
Genetic Dysruption of the Histaminergic Pathways: A Novel Deletion at the 15q21.2 locus Associated with Variable Expressivity of Neuropsychiatric Disorders
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Family Enrollment
2.2. Array CGH
2.3. Retrospective Study
3. Results
3.1. Clinical Presentation of the Patient and Neuropsychological Profile
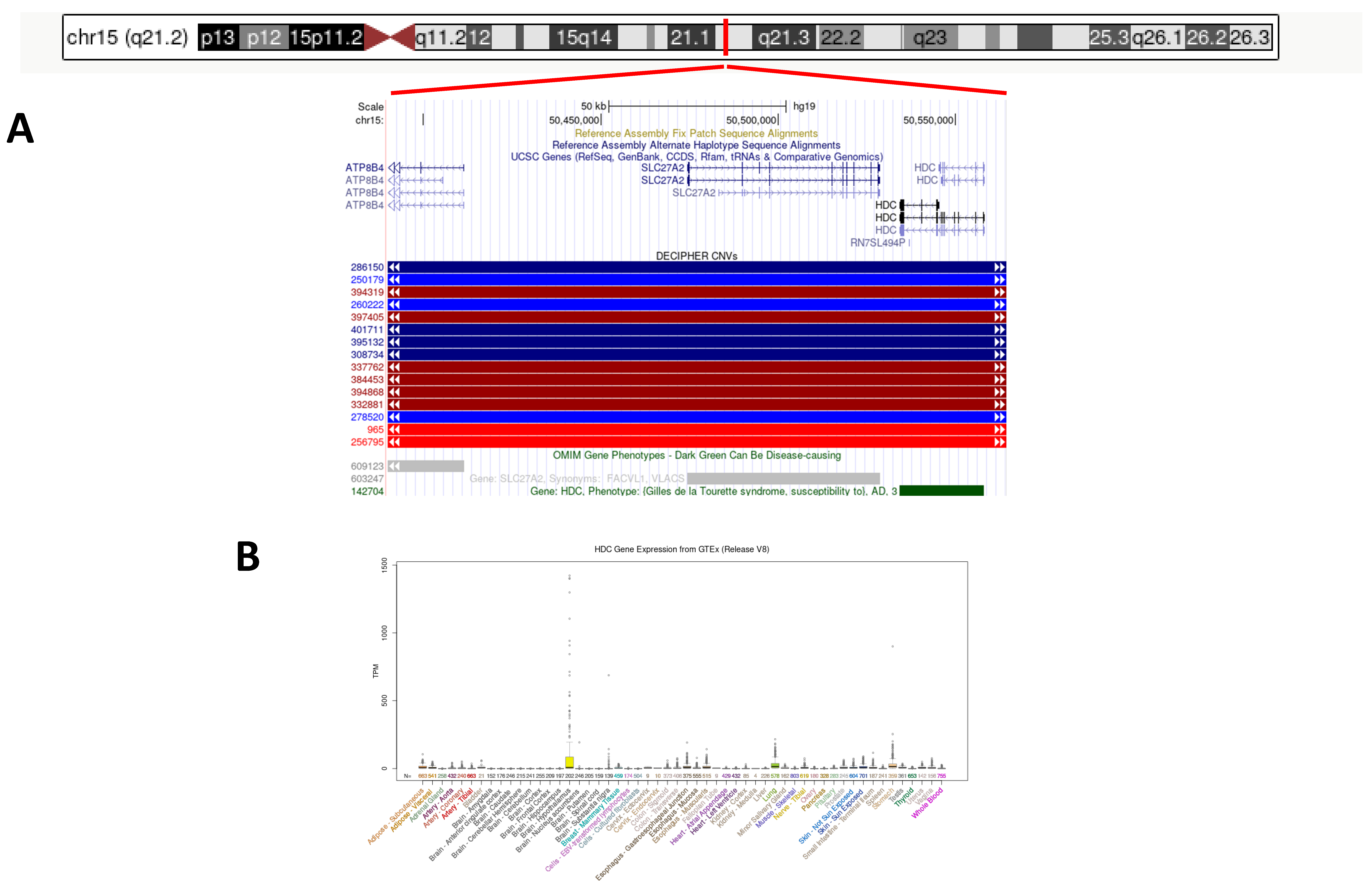
3.2. Genetic Findings
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Chaplin, E.; McCarthy, J.; Ali, S.; Marshall-Tate, K.; Xenitidis, K.; Harvey, D.; Childs, J.; Srivastava, S.; McKinnon, I.; Robinson, C.S.; et al. Severe mental illness, common mental disorders, and neurodevelopmental conditions amongst 9088 lower court attendees in London, UK. BMC Psychiatry 2022, 22, 551. [Google Scholar] [CrossRef] [PubMed]
- Jęśko, H.; Cieślik, M.; Gromadzka, G.; Adamczyk, A. Dysfunctional proteins in neuropsychiatric disorders: From neurodegeneration to autism spectrum disorders. Neurochem. Int. 2020, 141, 104853. [Google Scholar] [CrossRef]
- Huang, H.; Li, Y.; Liang, J.; Finkelman, F.D. Molecular Regulation of Histamine Synthesis. Front. Immunol. 2018, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Santos, T.; Gonçalves, J.; Baltazar, G.; Ferreira, L.; Agasse, F.; Bernardino, L. Histamine modulates microglia function. J. Neuroinflammation 2012, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Shin, J.H.; Rajpurohit, A.; Deep-Soboslay, A.; Collado-Torres, L.; Brandon, N.J.; Hyde, T.M.; Kleinman, J.E.; Jaffe, A.E.; Cross, A.J.; et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 2017, 7, e1126. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.V.; Sanders, S.J.; Yurkiewicz, I.R.; Ercan-Sencicek, A.G.; Kim, Y.S.; Fishman, D.O.; Raubeson, M.J.; Song, Y.; Yasuno, K.; Ho, W.S.C.; et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry 2012, 71, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ercan-Sencicek, A.G.; Stillman, A.A.; Ghosh, A.K.; Bilguvar, K.; O’Roak, B.J.; Mason, C.E.; Abbott, T.; Gupta, A.; King, R.A.; Pauls, D.L.; et al. L-histidine decarboxylase and Tourette’s syndrome. N. Engl. J. Med. 2010, 362, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Tanaka, S.; Terui, T.; Hori, Y.; Makabe-Kobayashi, Y.; Pejler, G.; Tchougounova, E.; Hellman, L.; Gertsenstein, M.; Hirasawa, N.; et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001, 502, 53–56. