kubicek, C.P.; Shen, Q.; Zhang, R. Comparative Genomic Analysis of Bacillus amyloliquefaciens and Bacillus subtilis Reveals Evolutional Traits for Adaptation to Plant-Associated Habitats. Front. Microbiol. 2016, 7, 2039. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Schmid, M.; van Tuinen, D.; Berg, G. Plant-driven selection of microbes. Plant. Soil 2009, 321, 235–257. [Google Scholar] [CrossRef]
- Hao, W.; Golding, G.B. The Fate of Laterally Transferred Genes: Life in the Fast Lane to Adaptation or Death. Genome Res. 2006, 16, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Richards, V.P.; Palmer, S.R.; Bitar, P.D.P.; Qin, X.; Weinstock, G.M.; Highlander, S.K.; Town, C.D.; Burne, R.A.; Stanhope, M.J. Phylogenomics and the Dynamic Genome Evolution of the Genus Streptococcus. Genome Biol. Evol. 2014, 6, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Perez, F.; Nataro, J.P. Bacterial Serine Proteases Secreted by Autotransporter Pathway: Classification, Specificity and Role in Virulence. Cell. Mol. Life Sci. 2014, 71, 745–770. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Qiu, Y.; Meng, X.; Feng, J.; Tao, J.; Liu, W. Correction to “A Heterotrimeric Dehydrogenase Complex Functions with 2 Distinct YcaO Proteins to Install 5 Azole Heterocycles in 35-Membered Sulfomycin Thiopeptides”. J. Am. Chem. Soc. 2020, 142, 8454–8463. [Google Scholar] [CrossRef]
- Deutscher, J.; Aké, F.M.D.; Derkaoui, M.; Zébré, A.C.; Cao, T.N.; Bouraoui, H.; Kentache, T.; Mokhtari, A.; Milohanic, E.; Joyet, P. The Bacterial Phosphoenolpyruvate:Carbohydrate Phosphotransferase System: Regulation by Protein Phosphorylation and Phosphorylation-Dependent Protein-Protein Interactions. Microbiol. Mol. Biol. Rev. 2014, 78, 231–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawaz, M.V.; Topper, M.E.; Firestine, S.M. The ATP-grasp enzymes. Bioorganic Chem. 2011, 39, 185–191. [Google Scholar] [CrossRef]
- Kikuma, T.; Ohtsu, M.; Utsugi, T.; Koga, S.; Okuhara, K.; Eki, T.; Fujimori, F.; Murakami, Y. Dbp9p, a Member of the DEAD Box Protein Family, Exhibits DNA Helicase Activity. J. Biol. Chem. 2004, 279, 20692–20698. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Sands, Z.A.; Biggin, P.C. A Numbering System for MFS Transporter Proteins. Front. Mol. Biosci. 2016, 3, 1–13. [Google Scholar] [CrossRef]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front. Microbiol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Zhou, S.; Raj, S.M.; Ashok, S.; Edwardraja, S.; Lee, S.-G.; Park, S. Cloning, Expression and Characterization of 3-Hydroxyisobutyrate Dehydrogenase from Pseudomonas denitrificans ATCC 13867. PLoS ONE 2013, 8, e62666. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.A.; Dantas, G. The Shared Antibiotic Resistome of Soil Bacteria and Human Pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [Green Version]
- Kananavičiūtė, R.; Kvederavičiūtė, K.; Dabkevičienė, D.; Mackevičius, G.; Kuisienė, N. Collagen-like sequences encoded by extremophilic and extremotolerant bacteria. Genomics 2020, 112, 2271–2281. [Google Scholar] [CrossRef]
