
Molecular Characterization of Choroideremia-Associated Deletions Reveals an Unexpected Regulation of CHM Gene Transcription

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
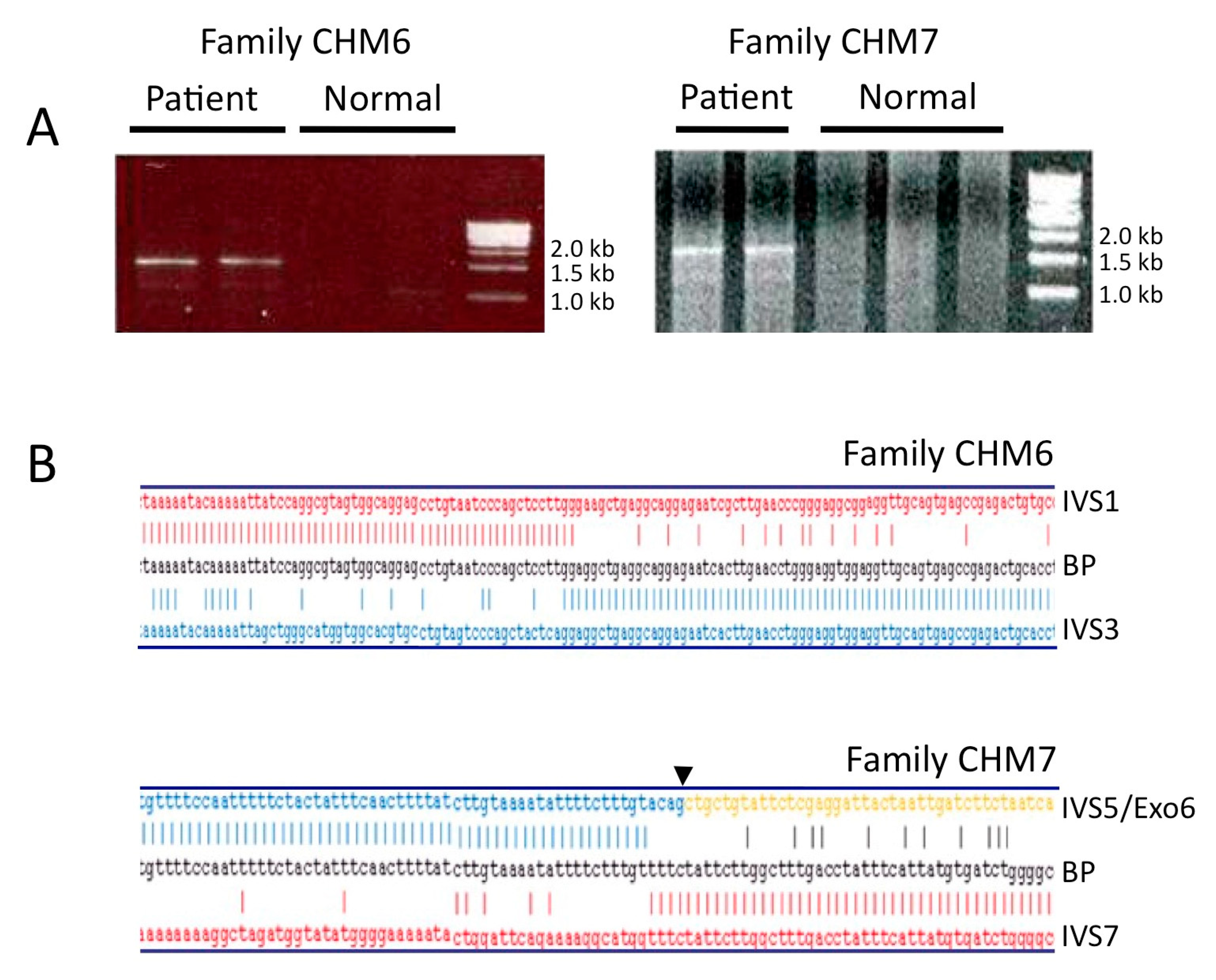
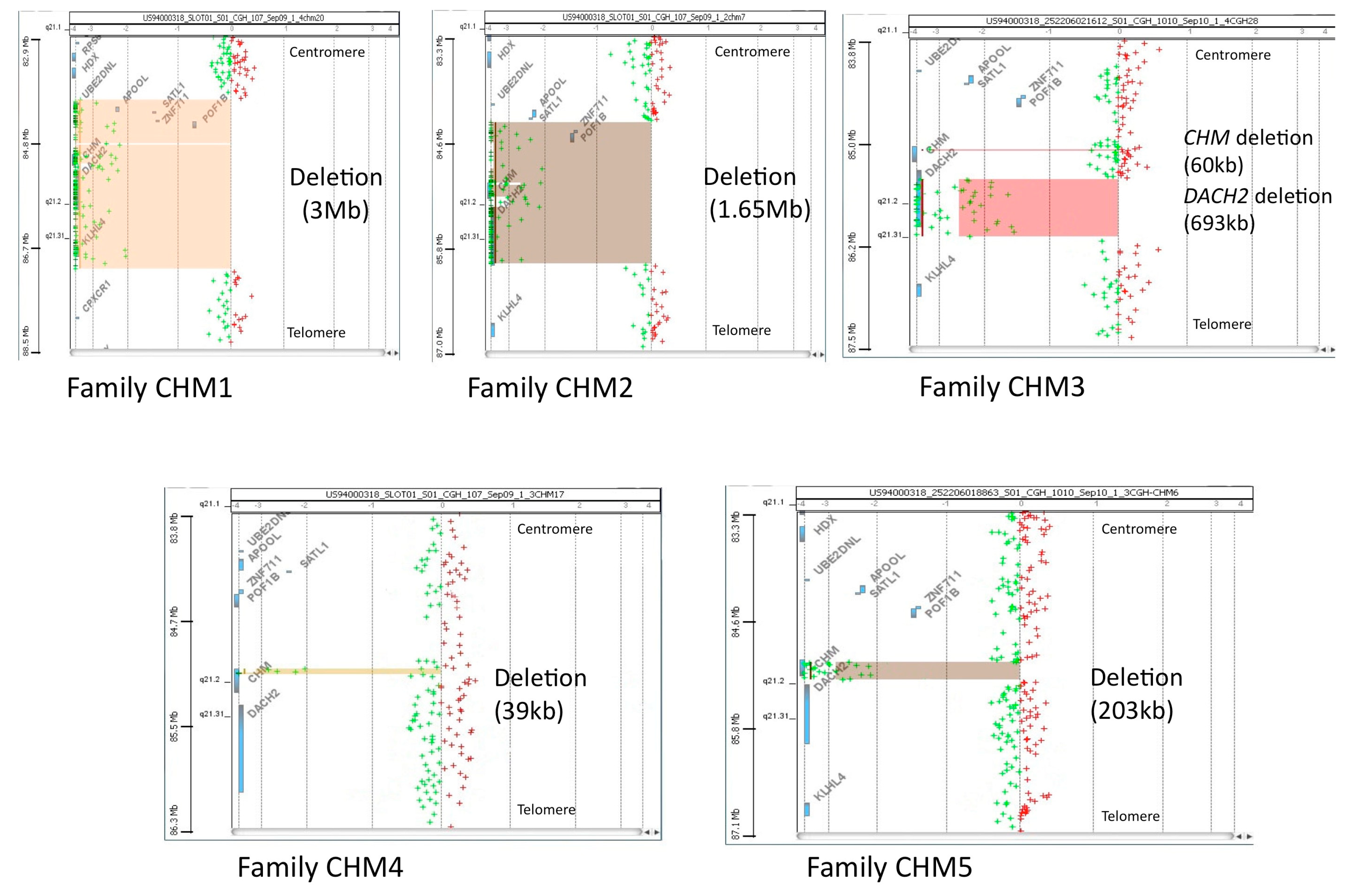
3.1. Molecular Characterization of CHM Deletions
3.2. Analysis of the Genomic Architecture Flanking the BP Junctions
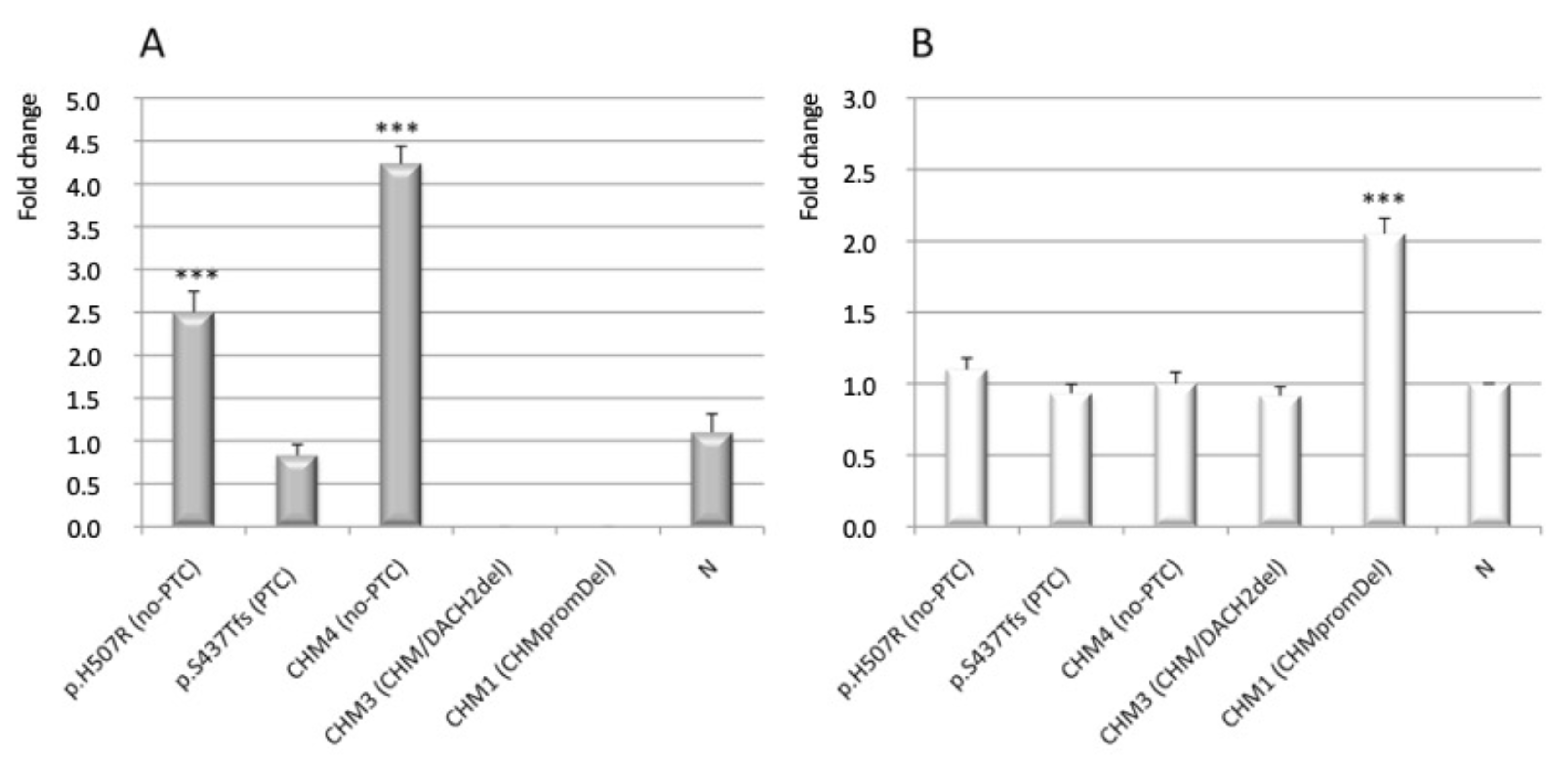
3.3. Analysis of CHM and CHML Transcript Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Coussa, R.G.; Traboulsi, E.I. Choroideremia, a review of general findings and pathogenesis. Ophthalmic Genet. 2012, 33, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Preising, M.N.; Wegscheider, E.; Friedburg, C.; Poloschek, C.M.; Wabbels, B.K.; Lorenz, B. Fundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical data. Ophthalmology 2009, 116, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, R.; Park, K.S.; Tanaka, A.J.; Cho, A.; Paavo, M.; Zernant, J.; Francis, J.H.; Allikmets, R.; Sparrow, J.R.; Tsang, S.H. Spectrum of disease severity and phenotype in choroideremia carriers. Am. J. Ophthalmol. 2019, 207, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Van Bokhoven, H.; Schwartz, M.; Andréasson, S.; van den Hurk, J.A.; Bogerd, L.; Jay, M.; Pawlowitzki, I.H.; Sankila, E.M.; Wright, A.; Ropers, H.H.; et al. Mutation spectrum in the CHM gene of Danish and Swedish choroideremia patients. Hum. Mol. Genet. 1994, 3, 1047–1051. [Google Scholar] [CrossRef]
- Andres, D.A.; Seabra, M.C.; Brown, M.S.; Armstrong, S.A.; Smeland, T.E.; Cremers, F.P.; Goldstein, J.L. cDNA cloning of component A of Rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell 1993, 73, 1091–1099. [Google Scholar] [CrossRef]
- Cremers, F.P.; Armstrong, S.A.; Seabra, M.C.; Brown, M.S.; Goldstein, J.L. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J. Biol. Chem. 1994, 269, 2111–2117. [Google Scholar] [CrossRef]
- Ali, B.R.; Seabra, M.C. Targeting of Rab GTPases to cellular membranes. Biochem. Soc. Trans. 2005, 33, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.Q.; Ali, B.R.; Ramalho, J.S.; Godfrey, R.F.; Barral, D.C.; Hume, A.N.; Seabra, M.C. Membrane targeting of Rab GTPases is influenced by the prenylation motif. Mol. Biol Cell 2003, 14, 1882–1899. [Google Scholar] [CrossRef] [Green Version]
