
SCN1A Variants as the Underlying Cause of Genetic Epilepsy with Febrile Seizures Plus in Two Multi-Generational Colombian Families

 , , , and
, , , and 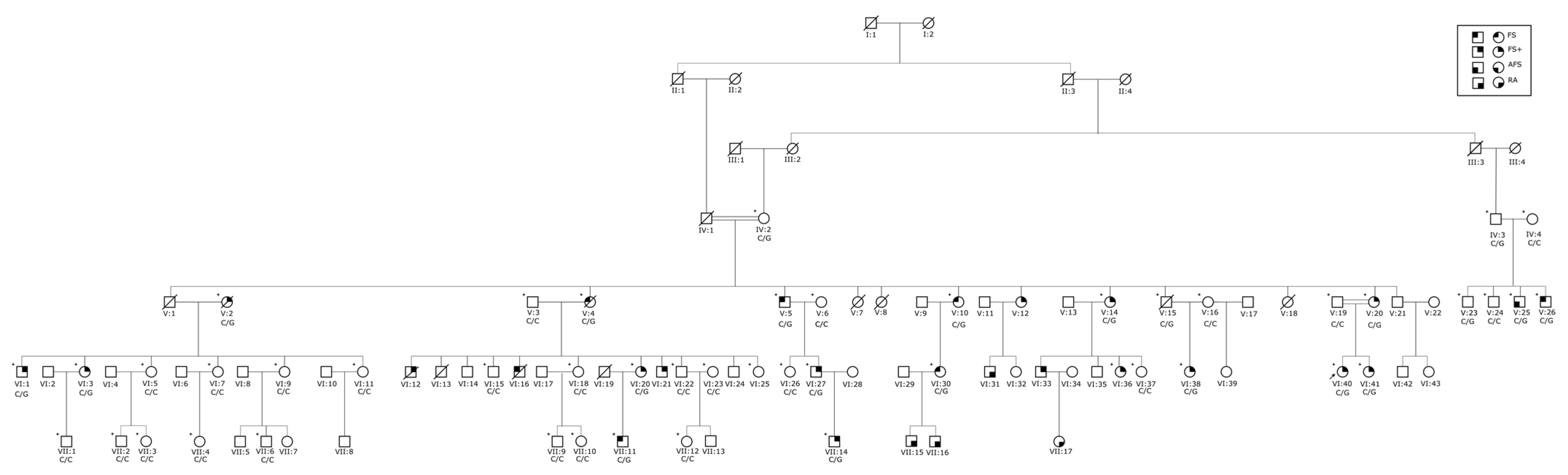{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standard Protocol Approvals, Registrations, and Patient Consents
2.2. Clinical Assessment
2.3. Exome Analysis
2.4. Sanger Sequencing
3. Results
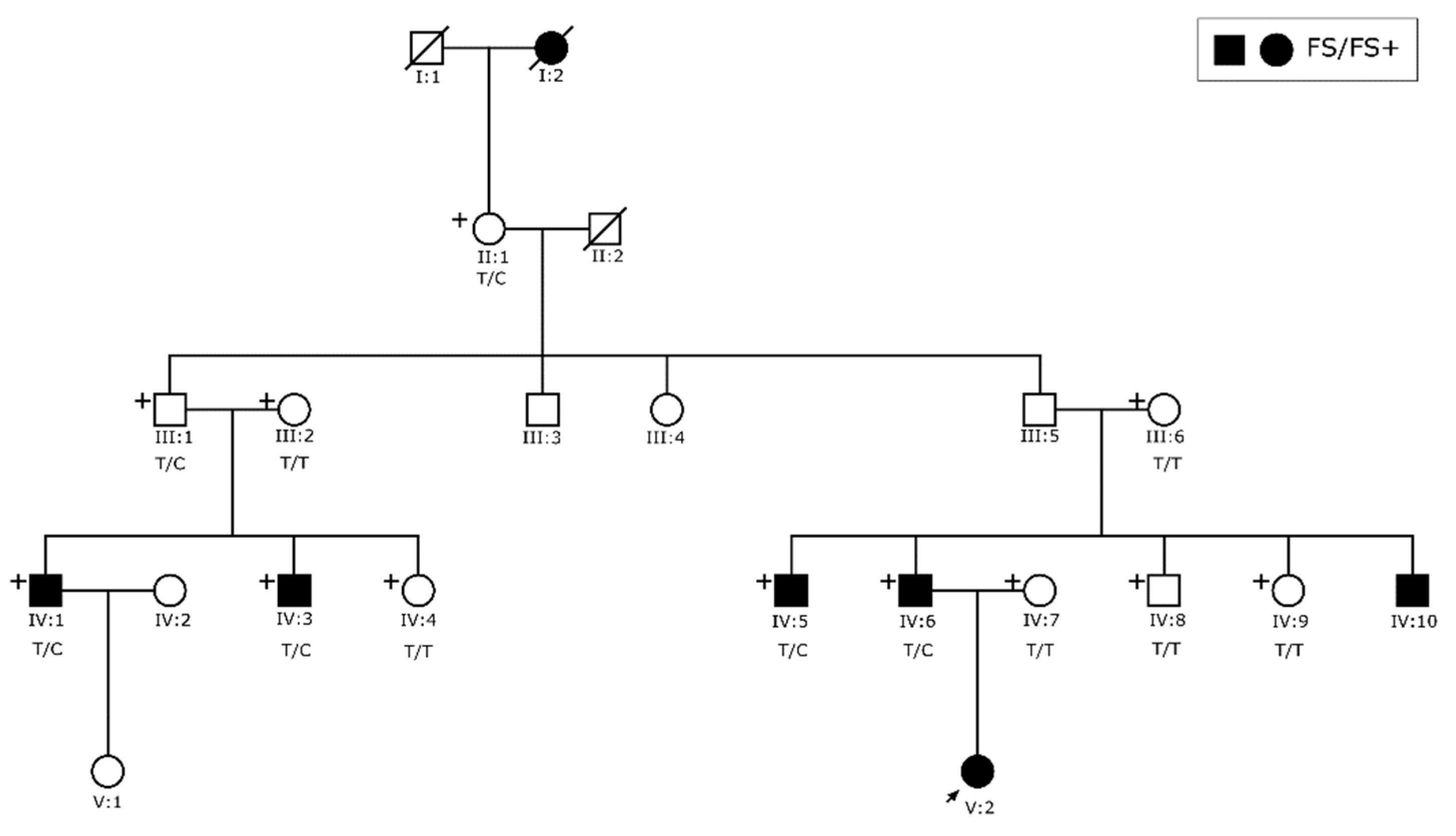
3.1. Clinical Findings
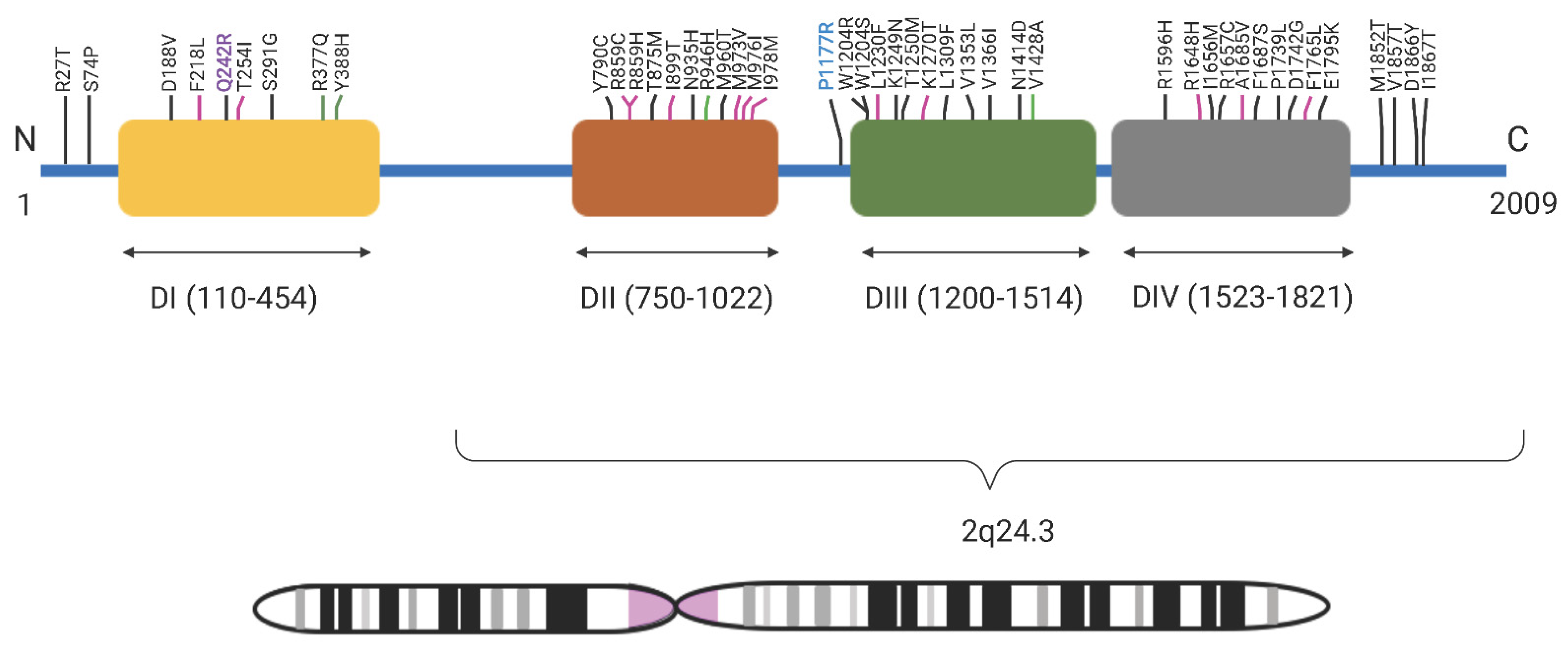
3.2. Exome Analysis
3.3. Sequencing Analysis and Penetrance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A Practical Clinical Definition of Epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabbout, R.; Kuchenbuch, M. Impact of Predictive, Preventive and Precision Medicine Strategies in Epilepsy. Nat. Rev. Neurol. 2020, 16, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Epilepsy: A Public Health Imperative; World Health Organization (WHO): Geneva, Switzerland, 2019; ISBN 978-92-4-151593-1.
- Banerjee, P.N.; Filippi, D.; Allen Hauser, W. The Descriptive Epidemiology of Epilepsy—A Review. Epilepsy Res. 2009, 85, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy Genetics: Clinical Impacts and Biological Insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Burgess, R.; Malone, J.P.; Glubb, G.C.; Helbig, K.L.; Vadlamudi, L.; Kivity, S.; Afawi, Z.; Bleasel, A.; Grattan-Smith, P.; et al. Genetic Epilepsy with Febrile Seizures plus: Refining the Spectrum. Neurology 2017, 89, 1210–1219. [Google Scholar] [CrossRef]
- Scheffer, I. Generalized Epilepsy with Febrile Seizures plus. A Genetic Disorder with Heterogeneous Clinical Phenotypes. Brain 1997, 120, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Nav1.1 Sodium Channel (SCN1A) Loss-of-Function Mutation Associated with Familial Simple Febrile Seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [Green Version]