- Bolotin, E.; Hershberg, R. Gene Loss Dominates As a Source of Genetic Variation within Clonal Pathogenic Bacterial Species. Genome Biol. Evol. 2015, 7, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of Nonribosomal Peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Koumoutsi, A.; Chen, X.-H.; Henne, A.; Liesegang, H.; Hitzeroth, G.; Franke, P.; Vater, J.; Borriss, R. Structural and Functional Characterization of Gene Clusters Directing Nonribosomal Synthesis of Bioactive Cyclic Lipopeptides in Bacillus amyloliquefaciens Strain FZB42. J. Bacteriol. 2004, 186, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Veith, B.; Herzberg, C.; Steckel, S.; Feesche, J.; Maurer, K.H.; Ehrenreich, P.; Bäumer, S.; Henne, A.; Liesegang, H.; Merkl, R.; et al. The Complete Genome Sequence of Bacillus licheniformis DSM13, an Organism with Great Industrial Potential. J. Mol. Microbiol. Biotechnol. 2004, 7, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Grangemard, I.; Wallach, J.; Maget-Dana, R.; Peypoux, F. Lichenysin: A More Efficient Cation Chelator Than Surfactin. Appl. Biochem. Biotechnol. Part A Enzym. Eng. Biotechnol. 2001, 90, 199–210. [Google Scholar] [CrossRef]
- Köcher, S.; Breitenbach, J.; Müller, V.; Sandmann, G. Structure, function and biosynthesis of carotenoids in the moderately halophilic bacterium Halobacillus halophilus. Arch. Microbiol. 2009, 191, 95–104. [Google Scholar] [CrossRef]
- Liu, X.; Gai, Z.; Tao, F.; Tang, H.; Xu, P. Carotenoids Play a Positive Role in the Degradation of Heterocycles by Sphingobium yanoikuyae. PLoS ONE 2012, 7, e39522. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Espariz, M.; Zuljan, F.A.; Esteban, L.; Magni, C. Taxonomic Identity Resolution of Highly Phylogenetically Related Strains and Selection of Phylogenetic Markers by Using Genome-Scale Methods: The Bacillus pumilus Group Case. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Auch, A.F.; Klenk, H.-P.; Göker, M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genom. Sci. 2010, 2, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Guoping, C. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef]
- Mehta, S.; Nautiyal, C.S. An Efficient Method for Qualitative Screening of Phosphate-Solubilizing Bacteria. Curr. Microbiol. 2001, 43, 51–56. [Google Scholar] [CrossRef]
- Sashidhar, B.; Podile, A.R. Mineral phosphate solubilization by rhizosphere bacteria and scope for manipulation of the direct oxidation pathway involving glucose dehydrogenase. J. Appl. Microbiol. 2010, 109, 1–12. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Jochimsen, B.; Lolle, S.; McSorley, F.R.; Nabi, M.; Stougaard, J.; Zechel, D.L.; Hove-Jensen, B. Five phosphonate operon gene products as components of a multi-subunit complex of the carbon-phosphorus lyase pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Kobayashi, Y.; Hulett, F.M. The pst operon of Bacillus subtilis has a phosphate-regulated promoter and is involved in phosphate transport but not in regulation of the pho regulon. J. Bacteriol. 1997, 179, 2534–2539. [Google Scholar] [CrossRef] [Green Version]
- Raza, W.; Shen, Q. Growth, Fe3+ Reductase Activity, and Siderophore Production by Paenibacillus polymyxa SQR-21 Under Differential Iron Conditions. Curr. Microbiol. 2010, 61, 390–395. [Google Scholar] [CrossRef]
