
The Benefit of Multigene Panel Testing for the Diagnosis and Management of the Genetic Epilepsies

 , and
, and
Abstract
:1. Introduction
2. Methods
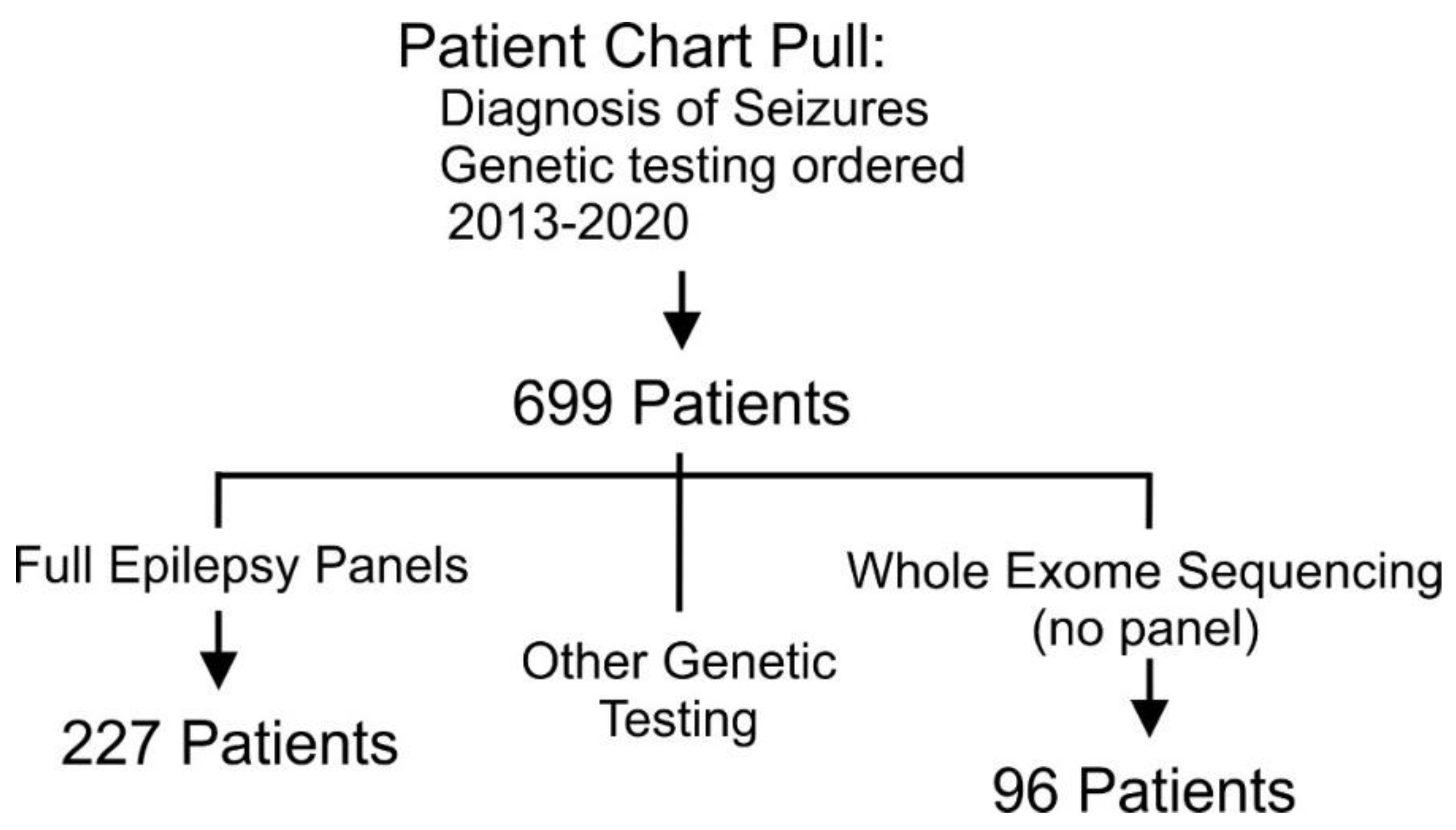
2.1. Inclusion Criteria and Cohort Identification
2.2. Chart Review
2.3. Data Analysis
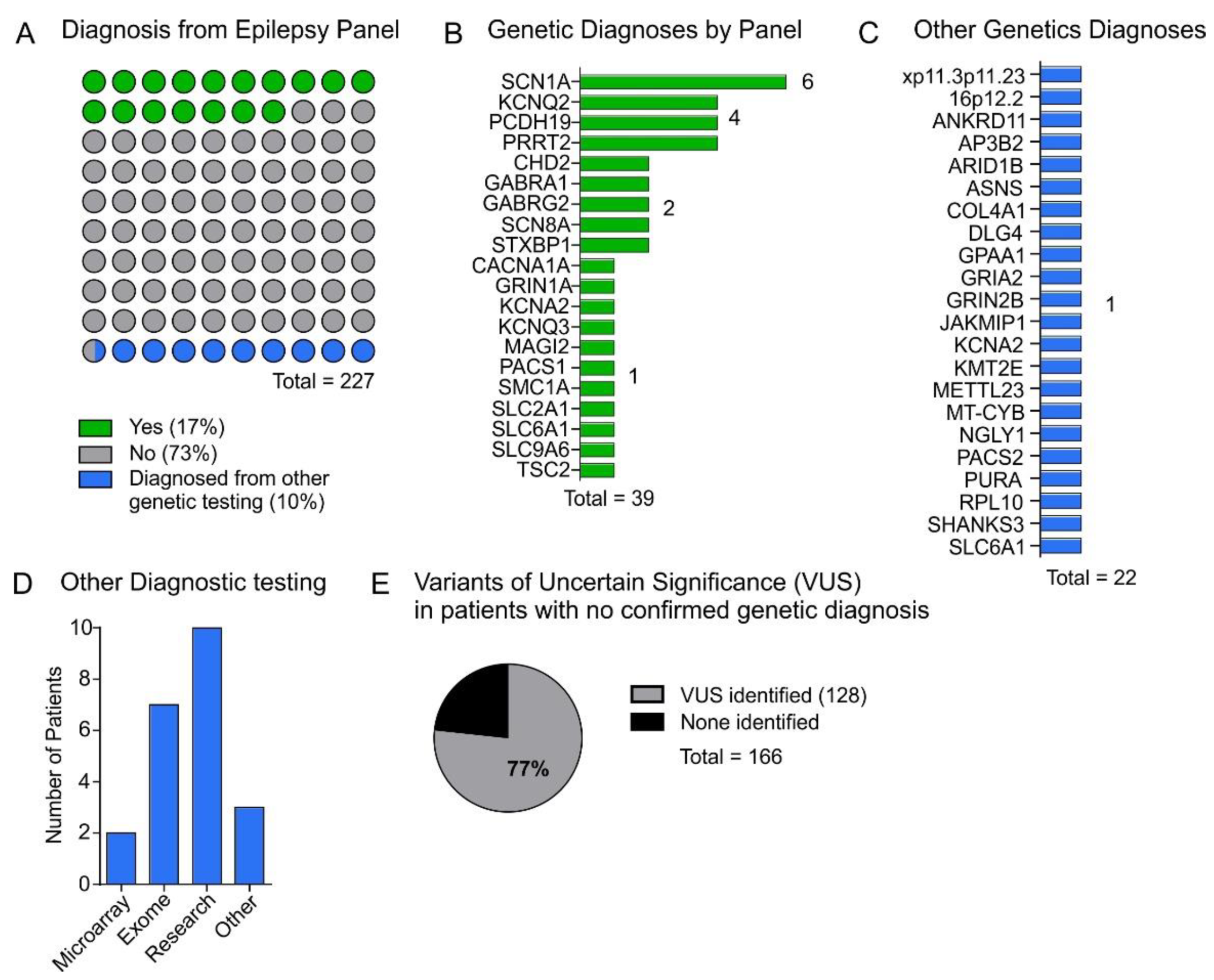
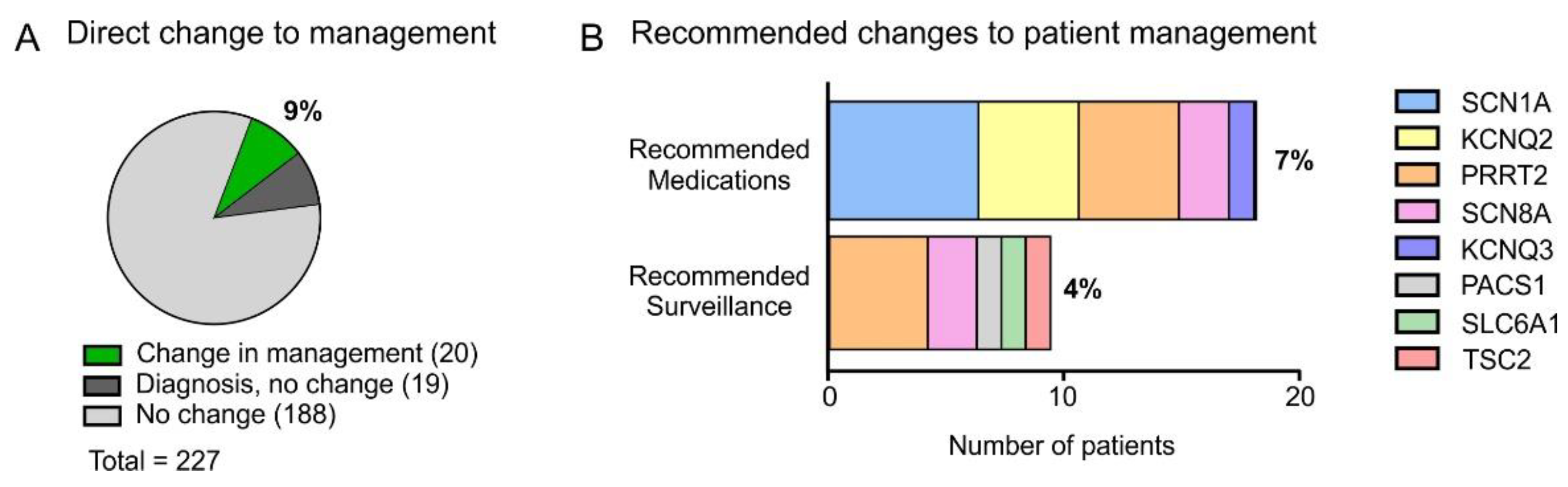
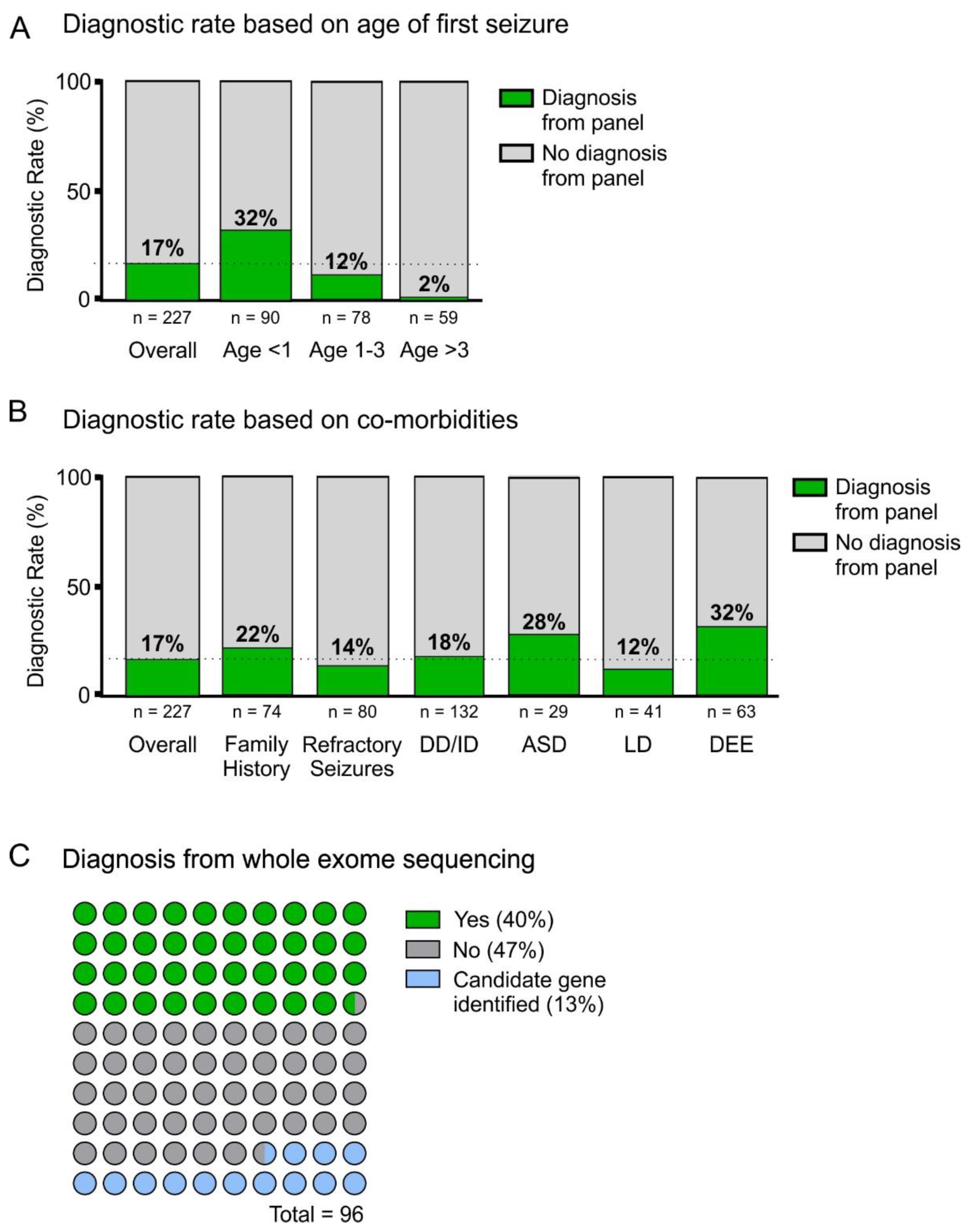
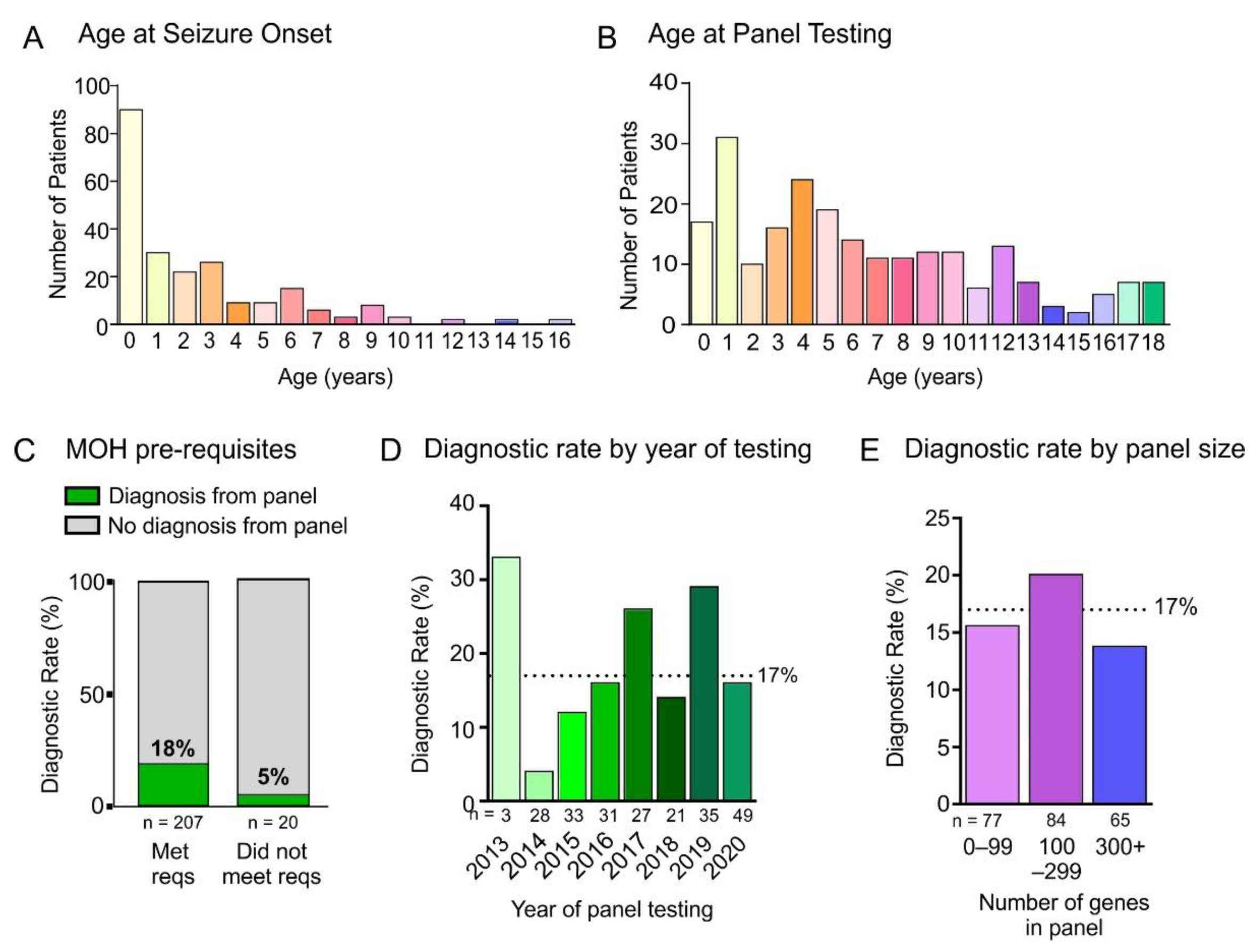
3. Results
4. Discussion
4.1. Panel Diagnostic Rate
4.2. Changes to Patient Management as a Result of Testing
4.3. Delay to Panel Testing
4.4. Limitations
4.5. Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Coding DNA Variant | Company | Interpretation on Report | Seizures/Clinical Diagnosis |
|---|---|---|---|---|---|
| SCN1A | NM_001165963.2 | c.662 T > C p.Leu221Pro | Fulgent | VUS | Febrile, focal, generalized |
| SCN1A | NM_001165963.1 | c.5260 G > C p.Gly1754Arg | Invitae | Pathogenic | Dravet |
| SCN1A | NM_001165963.1 | c.2975 dupT p.Ser993GlufsTer4 | GeneDx | Pathogenic | Dravet, Status |
| SCN1A | NM_001165963.1 | c.4223 G > A p.Trp1408Ter | Blueprint Genetics | Pathogenic | Atypical febrile, status, myoclonic |
| SCN1A | NM_001165963.1 | c.2846 G > A p.Cys949Tyr | GeneDx | Pathogenic | Dravet, generalized, myoclonus |
| SCN1A | NM_001165964 | Deletion exons 17–20 | MNG | Pathogenic | Febrile seizures, GTC |
| KCNQ2 | NM_172107.2 | c.436 T > A p.Trp146Arg | GeneDx | VUS | Neonatal seizures |
| KCNQ2 | NM_172107.2 | c.1023 + 1 G > A | GeneDx | Pathogenic | Infantile seizures |
| KCNQ2 | NM_172107.2 | c.1627 G > A p.Val543Met | GeneDx | VUS | Febrile seizures |
| KCNQ2 | NM_172107.2 | c.1057 C > T p.Arg353Cys | GeneDx | Pathogenic | Infantile seizures |