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Dehning, S.; Sandor, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; Madruga-Garrido, M.; Hebebrand, J.; Nöthen, M.M.; Lehmkuhl, G.; et al. Support of the histaminergic hypothesis in Tourette syndrome: Association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 2013, 50, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Heidari, A.; Tongsook, C.; Najafipour, R.; Musante, L.; Vasli, N.; Garshasbi, M.; Hu, H.; Mittal, K.; McNaughton, J.M.; Sritharan, K.; et al. Mutations in the histamine N-methyltransferase gene, HNMT, are associated with nonsyndromic autosomal recessive intellectual disability. Hum. Mol. Genet. 2015, 24, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, W.M.A.; Egger, J.I.M.; Janssen, P.K.C.; van Haeringen, A. Adult male patient with severe intellectual disability caused by a homozygous mutation in the HNMT gene. BMJ Case Rep. 2020, 13, e235972. [Google Scholar] [CrossRef] [PubMed]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.-H.; Jawoeski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Mulatinho, M.V.; de Carvalho Serao, C.L.; Scalco, F.; Hardekopf, D.; Pekova, S.; Mrasek, K.; Liehr, T.; Weise, A.; Rao, N.; Llerena, J.C., Jr. Severe intellectual disability, omphalocele, hypospadia and high blood pressure associated to a deletion at 2q22.1q22.3: Case report. Mol. Cytogenet. 2012, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Baldan, L.C.; Williams, K.A.; Gallezot, J.D.; Pogorelov, V.; Rapanelli, M.; Crowley, M.; Anderson, G.M.; Loring, E.; Gorczyca, R.; Billingslea, E.; et al. Histidine decarboxylase deficiency causes tourette syndrome: Parallel findings in humans and mice. Neuron 2014, 81, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, C.; Zhong, M.; Che, F.; Guan, C.; Zheng, X.; Liu, S. Role of histidine decarboxylase gene in the pathogenesis of Tourette syndrome. Brain Behav. 2022, 12, e2511. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cognitive Test | Score or Level of Performance |
|---|---|
| WAIS-IV | |
| - Verbal comprehension IQ - Visuo-perceptual reasoning IQ - Working memory IQ - Visuo-motor speed IQ - Full scale IQ | 84 (normal) 90 (normal) 72 (poor) 72 (poor) 75 (poor) |
| Executive functions | |
| - Trail making test - Stroop test - Wisconsin card sorting test/set-shifting ability - Tower of London - Phonological fluency | Poor Normal Normal Poor Poor |
| Visuo-constructional abilities | |
| - Rey complex figure | Normal |
| Arithmetical abilities | |
| - Mental calculus - Mathematical reasoning | Poor Poor |
| ADOS-2 | |
| - Language and communication | 1 |
| - Social interaction | 7 |
| Total score | 8 (ASD threshold = 7) |
| Gene | Function | CNV/SNV | Phenotype | Additional Phenotypic Features | Ref. |