- Strunnikova, N.V.; Barb, J.; Sergeev, Y.V.; Thiagarajasubramanian, A.; Silvin, C.; Munson, P.J.; Macdonald, I.M. Loss-of-function mutations in Rab escort protein 1 [REP-1] affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS ONE 2009, 4, e8402. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Mysore, N.; Vali, H.; Koenekoop, J.; Cao, S.N.; Li, S.; Ren, H.; Keser, V.; Lopez-Solache, I.; Siddiqui, S.N.; et al. Choroideremia is a systemic disease with lymphocyte crystals and plasma lipid and RBC membrane abnormalities. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8158–8165. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Falco, F.; Tinto, N.; Testa, F.; Vitagliano, L.; Tandurella, I.C.; Iannone, L.; Rossi, S.; Rinaldi, E.; Simonelli, F.; et al. Comprehensive mutation analysis [20 families] of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum. Mutat. 2011, 32, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Ruggiero, R.; Savarese, M.; Savarese, G.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin. Chem. Lab. Med. 2013, 51, 2239–2245. [Google Scholar] [CrossRef]
- MacDonald, I.M.; Moen, C.; Duncan, J.L.; Tsang, S.H.; Cehajic-Kapetanovic, J.; Aleman, T.S. Perspectives on Gene Therapy: Choroideremia Represents a Challenging Model for the Treatment of Other Inherited Retinal Degenerations. Transl Vis. Sci. Technol. 2020, 9, 17. [Google Scholar] [CrossRef]
- Abbouda, A.; Avogaro, F.; Moosajee, M.; Vingolo, E.M. Update on Gene Therapy Clinical Trials for Choroideremia and Potential Experimental Therapies. Medicina 2021, 57, 64. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, V.; Orrico, A.; Esposito, G.; Melillo, P.; Rossi, S.; Sbordone, S.; Auricchio, A.; Testa, F.; Simonelli, F. Association between genotype and disease progression in Italian Stargardt patients: A retrospective natural history study. Retina 2019, 39, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, T.; Ungari, S.; Savarese, M.; Cattaneo, F.; Pirozzi, E.; Esposito, G. A putative frameshift variant in the CHM gene is associated with an unexpected splicing alteration in a choroideremia patient. Mol. Genet. Genom. Med. 2020, 8, e1490. [Google Scholar] [CrossRef] [PubMed]