| PCDH19 | NM_001105243.1 | c.2515 C > T p.Arg839Ter | GeneDx | Disease-causing mutations | Status, generalized |
| PCDH19 | NM_001184880.1 | Whole gene deletion | GeneDx | Pathogenic | Status, focal seizures |
| PCDH19 | Not reported | c.47G > A p.Trp16Ter | Unknown | Likely pathogenic | Generalized, focal |
| PCDH19 | NM_001184880.1 | c.1465 T > C p.Ser489Pro | GeneDx | Likely pathogenic | Febrile, GTC |
| PRRT2 | NM_145239.2 | c.649del p.Arg217GlufsTer12 | Invitae | Pathogenic | Infantile seizures |
| PRRT2 | NM_145239.2 | c.649 C > T p.Arg217Ter | Fulgent | Pathogenic | Infantile seizures |
| PRRT2 | NM_145239.2 | c.649delC p.Arg217GlufsTer12 | GeneDx | Pathogenic | Infantile seizures |
| PRRT2 | NM_145239.2 | c.649dup p.Arg217ProfTer8 | Blueprint | Pathogenic | Infantile seizures |
| CHD2 | NM_001271.3 | c.4265 A > G p.Asp1422Gly | GeneDx | Likely pathogenic | Myoclonic |
| CHD2 | NM_001271.3 | c.2084dup p.Leu697ProfTer16 | Fulgent | Pathogenic | Myoclonic |
| GABRA1 | NM_000806.5 | c.641 G > A p.Arg214His | Prevention | Pathogenic | Febrile, myoclonic |
| GABRA1 | Reference GRCH37 | c.604 C > T p.Arg214Cys | MNG | VUS with pathogenic features | Myoclonic-astatic, generalized |
| GABRG2 | NM_198903.2 | c.452_455delTCTT p.Phe151SerfsTer19 | MNG | Likely pathogenic | Absence, febrile |
| GABRG2 | NM_000816.3 | c.529 C > T p.Arg177Ter | Blueprint | Likely pathogenic | Absence |
| KCNA2 | NM_00497.3 | c.193 C > T p.Arg65Ter | Fulgent | VUS-Likely pathogenic | Absence, GTC |
| SCN8A | NM_014191.3 | c.4351 G > A p.Gly1451Ser | Invitae | Pathogenic | Febrile, myoclonic, absence |
| SCN8A | NM_014191.3 | c.4850 G > A p.Arg1617Gln | GeneDx | Pathogenic | Infantile epilepsy |
