
A De Novo Missense Variant in TUBG2 in a Child with Global Developmental Delay, Microcephaly, Refractory Epilepsy and Perisylvian Polymicrogyria

 and
and 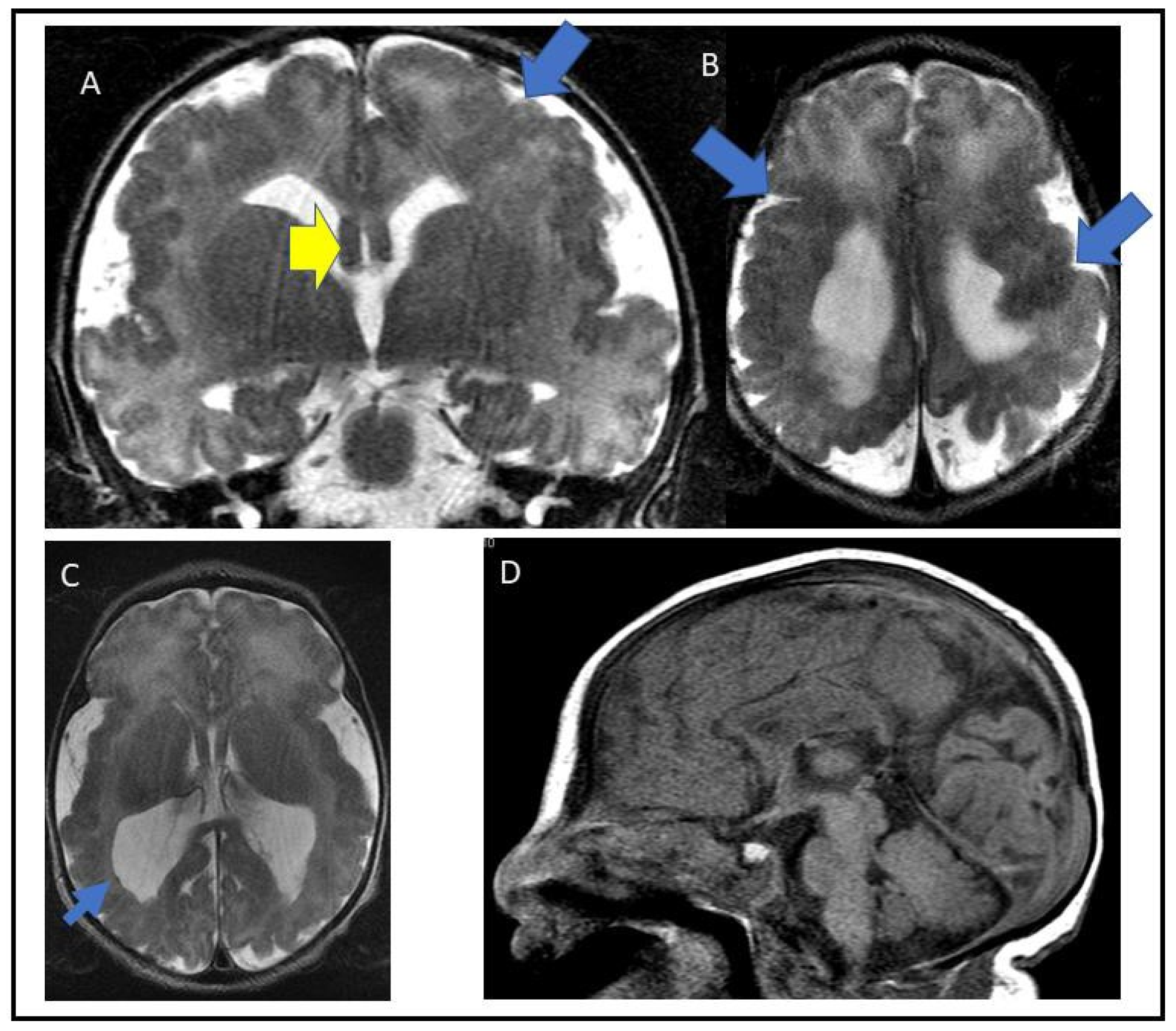{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Exome Sequencing and Analysis