- Van den Hurk, J.A.; Schwartz, M.; van Bokhoven, H.; van de Pol, T.J.; Bogerd, L.; Pinckers, A.J.; Bleeker-Wagemakers, E.M.; Pawlowitzki, I.H.; Rüther, K.; Ropers, H.H.; et al. Molecular basis of choroideremia [CHM] mutations involving the Rab escort protein-1 [REP-1] gene. Hum. Mutat 1997, 9, 110–117. [Google Scholar] [CrossRef]
- Furgoch, M.J.; Mewes-Arès, J.; Radziwon, A.; Macdonald, I.M. Molecular genetic diagnostic techniques in choroideremia. Mol. Vis. 2014, 20, 535–544. [Google Scholar]
- Zeitz, C.; Nassisi, M.; Laurent-Coriat, C.; Andrieu, C.; Boyard, F.; Condroyer, C.; Démontant, V.; Antonio, A.; Lancelot, M.E.; Frederiksen, H.; et al. CHM mutation spectrum and disease: An update at the time of human therapeutic trials. Hum. Mutat. 2021, 42, 323–341. [Google Scholar] [CrossRef]
- Freund, P.R.; Sergeev, Y.V.; MacDonald, I.M. Analysis of a large choroideremia dataset does not suggest a preference for inclusion of certain genotypes in future trials of gene therapy. Mol. Genet. Genom. Med. 2016, 4, 344–358. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; Diergaarde, P.J.; Wieringa, B.; Nussbaum, R.L.; Schwartz, M.; Ropers, H.H. Physical fine mapping of the choroideremia locus using Xq21 deletions associated with complex syndromes. Genomics 1989, 4, 41–46. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; de Silva, M.G.; Tan, T.Y.; Rose, E.; Nishimura, C.; Tolmachova, T.; Hulett, J.M.; White, S.M.; Silver, J.; Bahlo, M.; et al. Molecular characterization of a novel X-linked syndrome involving developmental delay and deafness. Am. J. Med. Genet. A 2007, 143, 2564–2575. [Google Scholar] [CrossRef]
- Poloschek, C.M.; Kloeckener-Gruissem, B.; Hansen, L.L.; Bach, M.; Berger, W. Syndromic choroideremia, sublocalization of phenotypes associated with Martin-Probst deafness mental retardation syndrome. Investig. Ophthalmol. Vis. Sci 2008, 49, 4096–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, E.T.; Schaefer, G.B. Identification of an X-linked deletion syndrome through comparative genomic hybridization microarray. Semin. Pediatr. Neurol. 