
Phenotypic Spectrum of NFIA Haploinsufficiency: Two Additional Cases and Review of the Literature
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Analysis
2.2. Bioinformatic Analyses
2.3. Patients
2.3.1. Patient 1
2.3.2. Patient 2
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, W.; Quintero-Rivera, F.; Fan, Y.; Alkuraya, F.S.; Donovan, D.J.; Xi, Q.; Turbe-Doan, A.; Li, Q.G.; Campbell, C.G.; Shanske, A.L.; et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007, 3, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.G.N.; Wang, H.; Hunter, G.W. Interstitial microdeletion of Chromosome 1p in two siblings. Am. J. Med. Genet. 2002, 111, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Koehler, U.; Holinski-Feder, E.; Ertl-Wagner, B.; Kunz, J.; von Moers, A.; von Voss, H.; Schell-Apacik, C. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. Case Rep. Eur. J. Pediatr. 2010, 169, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Su, Y.N.; Chen, Y.Y.; Chern, S.R.; Liu, Y.P.; Wu, P.C.; Lee, C.C.; Chen, Y.T.; Wang, W. Chromosome 1p32-p31 deletion syndrome: Prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Case Rep. Taiwan J. Obstet. Gynecol. 2011, 50, 345–352. [Google Scholar]
- Ji, J.; Salamon, N.; Quintero-Rivera, F. Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. Comment Eur. J. Med. Genet. 2014, 57, 267–268. [Google Scholar] [CrossRef]
- Labonne, J.D.J.; Shen, Y.; Kong, I.K.; Diamond, M.P.; Layman, L.C.; Kim, H.G. Comparative deletion mapping at 1p31.3-p32.2 implies NFIA responsible for intellectual disability coupled with macrocephaly and the presence of several other genes for syndromic intellectual disability. Mol. Cytogenet. 2016, 17, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Prontera, P.; Rogaia, D.; Mencarelli, A.; Ottaviani, V.; Sallicandro, E.; Guercini, G.; Esposito, S.; Bersano, A.; Merla, G.; Stangoni, G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. Int. J. Mol. Sci. 2017, 18, 1998. [Google Scholar] [CrossRef] [Green Version]
- Zenker, M.; Bunt, J.; Schanze, I.; Schanze, D.; Piper, M.; Priolo, M.; Gerkes, E.H.; Gronostajski, R.M.; Richards, L.J.; Vogt, J.; et al. Variants in nuclear factor I genes influence growth and development. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 611–626. [Google Scholar] [CrossRef]
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef]
- Singh, P.N.P.; Yadav, U.S.; Azad, K.; Goswami, P.; Kinare, V.; Bandyopadhyay, A. NFIA and GATA3 are crucial regulators of embryonic articular cartilage differentiation. Development 2018, 145, dev156554. [Google Scholar]