| STXBP1 | NM_003165.3 | c.1656 C > A p.Cys552Ter | GeneDx | Likely pathogenic | Neonatal seizures |
| STXBP1 | MIM: 602926 | c.1420C > T p.Gln474Ter | MNG | Pathogenic | Infantile, tonic, atonic, GTC |
| CACNA1A | NM_023035.3 | c.4188 G > A p.Val1396Met | MNG | VUS | Refractory seizures, GTC |
| GRIN1A | NM_000833.3 | c.1173delC p.R392GfsTer31 | GeneDx | Pathogenic | GTC |
| KCNQ3 | NM_004519.3 | c.988 C > T p.Arg330Cys | Fulgent | Pathogenic | Neonatal seizures, focal |
| MAGI2 | Not Reported | 7q11.23q21.13 deletion | GeneDx | Pathogenic | Infantile spasms |
| PACS1 | NM_018026.3 | c.607 C > T p.Arg203Trp | GeneDx | Pathogenic | Neonatal seizures |
| SMC1A | NM_006306.3 | c.2814dup p.Lys939Ter | Invitae | Pathogenic | Focal |
| SLC2A1 | NM_006516.2 | c.418 G > A p.Val140Met | GeneDx | Disease-causing | Generalized |
| SLC6A1 | NM_003042.3 | c.883dup p.Ser295PhefsTer61 | Fulgent | Pathogenic | Myoclonic-astatic, absence |
| SLC9A6 | NM_006359.2 | c.507 + 1G > T p.? | Fulgent | Likely pathogenic | Generalized |
| TSC2 | NM_000548.3 | c.1609_1653del45 p.Arg536_551del | GeneDx | Pathogenic | Infantile seizures, focal, TSC |
| Gene | Variant | Interpretation on Initial Report | Other Testing | Reasoning | ClinVar | gnomAd | Final Interpretation |
|---|---|---|---|---|---|---|---|
| SCN1A | c.662T > C, p.Leu221Pro | VUS | Parentals pending | PS1; PM2, PM5; PP3 | Reported pathogenic (2014) | Absent | Likely pathogenic |
| KCNQ2 | c.436T > A, p.Trp146Arg | VUS | Parentals performed (de novo) | PS2; PM2; PP3 | Reported as a VUS (2017) | Absent | Likely pathogenic |
| KCNQ2 | c.1627G > A; p.Val543Met | VUS | Parentals performed (inherited) | PS1; PM2; PP3 | Conflicting—most recent report as P/LP | Absent | Likely pathogenic |