- Pineda-Trujillo, N.; Carrizosa, J.; Cornejo, W.; Arias, W.; Franco, C.; Cabrera, D.; Bedoya, G.; Ruíz-Linares, A. A Novel SCN1A Mutation Associated with Severe GEFS+ in a Large South American Pedigree. Seizure 2005, 14, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Kluckova, D.; Kolnikova, M.; Lacinova, L.; Jurkovicova-Tarabova, B.; Foltan, T.; Demko, V.; Kadasi, L.; Ficek, A.; Soltysova, A. A Study among the Genotype, Functional Alternations, and Phenotype of 9 SCN1A Mutations in Epilepsy Patients. Sci. Rep. 2020, 10, 10288. [Google Scholar] [CrossRef]
- Escayg, A.; Goldin, A.L. Sodium Channel SCN1A and Epilepsy: Mutations and Mechanisms: Sodium Channel SCN1A and Epilepsy. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Sambrook, J. Isolation of High-Molecular-Weight DNA Using Organic Solvents. Cold Spring Harbor Protoc. 2017, 2017, pdb-prot093450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE Classification of the Epilepsies: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; NHLBI Exome Sequencing Project; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy Number Variation Detection and Genotyping from Exome Sequence Data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart Community Portal: An Innovative Alternative to Large, Centralized Data Repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A Curated Collection of Structural Variation in the Human Genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [Green Version]
- Genome Aggregation Database Consortium; Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to Be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. McKusick’s Online Mendelian Inheritance in Man (OMIM(R)). Nucleic Acids Res. 2009, 37, D793–D796. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; et al. Sequencing of 53,831 Diverse Genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Spector, E.; Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Gómez, M. Análisis De Ligamiento De Epilepsia Autosómica Dominante Con Convulsiones Febriles Plus En Familias Colombianas; Universidad De Antioquia: Medellin, Colombia, 2009. [Google Scholar]