- Hiraike, Y.; Waki, H.; Miyake, K.; Wada, T.; Oguchi, M.; Saito, K.; Tsutsumi, S.; Aburatani, H.; Yamauchi, T.; Kadowaki, T. NFIA differentially controls adipogenic and myogenic gene program through distinct pathways to ensure brown and beige adipocyte differentiation. PLoS Genet. 2020, 16, e1009044. [Google Scholar] [CrossRef] [PubMed]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sagner, A.; Zhang, I.; Watson, T.; Lazaro, J.; Melchionda, M.; Briscoe, J. A shared transcriptional code orchestrates temporal patterning of the central nervous system. PLoS Biol. 2021, 19, e3001450. [Google Scholar] [CrossRef] [PubMed]
- Santo, M.; Rigoldi, L.; Falcone, C.; Tuccillo, M.; Calabrese, M.; Martínez-Cerdeño, V.; Mallamaci, A. Spatial control of astrogenesis progression by cortical arealization genes. Cereb. Cortex. 2022, 12, mikabhac264. [Google Scholar] [CrossRef] [PubMed]
- Quist, E.; Trovato, F.; Avaliani, N.; Zetterdahl, O.G.; Gonzalez-Ramos, A.; Hansen, M.G.; Kokaia, M.; Canals, I.; Ahlenius, H. Transcription factor-based direct conversion of human fibroblasts to functional astrocytes. Stem Cell Rep. 2022, 17, 1620–1635. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, F.M.M.; Lose, E.J.; Robin, N.H.; Descartes, M.D.; Rutledge, K.D.; Rutledge, S.L.; Korf, B.R.; Carroll, A.J. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med. Genet. A 2011, 155, 2386–2396. [Google Scholar] [CrossRef]
- Hollenbeck, D.; Williams, C.L.; Drazba, K.; Descartes, M.; Korf, B.R.; Rutledge, S.L.; Lose, E.J.; Robin, N.H.; Carroll, A.J.; Mikhail, F.M. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet. Med. 2016, 19, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.; O’Donnell, S.; Bain, N.; Meldrum, C.; Shorter, D.; Goel, H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur. J. Med. Genet. 2014, 57, 65–70. [Google Scholar] [CrossRef]
- Nyboe, D.; Kreiborg, S.; Kirchhoff, M.; Hove, H.B. Familial craniosynostosis associated with a microdeletion involving the NFIA gene. Case Rep. Clin. Dysmorphol. 2015, 24, 109–112. [Google Scholar] [CrossRef]
- Bayat, A.; Kirchhoff, M.; Madsen, C.G.; Roos, L.; Kreiborg, S. Familial craniofacial abnormality and polymicrogyria associated with a microdeletion affecting the NFIA gene. Case Rep. Clin. Dysmorphol. 2017, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Miya, F.; Hattori, A.; Mizuno, K.; Hori, I.; Ando, N.; Okamoto, N.; Kato, M.; Tsunoda, T.; Yamasaki, M.; et al. Truncating mutation in NFIA causes brain malformation and urinary tract defects. Hum. Genome Var. 2015, 2, 15007. [Google Scholar] [CrossRef]