2010, 17, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.J.; Lupski, J.R. Implications of human genome architecture for rearrangement-based disorders, the genomic basis of disease. Hum. Mol. Genet. 2004, 13, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, V.; Esposito, G.; De Falco, F.; Boccia, R.; Fioretti, T.; Colucci, R.; De Rosa, G.; Melillo, P.; Salvatore, F.; Simonelli, F.; et al. CHM/REP1 transcript expression and loss of visual function in patients affected by choroideremia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Coussa, R.G.; Kim, J.; Traboulsi, E.I. Choroideremia, effect of age on visual acuity in patients and female carriers. Ophthalmic Genet. 2012, 33, 66–73. [Google Scholar] [CrossRef]
- Esposito, G.; Imperato, M.R.; Ieno, L.; Sorvillo, R.; Benigno, V.; Parenti, G.; Parini, R.; Vitagliano, L.; Zagari, A.; Salvatore, F. Hereditary fructose intolerance, functional study of two novel ALDOB natural variants and characterization of a partial gene deletion. Hum. Mutat 2010, 31, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Tremolaterra, M.R.; Marsocci, E.; Tandurella, I.C.; Fioretti, T.; Savarese, M.; Carsana, A. Precise mapping of 17 deletion breakpoints within the central hotspot deletion region [introns 50 and 51] of the DMD gene. J. Hum. Genet. 2017, 62, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Carsana, A.; Frisso, G.; Intrieri, M.; Tremolaterra, M.R.; Savarese, G.; Scapagnini, G.; Esposito, G.; Santoro, L.; Salvatore, F. A 15-year molecular analysis of DMD/BMD, genetic features in a large cohort. Front. Biosci. 2010, 2, 547–558. [Google Scholar]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxid. Med. Cell Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; Tremolaterra, M.R.; Savarese, M.; Spiniello, M.; Patrizio, M.P.; Lombardo, B.; Pastore, L.; Salvatore, F.; Carsana, A. Unraveling unusual X-chromosome patterns during fragile-X syndrome genetic testing. Clin. Chim. Acta 2018, 476, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Iossa, S.; Costa, V.; Corvino, V.; Auletta, G.; Barruffo, L.; Cappellani, S.; Ceglia, C.; Cennamo, G.; D’Adamo, A.P.; D’Amico, A.; et al. Phenotypic and genetic characterization of a family carrying two Xq21.1-21.3 interstitial deletions associated with syndromic hearing loss. Mol. Cytogenet. 