- Spampanato, J.; Escayg, A.; Meisler, M.H.; Goldin, A.L. Generalized Epilepsy with Febrile Seizures plus Type 2 Mutation W1204R Alters Voltage-Dependent Gating of Nav1.1 Sodium Channels. Neuroscience 2003, 116, 37–48. [Google Scholar] [CrossRef]
- Bechi, G.; Rusconi, R.; Cestèle, S.; Striano, P.; Franceschetti, S.; Mantegazza, M. Rescuable Folding Defective NaV1.1 (SCN1A) Mutants in Epilepsy: Properties, Occurrence, and Novel Rescuing Strategy with Peptides Targeted to the Endoplasmic Reticulum. Neurobiol. Dis. 2015, 75, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Schorge, S.; Smith, A.D.; Ghanty, I.; Stewart, K.; Gardiner, S.; Du, J.; Pérez-Palma, E.; Symonds, J.D.; Collier, A.C.; et al. SCN1A Variants from Bench to Bedside—Improved Clinical Prediction from Functional Characterization. Hum. Mutat. 2020, 41, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Veneruzzo, G.; Loos, M.; Reyes, G.; Araoz, H.V.; Garcia, F.M.; Gomez, G.; Alonso, C.N.; Chertkoff, L.P.; Caraballo, R. Molecular Diagnosis of Epileptic Encephalopathy of the First Year of Life Applying a Customized Gene Panel in a Group of Argentinean Patients. Epilepsy Behav. 2020, 111, 107322. [Google Scholar] [CrossRef] [PubMed]
- Gonsales, M.C.; Montenegro, M.A.; Preto, P.; Guerreiro, M.M.; Coan, A.C.; Quast, M.P.; Carvalho, B.S.; Lopes-Cendes, I. Multimodal Analysis of SCN1A Missense Variants Improves Interpretation of Clinically Relevant Variants in Dravet Syndrome. Front. Neurol. 2019, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Arredondo, R.E.; Brambila-Tapia, A.J.L.; Mercado-Silva, F.M.; Magaña-Torres, M.T.; Figuera, L.E. Determination of SCN1A Genetic Variants in Mexican Patients with Refractory Epilepsy and Dravet Syndrome. Genet. Mol. Res. 2017, 16, 1–5. [Google Scholar] [CrossRef]