- Revah-Politi, A.; Ganapathi, M.; Bier, L.; Cho, M.T.; Goldstein, D.B.; Hemati, P.; Iglesias, A.; Juusola, J.; Pappas, J.; Petrovski, S.; et al. Loss-of-function variants in NFIA provide further support that NFIA is a critical gene in 1p32-p31 deletion syndrome: A four patient series. Am. J. Med. Genet. 2017, 173, 3158–3164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, C.M.; Zheng, X.L.; Abuduxikuer, K. A novel NFIA gene nonsense mutation in a Chinese patient with macrocephaly, corpus callosum hypoplasia, developmental delay, and dysmorphic features. Mol. Genet. Genomic. Med. 2020, 8, e1492. [Google Scholar] [CrossRef]
- Uehara, T.; Sanuki, R.; Ogura, Y.; Yokoyama, A.; Yoshida, T.; Futagawa, H.; Yoshihashi, H.; Yamada, M.; Suzuki, H.; Takenouchi, T.; et al. Recurrent NFIA K125E substitution represents a loss-of-function allele: Sensitive in vitro and in vivo assays for nontruncating alleles. Am. J. Med. Genet. A 2021, 185, 2084–2093. [Google Scholar] [CrossRef]
- Coci, E.G.; Koehler, U.; Liehr, T.; Stelzner, A.; Fink, C.; Langen, H.; Riedel, J. CANPMR syndrome and chromosome 1p32-p31 deletion syndrome coexist in two related individuals affected by simultaneous haplo-insufficiency of CAMTA1 and NIFA genes. Mol. Cytogenet. 2016, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Uehara, T.; Ujibe, K.; Yoshihashi, H.; Yamada, M.; Suzuki, H.; Takenouchi, T.; Kosaki, K.; Hirata, H. The p.Thr395Met missense variant of NFIA found in a patient with intellectual disability is a defective variant. Am. J. Med. Genet. A 2022, 188, 1184–1192. [Google Scholar] [CrossRef]
- Wongkittichote, P.; Kondis, J.S.; Peglar, L.M.; Strahle, J.M.; Miller-Thomas, M.; Abell, K.B. Pathogenic variant in NFIA associated with subdural hematomas mimicking nonaccidental trauma. Am. J. Med. Genet. A 2022, 188, 1538–1544. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Bertini, V.; Milone, R.; Cristofani, P.; Cambi, F.; Bosetti, C.; Barbieri, F.; Bertelloni, S.; Cioni, G.; Valetto, A.; Battini, R. Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes 2022, 13, 859. [Google Scholar] [CrossRef]


{kind=link}
| DELETIONS | ||||||
|---|---|---|---|---|---|---|
| Reference | Subjects | Sex/Age | Position (GRCh37/hg19) | Extent (kb) | NCBI RefSeq Genes (UCSC) NFIA-207 | Inheritance |
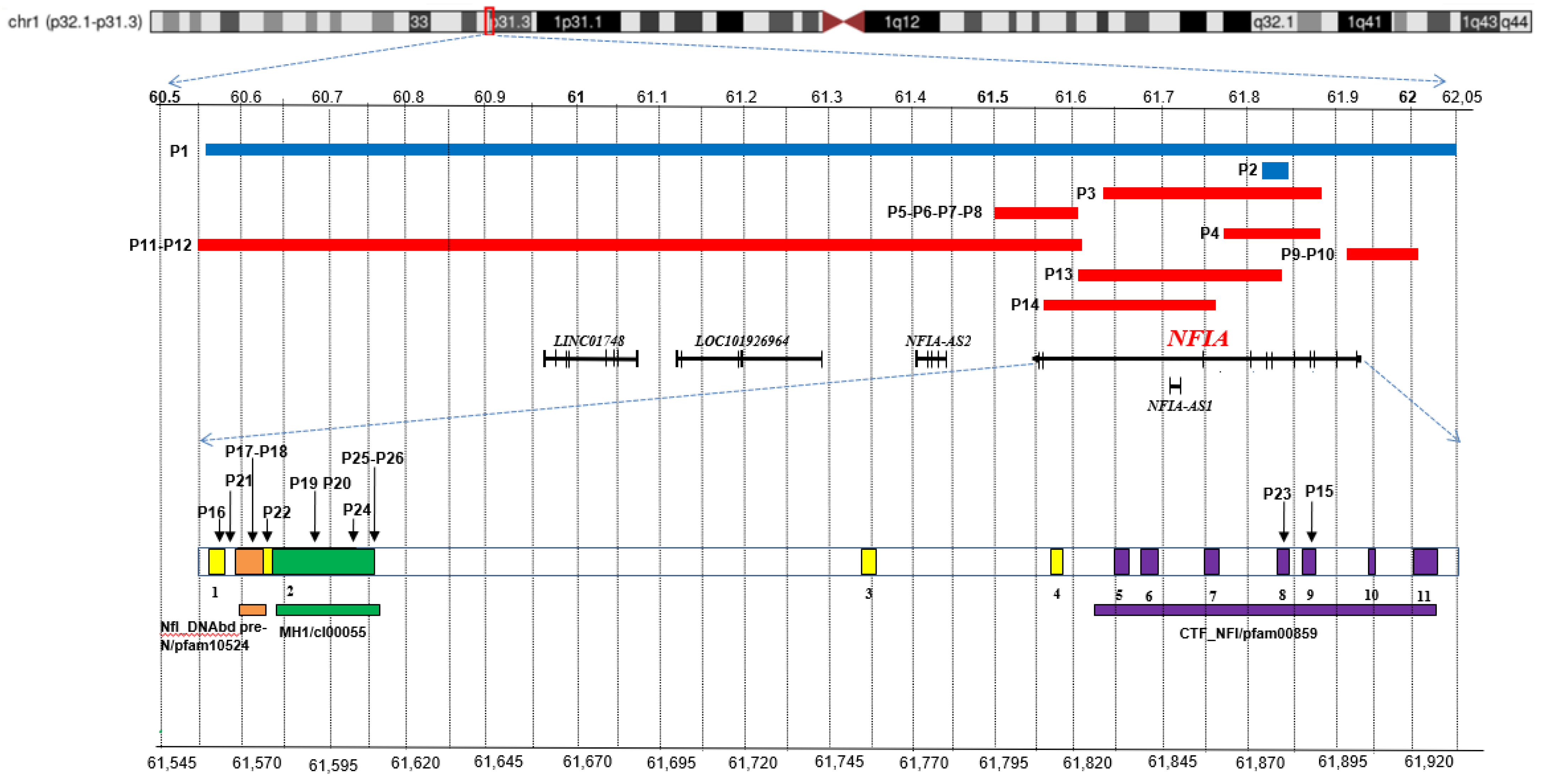
| In this report | P1 | M/2 year | 1:60,568,797–62,052,980 | 1.484 | LINC01748, LOC101926964, NFIA-AS2, NFIA (ex 1–11), NFIA-AS1 | dn |
| In this report | P2 | F/22 year | 1:61,818,169–61,845,524 | 27 | NFIA (ex 5–6) | NR |
| Mikhail et al., 2011 [17]; Hollenbeck et al., 2016 [18] (Patient 99199) | P3 | F/25 year | 1:61,632,666–61,886,758 | 254 | NFIA (ex 3–9) NFIA-AS1 | not mat NR pat |
| Rao et al., 2014 [19] | P4 | F/8 year | NR | 120 | NFIA (ex 4–9) | dn |