2015, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, B.; Ceglia, C.; Tarsitano, M.; Pierucci, I.; Salvatore, F.; Pastore, L. Identification of a deletion in the NDUFS4 gene using array-comparative genomic hybridization in a patient with suspected mitochondrial respiratory disease. Gene 2014, 535, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cano, H.J.; Garnica-Hayashi, R.E.; Zenteno, J.C. CHM gene molecular analysis and X-chromosome inactivation pattern determination in two families with choroideremia. Am. J. Med. Genet. A 2009, 149, 2134–2140. [Google Scholar] [CrossRef]
- Radziwon, A.; Arno, G.; Wheaton, K.D.; McDonagh, E.M.; Baple, E.L.; Webb-Jones, K.; Birch, G.D.; Webster, A.R.; MacDonald, I.M. Single-base substitutions in the CHM promoter as a cause of choroideremia. Hum. Mutat. 2017, 38, 704–715. [Google Scholar] [CrossRef]
- Chi, J.Y.; MacDonald, I.M.; Hume, S. Copy number variant analysis in CHM to detect duplications underlying choroideremia. Ophthalmic Genet. 2013, 34, 229–233. [Google Scholar] [CrossRef]
- Simunovic, M.P.; Jolly, J.K.; Xue, K.; Edwards, T.L.; Groppe, M.; Downes, S.M.; MacLaren, R.E. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6033–6039. [Google Scholar] [CrossRef] [Green Version]
- Kleine-Kohlbrecher, D.; Christensen, J.; Vandamme, J.; Abarrategui, I.; Bak, M.; Tommerup, N.; Shi, X.; Gozani, O.; Rappsilber, J.; Salcini, A.E.; et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 2010, 38, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Bione, S.; Rizzolio, F.; Sala, C.; Ricotti, R.; Goegan, M.; Manzini, M.C.; Battaglia, R.; Marozzi, A.; Vegetti, W.; Dalprà, L.; et al. Mutation analysis of two candidate genes for premature ovarian failure; DACH2 and POF1B. Hum. Reprod. 2004, 19, 2759–2766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Y.; Li, Y.; Duan, Y.; Zhang, K.; Wang, J.; Dai, Y. Exome sequencing identifies mutations in ABCD1 and DACH2 in two brothers with a distinct phenotype. BMC Med. Genet. 2014, 15, 105. [Google Scholar] [CrossRef]