- Møller, R.S.; Larsen, L.H.G.; Johannesen, K.M.; Talvik, I.; Talvik, T.; Vaher, U.; Miranda, M.J.; Farooq, M.; Nielsen, J.E.K.; Lavard Svendsen, L.; et al. Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 2016, 7, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Niibori, Y.; Lee, S.J.; Minassian, B.A.; Hampson, D.R. Sexually Divergent Mortality and Partial Phenotypic Rescue After Gene Therapy in a Mouse Model of Dravet Syndrome. Hum. Gene Ther. 2020, 31, 339–351. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornejo-Sanchez, D.M.; Acharya, A.; Bharadwaj, T.; Marin-Gomez, L.; Pereira-Gomez, P.; Nouel-Saied, L.M.; University of Washington Center for Mendelian Genomics; Nickerson, D.A.; Bamshad, M.J.; Mefford, H.C.; et al. SCN1A Variants as the Underlying Cause of Genetic Epilepsy with Febrile Seizures Plus in Two Multi-Generational Colombian Families. Genes 2022, 13, 754. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050754
Cornejo-Sanchez DM, Acharya A, Bharadwaj T, Marin-Gomez L, Pereira-Gomez P, Nouel-Saied LM, University of Washington Center for Mendelian Genomics, Nickerson DA, Bamshad MJ, Mefford HC, et al. SCN1A Variants as the Underlying Cause of Genetic Epilepsy with Febrile Seizures Plus in Two Multi-Generational Colombian Families. Genes. 2022; 13(5):754. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050754
Chicago/Turabian StyleCornejo-Sanchez, Diana M., Anushree Acharya, Thashi Bharadwaj, Lizeth Marin-Gomez, Pilar Pereira-Gomez, Liz M. Nouel-Saied, University of Washington Center for Mendelian Genomics, Deborah A. Nickerson, Michael J. Bamshad, Heather C. Mefford, and et al. 2022. "SCN1A Variants as the Underlying Cause of Genetic Epilepsy with Febrile Seizures Plus in Two Multi-Generational Colombian Families" Genes 13, no. 5: 754. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050754