| GABRA1 | c.640C > T; p.Arg214Cys | VUS | Parentals performed (de novo) | PS1, PS2; PM2; PP3 | Pathogenic (2019) | Absent | Pathogenic |
| KNCA2 | c.193C > T, p.Arg65Ter | VUS | Segregation in multiple affected family members | PVS1; PM2, PM4, PP1 | Conflicting | Absent | Likely pathogenic |
| CACNA1A | c.4186G > A, p.Val1396Met | VUS | Parentals performed (de novo) | PS1; PM2; PP3 | Absent | Absent | Likely pathogenic |
References
- Helbig, I.; Tayoun, A.A.N. Understanding Genotypes and Phenotypes in Epileptic Encephalopathies. Mol. Syndromol. 2016, 7, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheidley, B.R.; Malinowski, J.; Bergner, A.L.; Bier, L.; Gloss, D.S.; Mu, W.; Mulhern, M.M.; Partack, E.J.; Poduri, A. Genetic testing for the epilepsies: A systematic review. Epilepsia 2022, 63, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Johnstone, D.L.; Dyment, D.A. Epilepsy genetics: Current knowledge, applications, and future directions. Clin. Genet. 2019, 95, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poduri, A. When Should Genetic Testing Be Performed in Epilepsy Patients? Epilepsy Curr. 2017, 17, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Demos, M.; Guella, I.; DeGuzman, C.; McKenzie, M.B.; Buerki, S.E.; Evans, D.M.; Toyota, E.B.; Boelman, C.; Huh, L.L.; Datta, A.; et al. Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front. Neurol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Dhiman, V. Molecular Genetics of Epilepsy: A Clinician’s Perspective. Ann. Indian Acad. Neurol. 2017, 20, 96–102. [Google Scholar] [CrossRef]