- Lee, S.Y.; Yu, W.K.; Lin, P.K. Large gene deletion and changes in corneal endothelial cells in a family with choroideremia. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1887–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Hoyos, M.; Lorda-Sanchez, I.; Gómez-Garre, P.; Villaverde, C.; Cantalapiedra, D.; Bustamante, A.; Diego-Alvarez, D.; Vallespin, E.; Gallego-Merlo, J.; Trujillo, M.J.; et al. New type of mutations in three Spanish families with choroideremia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, H.; Mitsios, A.; Smart, M.; Skinner, J.; Welch, A.A.; Kalatzis, V.; Coffey, P.J.; Dubis, A.M.; Webster, A.R.; Moosajee, M. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum. Mol. Genet. 2019, 28, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Rosenberg, T.; van den Hurk, J.A.; van de Pol, D.J.; Cremers, F.P. Identification of mutations in Danish choroideremia families. Hum. Mutat 1993, 2, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; de Marco, R.; Damante, G.; Grainger, R.; van Heyningen, V.; Kleinjan, D.A. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Köhnke, M.; Delon, C.; Hastie, M.L.; Nguyen, U.T.; Wu, Y.W.; Waldmann, H.; Goody, R.S.; Gorman, J.J.; Alexandrov, K. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS ONE 2013, 8, e81758. [Google Scholar] [CrossRef] [Green Version]
- Cehajic Kapetanovic, J.; Barnard, A.R.; MacLaren, R.E. Molecular therapies for choroideremia. Genes 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsios, A.; Dubis, A.M.; Moosajee, M. Choroideremia: From genetic and clinical phenotyping to gene therapy and future treatments. Adv. Ophthalmol. 2018, 10, 7490. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family/Proband | Deleted Exon | Genomic Deletion | Protein a |
|---|---|---|---|
| CHM1 | 1–15 | chrX:g.(84,831,307_84,886,530)_(87,891,525_87,941,186)del | p.0 |
| CHM2 | 1–15 | chrX:g.(85,123,800_85,156,157)_(86,816,070_86,832,590)del | p.0 |