| Nyboe et al., 2015 [20] | P5 (I-1) P6 (II-1) P7 (II-2) P8 (II-3) | P5: M/42 year P6: F/13 year P7: M/10 year P8: M/6 year | 1:61,497,698–61,607,171 | 109 | NFIA (ex 1–2) | P5: NR P6-P7-P8: pat |
| Hollenbeck et al., 2016 [18] (Patient 13857) | P9 P10 (father) | P9: M/1.5 year P10: M/NR | NR | 99 | NFIA (ex 11) | P9: pat P10: NR |
| Bayat et al., 2017 [21] | P11 P12 (mother) | P11: F/9 year P12: M/37 year | 1:60,549,342–61,614,478 | 1.065 | LINC01748, LOC101926964 NFIA-AS2, NFIA (ex 1–2) | P11: mat P12: NR |
| 288170 DECIPHER | P13 | F/NR | 1:61,616,698–61,845,603 | 229 | NFIA-AS1, NFIA (ex 3–6) | dn |
| 358646 DECIPHER | P14 | F/1 year | 1:61,557,457–61,770,053 | 213 | NFIA-AS1, NFIA (ex 3) | dn |
| SNVs | ||||||
| Subjects | Sex/Age | NM_001134673 | NP_001128145.1 | Type of Mutation | Inheritance | |
| Negishi et al., 2015 [22] | P15 | M/5 year | c.1094delC | p.Pro365-HisfsTer32 | stop_gained/pathogenic | dn |
| Revah-Politi et al., 2017 [23] (patient 1) | P16 | F/18 year | c.25_26dupCC | p.Gln9ProfsTer49 | stop_gained/likely pathogenic | dn |
| Revah-Politi et al., 2017 [23] (patient 2), (patient 3) | P17 P18 | P17: F/7 year P18: F/35 year | c.70C > T | p.Arg24Ter | stop_gained/pathogenic | P17: matP18: NR |
| Zhang et al., 2020 [24] | P19 | M/3 month | c.220C > T | p.Arg74Ter | stop_gained/pathogenic | dn |
| 264161 DECIPHER | P20 | F/2 year 9 month | c.224T > C | p.Leu75Pro | missense/likely pathogenic | dn |
| 282711 DECIPHER | P21 | F/13 year | c.28-2A > G | SpliceAI: ΔS acceptor loss | splice_acceptor/pathogenic | dn |
| 291368 DECIPHER | P22 | M/2 year 9 month | c.112C > T | p.Arg38Ter | stop_gained/likely pathogenic | dn |
| 305439 DECIPHER | P23 | M/1 year | c.1051C > T | p.Arg351Ter | stop_gained/pathogenic | NR |
| 435805 DECIPHER | P24 | F/13 year | c.500A > G | p.His167Arg | missense/ likely pathogenic | mat |
| Uehara et al., 2021 [25] (patient 1) | P25 | F/6 year | c.373A > G | p.Lys125Glu | missense/ likely pathogenic | dn |
| Uehara et al., 2021 [25] (patient 2) | P26 | M/14 month | c.373A > G | p.Lys125Glu | missense/ likely pathogenic | dn |
| Patient | Macrocephaly | High Forehead | Low- Set Ears | Facial Dysmorphic Features | CraniofacialAnomalies | Hands and Feet Anomalies | Other Anomalies | |
|---|---|---|---|---|---|---|---|---|
| Bilateral Proximally Placed First Fingers | Other | |||||||
| P1 | Y | Y | Y | frontal bossingmild strabismus | sagittal synostosis scaphocephaly | Y | brachydactyly | short lower limbs |
| P2 | N | Y | Y | downslanting palpebral fissures, posteriorly rotated auricles, small and poorly structured philtrum, open bite | NO craniosynostosis | Y | hypotrophy of thenar and hypothenar eminences | - |
| P3 [17,18] | Y | Y | Y (left) | high palate, pointed chin hypotelorism | - | - | - | scarce hair, scoliosis, webbed neck |