- Jain, P.; Andrade, D.; Donner, E.; Dyment, D.; Prasad, A.N.; Goobie, S.; Boycott, K.; Lines, M.; Snead, O.C. Development of Criteria for Epilepsy Genetic Testing in Ontario, Canada. Can. J. Neurol. Sci. 2019, 46, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Dyment, D.A.; Prasad, A.N.; Boycott, K.M.; Ediae, G.U.; Hartley, T.; Hassan, A.; Muir, K.E.; Potter, M.; Boisse Lomax, L.; Jarinova, O.; et al. Implementation of Epilepsy Multigene Panel Testing in Ontario, Canada. Can. J. Neurol. Sci. 2020, 47, 61–68. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, A.T.; Coryell, J.; Saneto, R.P.; Grinspan, Z.M.; Alexander, J.J.; Kekis, M.; Sullivan, J.E.; Wirrell, E.C.; Shellhaas, R.A.; Mytinger, J.R.; et al. Early-Life Epilepsies and the Emerging Role of Genetic Testing. JAMA Pediatr. 2017, 171, 863–871. [Google Scholar] [CrossRef]
- Truty, R.; Patil, N.; Sankar, R.; Sullivan, J.; Millichap, J.; Carvill, G.; Entezam, A.; Esplin, E.D.; Fuller, A.; Hogue, M.; et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open 2019, 4, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gotway, G.; Pascual, J.M.; Park, J.Y. Diagnostic Yield of Clinical Next-Generation Sequencing Panels for Epilepsy. JAMA Neurol. 2014, 71, 650–651. [Google Scholar] [CrossRef] [Green Version]
- Herington, E.; McCormack, S. Genome-Wide Sequencing for Unexplained Developmental Delays and Multiple Congenital Anomalies: A Rapid Qualitative Review; CADTH Rapid Response Reports; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2019. [Google Scholar]
- Berkovic, S.F. Genetics of Epilepsy in Clinical Practice. Epilepsy Curr. 2015, 15, 192–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Kong, Y.; Dong, X.; Hu, L.; Lin, Y.; Chen, X.; Ni, Q.; Lu, Y.; Wu, B.; Wang, H.; et al. Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet. Med. 2019, 21, 564–571. [Google Scholar] [CrossRef]