- Zeng, Q.; Xie, J.; Li, Y.; Gao, T.; Xu, C.; Wang, Q. Comparative genomic and functional analyses of four sequenced Bacillus cereus genomes reveal conservation of genes relevant to plant-growth-promoting traits. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Veter. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Miethke, M.; Marahiel, M.A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, F.M.; Mercado-Blanco, J. Biological control of tree and woody plant diseases: An impossible task? BioControl 2016, 61, 233–242. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Length | Completeness | Score | Proteins | Location | Prophage | GC % | Phage Components |
|---|---|---|---|---|---|---|---|---|
| 1 | 59.9 kb | Complete | 140 | 76 | 513064-572967 | Brevib_Jimmer1 (NC_029104) | 41.22 | Capsid, integrase, terminase, tail, and head |
| 2 | 19.5 kb | Incomplete | 10 | 11 | 1680797-1700309 | Bacill_SP_10 (NC_019487) | 40.63 | NA |
| 3 | 27.3 kb | Incomplete | 50 | 36 | 3559626-3586998 | Brevib_Jimmer1 (NC_029104) | 40.79 | Tail, head, terminase |
| 4 | 14 kb | Incomplete | 30 | 16 | 2744188-2758208 | Bacilli- SPbeta (NC_001884) | 36.52 | Tail and transposase |
| 5 | 27.8 kb | Incomplete | 50 | 30 | 3746776-3774609 | Staphy_SPbeta_like (NC_029119) | 48.27 | Transposase and integrase |
| Strain | Accession | Size (Mb) | GC% | Protein | rRNA | tRNA | Other RNA | Gene | Pseu | Contamination (%) | Completeness (%) | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SH-B9 | CP011007.1 | 3.79 | 41.6 | 3727 | 24 | 81 | 5 | 3887 | 50 | 1.98 | 95.49 | Sugar beet rhizosphere, The Netherlands |
| NCTC10337 | LT906438.1 | 3.86 | 41.7 | 3775 | 24 | 81 | 5 | 3950 | 65 | 3.35 | 95.83 | NCTC United Kingdom |
| 145 | CP027116.1 | 3.94 | 41.2 | 3880 | 24 | 81 | 5 | 4064 | 74 | 2.71 | 92.62 | Sediment top, Mexico |
| SH-B11 | CP010997.1 | 3.86 | 41.3 | 3776 | 24 | 81 | 5 | 3913 | 27 | 5.27 | 95.30 | Sugar beet rhizosphere, Netherlands |
| MTCC B6033 | CP007436.1 | 3.76 | 41.4 | 3708 | 24 | 81 | 5 | 3881 | 63 | 3.12 | 95.06 | Culture collection Canada |
| 150a | CP027034. | 3.75 | 41.4 | 3643 | 24 | 82 | 5 | 3821 | 67 | 1.93 | 95.64 | Sediment top, Mexico |
| TUAT1 | AP014928.1 | 3.72 | 41.4 | 3688 | 24 | 81 | 5 | 3817 | 19 | 1.24 | 96.49 | Field soil, Japan |
| SAFR-032 | CP000813.4 | 3.7 | 41.3 | 3588 | 21 | 72 | 5 | 3764 | 78 | 0.17 | 98.49 | Spacecraft |
| PDSLzg-1 | CP016784.1 | 3.7 | 42.0 | 3600 | 24 | 81 | 5 | 3761 | 51 | 2.62 | 93.07 | Oil Sands, China |
| ZB201701 | CP029464.1 | 3.64 | 41.9 | 3545 | 24 | 81 | 5 | 3723 | 68 | 1.78 | 93.24 | Rhizosphere soil, China |
| SF-4 | CP047089.1 | 3.77 | 41.2 | 3669 | 10 | 75 | 5 | 3845 | 86 | 2.28 | 96.36 | Soil field, Pakistan |
| C4 | CP011109.1 | 3.66 | 41.4 | 3622 | 10 | 59 | 5 | 3728 | 32 | 1.42 | 94.74 | Compost, Egypt |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, S.; Vollmers, J.; Janjua, H.A. Genome Mining and Comparative Genome Analysis Revealed Niche-Specific Genome Expansion in Antibacterial Bacillus pumilus Strain SF-4. Genes 2021, 12, 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071060
Iqbal S, Vollmers J, Janjua HA. Genome Mining and Comparative Genome Analysis Revealed Niche-Specific Genome Expansion in Antibacterial Bacillus pumilus Strain SF-4. Genes. 2021; 12(7):1060. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071060
Chicago/Turabian StyleIqbal, Sajid, John Vollmers, and Hussnain Ahmed Janjua. 2021. "Genome Mining and Comparative Genome Analysis Revealed Niche-Specific Genome Expansion in Antibacterial Bacillus pumilus Strain SF-4" Genes 12, no. 7: 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071060