| P4 [19] | Y | Y * | Y * | upslanting palpebral fissures, broad anteverted nose, overfolded helices | metopic synostosis | - | - | cutis marmorata |
| P5 [20] | Y | - | - | - | sagittal synostosis | - | - | overgrowth |
| P6 [20] | Y | Y * | Y | downslanting palpebral fissures | lambdoid synostosis | Y | - | overgrowth |
| P7 [20] | Y | - | Y | - | - | - | - | overgrowth |
| P8 [20] | Y | - | - | - | lambdoid synostosis | Y | - | overgrowth |
| P9 [18] | - | - | - | right eye exotropia, mild ptosis | - | - | - | - |
| P11 [21] | Y | Y * | Y | downslanting palpebral fissures | craniofacial asymmetry | Y | bilateral slightly broad first fingers | - |
| P12 [21] | Y | Y * | Y | high palate | craniofacial asymmetry | Y | - | - |
| P13 | Y | - | - | - | - | - | - | - |
| P14 | Y | - | - | - | - | - | - | - |
| P15 [22] | Y | Y | - | - | - | - | - | - |
| P16 [23] | Y | - | - | - | - | - | small hands and feet | obesity |
| P17 [23] | Y | Y | - | frontal bossing | - | - | - | bilateral radioulnar synostoses |
| P18 [23] | Y | - | - | - | - | - | - | |
| P19 [24] | Y | Y * | Y * | hypertelorism, slightly pointed chin, large ears | - | - | - | - |
| P20 | - | - | - | - | abnormal facial shape | - | - | - |
| P21 | Y | Y | - | short nose, wide nasal bridge | - | - | - | melanocytic nevus, papule, supernumerary nipple |
| P22 | Y | - | - | - | abnormal facial shape | - | - | genu valgum, pes planus |
| P23 | Y | - | - | - | - | - | broad hallux, broad thumb, short distal phalanx of the thumb | - |
| P24 | Y | - | - | - | - | - | - | - |
| P25 [25] | Y | Y * | - | small eyes, anteverted nares, depressed nasal bridge, a broad columella, a thin upperlip, high arched palate, exotropia | - | - | - | - |
| P26 [25] | Y | Y | - | thick eyebrow, short nose, anteverted nares, long philtrum, thin upperlip, retrognathia | - | - | - | - |
| Neurological/Behavioral Abnormalities | Renal/Urinary Tract Defects | |||||
|---|---|---|---|---|---|---|
| Patient | ID | DD | Seizure | NDDs | Other | |
| P1 | Y | Y | N | hyperactivity disorder, language and speech delay | N | |
| P2 | Y | - | N | ODD | trichotillomania | N |
| P3 [17,18] | Y | - | - | - | bipolar disorder/depression, incapability of making her own decisions | N |
| P4 [19] | Y | Y | - | PDDNOS, ADHD-Combined Type | hypotonia | hydronephrosis, renal calculus, kinking of the pelvic–ureteric junction |
| P5 [20] | N | N | - | - | - | two renal cysts |
| P6 [20] | Y | Y | - | - | - | N |
| P7 [20] | Y | Y | - | - | - | right hydronephrosis and hydrourethra, small ureterocele, frequent urinary tract infections |
| P8 [20] | Y | Y | - | - | - | N |