- McKnight, D.; Bristow, S.L.; Truty, R.M.; Morales, A.; Stetler, M.; Westbrook, M.J.; Robinson, K.; Riethmaier, D.; Borlot, F.; Kellogg, M.; et al. Multigene Panel Testing in a Large Cohort of Adults with Epilepsy: Diagnostic Yield and Clinically Actionable Genetic Findings. Neurol. Genet. 2022, 8, e650. [Google Scholar] [CrossRef]
- Chiron, C.; Dulac, O. The pharmacologic treatment of Dravet syndrome. Epilepsia 2011, 52 (Suppl. S2), 72–75. [Google Scholar] [CrossRef]
- Brunklaus, A.; Dorris, L.; Ellis, R.; Reavey, E.; Lee, E.; Forbes, G.; Appleton, R.; Cross, J.H.; Ferrie, C.; Hughes, I.; et al. The clinical utility of an SCN1A genetic diagnosis in infantile-onset epilepsy. Dev. Med. Child Neurol. 2013, 55, 154–161. [Google Scholar] [CrossRef]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.K.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Joshi, N.; Weckhuysen, S.; Cooper, E.C.; Taglialatela, M. KCNQ3-Related Disorders. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK201978/ (accessed on 10 March 2022).
- Gardella, E.; Møller, R.S. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 2019, 60, S77–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, M.F.; Wagnon, J.L.; Mefford, H.C.; Meisler, M.H. SCN8A-Related Epilepsy with Encephalopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK379665/ (accessed on 10 March 2022).
- Landolfi, A.; Barone, P.; Erro, R. The Spectrum of PRRT2-Associated Disorders: Update on Clinical Features and Pathophysiology. Front. Neurol. 2021, 12, 629747. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Northrup, H. Tuberous Sclerosis Complex Surveillance and Management: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusk, L.; Smith, S.; Martin, C.; Taylor, C.; Chung, W. PACS1 Neurodevelopmental Disorder. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559434/ (accessed on 10 March 2022).