|---|---|---|---|---|---|
| HDC | Conversion of histidine into histamine | p.Trp317Stop (heterozygous) | Tourette syndrome (two-generation pedigree) | Two children and their father had also obsessive compulsive disorder | [8] |
| Family study rs854150 and rs1894236 520 European trios | Tourette syndrome | Attention deficit Hyperactivity Disorder, obsessive compulsive disorder | [10] | ||
| Inherited CNV (loss, heterozygous) chr15:50,389,918-50,564,328 (hg19) | Mild intellectual disability | Emotional dysregulation, excessive daytime sleepness, constipation, autistic traits, dyspraxia, learning disorder, sleep disturbance, pervasive and perservant behaviour | This study | ||
| HNMT | Inactivation of histamine | de novo CNV (loss, heterozygous) chr2:137,234,086–144,547,896 (hg18) | Autism | - | [13] |
| de novo CNV (loss, heterozygous) chr2:138,750,000-144,750,000 (hg19) | Severe intellectual disability, autism, essential hypertension, congenital malformations | - | [14] | ||
| p.Gly60Asp (homozygous) p.Leu208Pro (homozygous) | Intellectual disability ranging from mild to severe in two not related pedigrees | Delayed speech development in the Iranian pedigree and mild regression from the age of 5 years in the Kurdish pedigree. No dysmorphisms | [11] | ||
| p.Gln30Stop(homozygous) | Severe intellectual disability | Autism, aggression, sleep disturbance, global regression at the age of 4 years, delayed speech, gastrointestinal problems. No dysmorphisms | [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lintas, C.; Sacco, R.; Azzarà, A.; Cassano, I.; Laino, L.; Grammatico, P.; Gurrieri, F. Genetic Dysruption of the Histaminergic Pathways: A Novel Deletion at the 15q21.2 locus Associated with Variable Expressivity of Neuropsychiatric Disorders. Genes 2022, 13, 1685. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13101685
Lintas C, Sacco R, Azzarà A, Cassano I, Laino L, Grammatico P, Gurrieri F. Genetic Dysruption of the Histaminergic Pathways: A Novel Deletion at the 15q21.2 locus Associated with Variable Expressivity of Neuropsychiatric Disorders. Genes. 2022; 13(10):1685. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13101685
Chicago/Turabian StyleLintas, Carla, Roberto Sacco, Alessia Azzarà, Ilaria Cassano, Luigi Laino, Paola Grammatico, and Fiorella Gurrieri. 2022. "Genetic Dysruption of the Histaminergic Pathways: A Novel Deletion at the 15q21.2 locus Associated with Variable Expressivity of Neuropsychiatric Disorders" Genes 13, no. 10: 1685. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13101685