| P9 [18] | - | Y | mild lower-extremity spasticity, asymmetric movement of facial muscles | renal cysts | ||
| P11 [21] | Y | Y | - | - | - | - |
| P12 [21] | Y | Y | - | - | - | N |
| P13 | Y | - | - | - | - | N |
| P14 | Y | - | - | - | - | - |
| P15 [22] | - | Y | - | - | - | - |
| P16 [23] | Y | Y | - | - | - | left hydronephrosis, cystectasia, bilateral grade IV vesicoureteral reflux |
| P17 [23] | N | Y | Y | extremely low PSI | hypotonia | N |
| P18 [23] | - | Y | Y | - | bilateral hearing loss, photophobia headaches, nonspecific complaints of arm and leg pain | renal cyst, urinary retention, frequent urinary tract infections |
| P19 [24] | N | N | Y | - | headaches and/or migraines, depression | N |
| P20 | Y | Y | - | - | impaired left ear auditory brainstem response, bilateral ametropia | N |
| P21 | Y | - | - | - | sleep disturbance | nephrolithiasis |
| P22 | - | Y | Y | - | hypotonia | - |
| P23 | - | Y | - | - | - | - |
| P24 | - | Y | - | - | - | - |
| P25 [25] | Y | Y | - | - | congenital hearing loss | N |
| P26 [25] | - | Y | - | - | mild congenital hearing impairment | N |
| Patient | Corpus Callosum Anomalies | Ventricular Anomalies | Other |
|---|---|---|---|
| P1 | hypoplasia | dysmorphic aspect of the cerebral ventriculus with widening of the anterior portions of the horns | no Chiari I malformation |
| P2 | thin | N | Chiari I malformation, cerebral vascular malformations |
| P3 [17,18] | hypoplasia | mild hydrocephalus | diffusely decreased volume of white matter, mild tonsillar ectopia, no Chiari I malformation |
| P4 [19] | hypoplasia | ventriculomegaly, partial fusion of lateral ventricles | partial absence of mid/posterior septum pellucidum |
| P5 [20] | hypoplasia | N | absent falx cerebri, partial incomplete inversion of the left hippocampi |
| P6 [20] | hypoplasia | ventriculomegaly | - |
| P8 [20] | hypoplasia | ventriculomegaly | herniation of cerebellar tonsils |
| P9 [18] | - | - | prominent cavum septum pellucidum and cavum vergae, tethered spinal cord |
| P11 [21] | thin | ventriculomegaly with asymmetric widened, edged lateral ventricles, widened third ventricle | loss of/missing white matter, frontalcortical malformation with polymicrogyria, hypoplastic falx cerebri with interdigitated frontal gyri, bilateral partial incomplete inversion of the hippocampi, arachnoid cysts in the posterior fossa |
| P12 [21] | hypoplasia | mild ventriculomegaly | mild frontoparietal atrophy/hypoplasia, slight asymmetry of frontal gyri |
| P14 | agenesis | - | - |
| P15 [22] | agenesis | ventricular enlargement | interhemispheric cysts, polymicrogyria in the right frontal lobe |
| P16 [23] | agenesis of the rostral part | ventriculomegaly | Chiari I malformation |