| CHM3 | 12 | chrX: g.[(85,879,855_85,894,160)_(85,894,219_85,901,695)del; (86,233,417_86,253,764)_(86,946,402–87,002,613)del] b | p.0 |
| CHM4 | 10–15 | chrX:g.(85,862,591_85,864,525)_(85,903,612_85,909,335)del | p.Cys416Ter |
| CHM5 | 1–12 | chrX:g.(85,890,383_85,894,156)_(86,097,388_86,111,691)del | p.0 |
| CHM6 | 2–3 | chrX:g.85,981,356_86,037,319del | p.Leu18Lysfs*10 |
| CHM7 | 6–7 | chrX:g.85,957,346_85,958,981del | p.Leu234Aspfs*4 |
| Family/Proband | Deletion Extent | Lost Exons | Proximal RE | Distal RE | RE Class | Sequence Identity (%) |
|---|---|---|---|---|---|---|
| CHM1 | ~3 Mb | 1–15 | L1PA10, L1PA12 | 2L1PA14, STR | LINE, STR | 78 |
| CHM2 | ~1.65 Mb | 1–15 | AluY, AluJb, STR | AluSx | SINE, STR | 70–80 |
| CHM3 | ~60 kb | 12 | L1MA9 | L1PA16 | LINE | 69 |
| ~670 kb [DACH2] | 2–12 * | L1PA15, STR | L1PA2, STR | LINE, STR | 76 | |
| CHM4 | ~34 kb | 10–15 | AluSx | AluSx1 | SINE | 81 |
| CHM5 | ~235 kb | 1–12 | THE1D-int | THE1A-int, STR | LTR, STR | 78 |
| CHM6 | ~55 kb | 2–3 | AluSq | AluSx | SINE | 84 |
| CHM7 | ~1.6 kb | 6–7 | MER46C | MER3, MIRc | SINE, STR | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fioretti, T.; Di Iorio, V.; Lombardo, B.; De Falco, F.; Cevenini, A.; Cattaneo, F.; Testa, F.; Pastore, L.; Simonelli, F.; Esposito, G. Molecular Characterization of Choroideremia-Associated Deletions Reveals an Unexpected Regulation of CHM Gene Transcription. Genes 2021, 12, 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081111
Fioretti T, Di Iorio V, Lombardo B, De Falco F, Cevenini A, Cattaneo F, Testa F, Pastore L, Simonelli F, Esposito G. Molecular Characterization of Choroideremia-Associated Deletions Reveals an Unexpected Regulation of CHM Gene Transcription. Genes. 2021; 12(8):1111. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081111
Chicago/Turabian StyleFioretti, Tiziana, Valentina Di Iorio, Barbara Lombardo, Francesca De Falco, Armando Cevenini, Fabio Cattaneo, Francesco Testa, Lucio Pastore, Francesca Simonelli, and Gabriella Esposito. 2021. "Molecular Characterization of Choroideremia-Associated Deletions Reveals an Unexpected Regulation of CHM Gene Transcription" Genes 12, no. 8: 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081111