- Kahen, A.; Kavus, H.; Geltzeiler, A.; Kentros, C.; Taylor, C.; Brooks, E.; Snyder, L.G.; Chung, W. Neurodevelopmental phenotypes associated with pathogenic variants in SLC6A1. J. Med. Genet. 2021. published online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, K.M.; Gardella, E.; Linnankivi, T.; Courage, C.; de Saint Martin, A.; Lehesjoki, A.-E.; Mignot, C.; Afenjar, A.; Lesca, G.; Abi-Warde, M.-T.; et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia 2018, 59, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Genetics of Epilepsy Syndromes Starting in the First Year of Life Symbol. Available online: https://oce-ovid-com.proxy.bib.uottawa.ca/article/00006114-200901200-00014/HTML (accessed on 23 January 2022).
- Wirrell, E.C.; Grossardt, B.R.; Wong-Kisiel, L.C.-L.; Nickels, K.C. Incidence and Classification of New-Onset Epilepsy and Epilepsy Syndromes in Children in Olmsted County, Minnesota from 1980–2004: A population-based study. Epilepsy Res. 2011, 95, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Balciuniene, J.; DeChene, E.T.; Akgumus, G.; Romasko, E.J.; Cao, K.; Dubbs, H.A.; Mulchandani, S.; Spinner, N.B.; Conlin, L.K.; Marsh, E.D.; et al. Use of a Dynamic Genetic Testing Approach for Childhood-Onset Epilepsy. JAMA Netw. Open 2019, 2, e192129. [Google Scholar] [CrossRef]
- Criteria for Genetic Testing Related to Epilepsy. Available online: https://www.health.gov.on.ca/en/pro/programs/gtac/docs/gtac_report_criteria_genetic_testing_related_epilepsy.pdf (accessed on 8 April 2022).