| P17 [23] | dysgenesis | hydrocephalus | - |
| P18 [23] | N | N | Chiari I malformation |
| P19 [24] | thin | enlarged bilateral cerebral ventricles | bilateral frontal and temporal brain atrophy |
| P21 | hypoplasia | ventriculomegaly | abnormality of neuronal migration |
| P23 | ventriculomegaly | - | |
| P25 [25] | thin | ventricular enlargement, ventricular wall irregularity | cyst of septipellucidi, decreased white matter volume |
| P26 [25] | hypoplasia | - | polycerebral gyrus at parasylvius fissures, cortical dysplasia of bilateral cerebral hemisphere, partial myelination delay |
| CLINICAL FEATURES | A | B |
|---|---|---|
| DYSMORPHISMS | ||
| Macrocephaly | 22/23 (96%) | 22/26 (85%) |
| High forehead | 13/13 (100%) | 13/26 (50%) |
| Low-set ears | 9/9 (100%) | 9/26 (35%) |
| Facial dysmorphic features | 13/13 (100%) | 13/26 (50%) |
| Craniofacial anomalies | 9/10 (90%) | 9/26 (35%) |
| Bilateral proximally placed first fingers | 6/6 (100%) | 6/26 (26%) |
| Other hands and feet anomalies | 5/5 (100%) | 5/26 (19%) |
| Other | 11/11 (100%) | 11/26 (42%) |
| NEUROLOGICAL/BEHAVIORAL ABNORMALITIES | ||
| ID | 15/18 (83%) | 15/26 (58%) |
| DD | 18/20 (90%) | 18/26 (69%) |
| Seizure | 4/6 (67%) | 4/26 (15%) |
| NDDs | 5/5 (100%) | 5/26 (19%) |
| Other | 11/11 (100%) | 11/26 (42%) |
| RENAL/URINARY TRACT DEFECTS | ||
| Hydronephrosis | 3/19 (16%) | 3/26 (11%) |
| Renal cysts | 3/19 (16%) | 3/26 (11%) |
| Other | 5/19 (26%) | 5/26 (19%) |
| CNS DEFECTS | ||
| Corpus callosum anomalies | 17/18 (94%) | 17/26 (65%) |
| Ventricular anomalies | 14/17 (82%) | 14/26 (54%) |
| Other | 16/16 100%) | 16/26 (61%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, V.; Cambi, F.; Orsini, A.; Bonuccelli, A.; Fiorini, A.; Santangelo, A.; Scacciati, M.; Elia, M.; Galesi, O.; Peroni, D.; et al. Phenotypic Spectrum of NFIA Haploinsufficiency: Two Additional Cases and Review of the Literature. Genes 2022, 13, 2249. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122249
Bertini V, Cambi F, Orsini A, Bonuccelli A, Fiorini A, Santangelo A, Scacciati M, Elia M, Galesi O, Peroni D, et al. Phenotypic Spectrum of NFIA Haploinsufficiency: Two Additional Cases and Review of the Literature. Genes. 2022; 13(12):2249. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122249
Chicago/Turabian StyleBertini, Veronica, Francesca Cambi, Alessandro Orsini, Alice Bonuccelli, Aureliano Fiorini, Andrea Santangelo, Massimo Scacciati, Maurizio Elia, Ornella Galesi, Diego Peroni, and et al. 2022. "Phenotypic Spectrum of NFIA Haploinsufficiency: Two Additional Cases and Review of the Literature" Genes 13, no. 12: 2249. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122249