- El Achkar, C.M.; Olson, H.E.; Poduri, A.; Pearl, P.L. The genetics of the epilepsies. Curr. Neurol. Neurosci. Rep. 2015, 15, 39. [Google Scholar] [CrossRef]
- Home. Project ECHO Ontario. Available online: https://oen.echoontario.ca/ (accessed on 10 March 2022).





| All Panel Testing n (%) | Positive Diagnosis n (%) | |
|---|---|---|
| Participants | 227 | 39 |
| Sex (%) | ||
| Male | 117 (51) | 19 (49) |
| Female | 110 (49) | 20 (51) |
| Panel Company | ||
| GeneDx | 92 (40.5) | 17 (43) |
| Fulgent | 45 (20) | 7 (18) |
| MNG | 20 (9) | 5 (13) |
| Invitae | 27 (12) | 4 (10) |
| Blueprint Genetics | 14 (6) | 3 (8) |
| Prevention Genetics | 8 (3.5) | 1 (2.5) |
| Transgenomics | 13 (6) | 0 (0) |
| Other | 8 (3) | 2 (5) |
| Age at seizure onset (years) | ||
| 0 | 90 (40) | 29 (74) |
| 1 | 30 (13) | 4 (10) |
| 2 | 22 (10) | 5 (13) |
| 3 | 26 (11) | 0 (0) |
| 4 | 9 (4) | 0 (0) |
| ≥5 | 50 (22) | 1 (2.5) |
| Mean | 2.5 | 0.5 |
| Age at testing (years) | ||
| 0 | 17 (7) | 8 (20.5) |
| 1 | 31 (14) | 10 (26) |
| 2 | 10 (4) | 1 (2.5) |
| 3 | 16 (7) | 4 (10) |
| 4 | 24 (11) | 3 (8) |
| ≥5 | 129 (57) | 13 (33) |
| Mean | 6.5 | 4.5 |
| Investigations | ||
| Microarray | 149 (66) | 23 (59) |
| Parental testing | 92 (41) | 25 (64) |
| Whole exome sequencing | 47 (21) | 0 (0) |
| MRI | 207 (91) | 38 (97) |
| EEG | 227 (100) | 39 (100) |
| Co-morbidities and family history | ||
| Refractory Seizures | 80 (35) | 11 (28) |
| Developmental Delay/Intellectual Disability | 132 (58) | 24 (62) |
| Autism Spectrum Disorder | 29 (13) | 8 (20.5) |
| Learning Disability | 41 (18) | 5 (13) |
| Dysmorphisms or multi-system involvement | 33 (15) | 3 (8) |
| Epileptic encephalopathy | 63 (28) | 19 (49) |
| Family History of Epilepsy | 74 (33) | 16 (41) |
| All Exome Testing n (%) | Positive Diagnosis n (%) | |
|---|---|---|
| Participants | 96 | 38 |
| Age at seizure onset (years) | ||
| 0 | 18 (19) | 8 (21) |
| 1 | 9 (9) | 6 (16) |
| 2 | 12 (13) | 3 (80) |
| 3 | 8 (8) | 3 (8) |
| 4 | 12 (13) | 6 (16) |
| ≥5 | 27 (28) | 9 (24) |
| Unknown | 9 (9) | 3 (8) |
| Mean | 4.2 | 4.1 |
| Age at testing (years) | ||
| 0 | 4 (4) | 2 (5) |
| 1 | 7 (7) | 4 (11) |
| 2 | 4 (4) | 2 (5) |
| 3 | 8 (8) | 3 (8) |
| 4 | 8 (8) | 3 (8) |
| 5–8 | 13 (14) | 4 (11) |
| ≥9 | 52 (54) | 20 (53) |
| Mean | 8.7 | 8.6 |
| Co-morbidities | ||
| Developmental Delay/Intellectual Disability | 77 (80) | 34 (89) |
| Autism Spectrum Disorder | 18 (19) | 6 (15) |
| Learning Disability | 2 (2) | 0 (0) |
| Dysmorphisms or multi-system involvement | 96 (100) | 38 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leduc-Pessah, H.; White-Brown, A.; Hartley, T.; Pohl, D.; Dyment, D.A. The Benefit of Multigene Panel Testing for the Diagnosis and Management of the Genetic Epilepsies. Genes 2022, 13, 872. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050872
Leduc-Pessah H, White-Brown A, Hartley T, Pohl D, Dyment DA. The Benefit of Multigene Panel Testing for the Diagnosis and Management of the Genetic Epilepsies. Genes. 2022; 13(5):872. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050872
Chicago/Turabian StyleLeduc-Pessah, Heather, Alexandre White-Brown, Taila Hartley, Daniela Pohl, and David A. Dyment. 2022. "The Benefit of Multigene Panel Testing for the Diagnosis and Management of the Genetic Epilepsies" Genes 13, no. 5: 872. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050872