2.2. Expression of Recombinant TUBG2
2.3. Western Blot
2.4. Growth Curve of Patient and Control Lymphoblasts
2.5. Isolation of Total RNA, Reverse-Transcription PCR and qPCR
2.6. Co-Immunoprecipitation
3. Results
3.1. Case Report
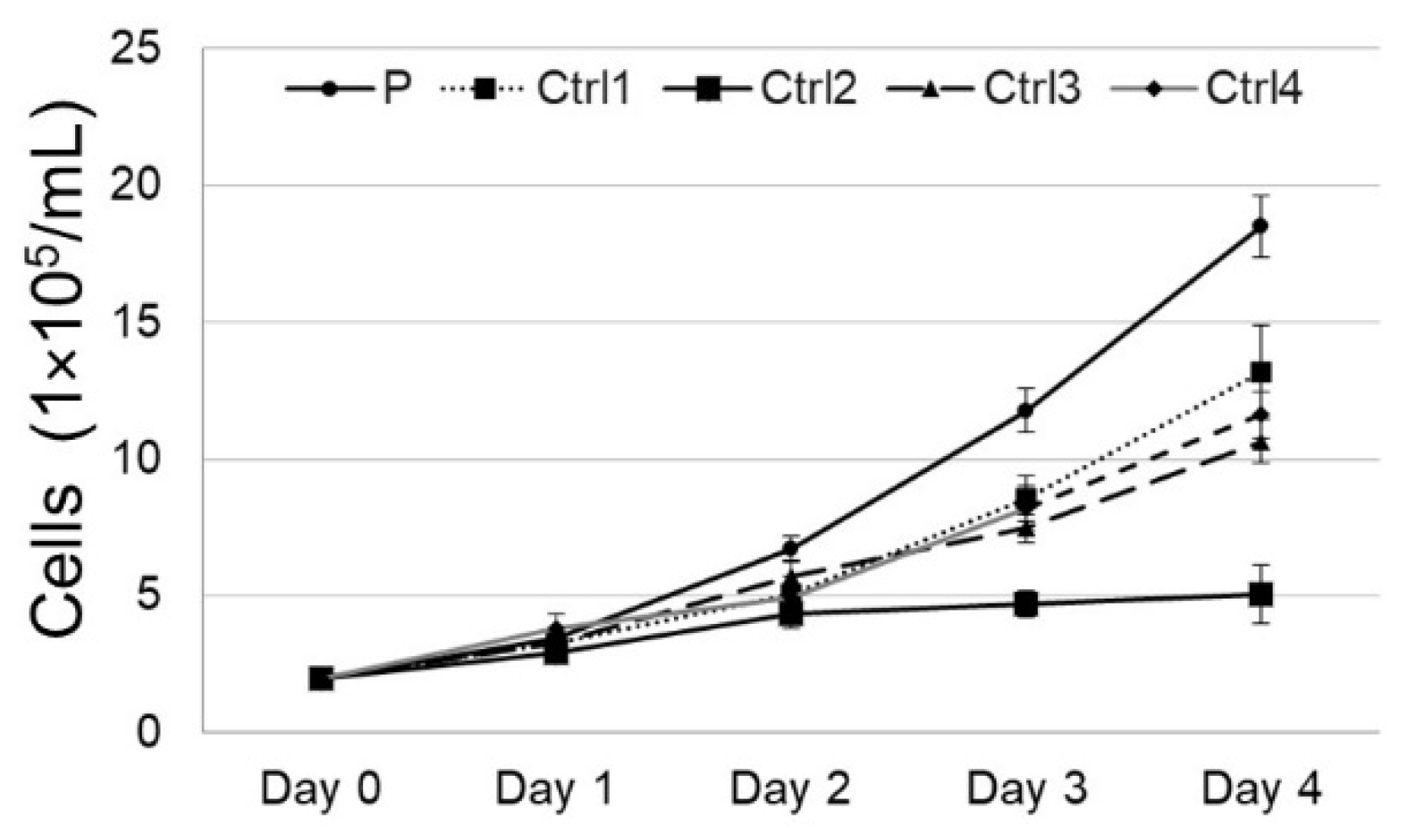
3.2. Cell Growth
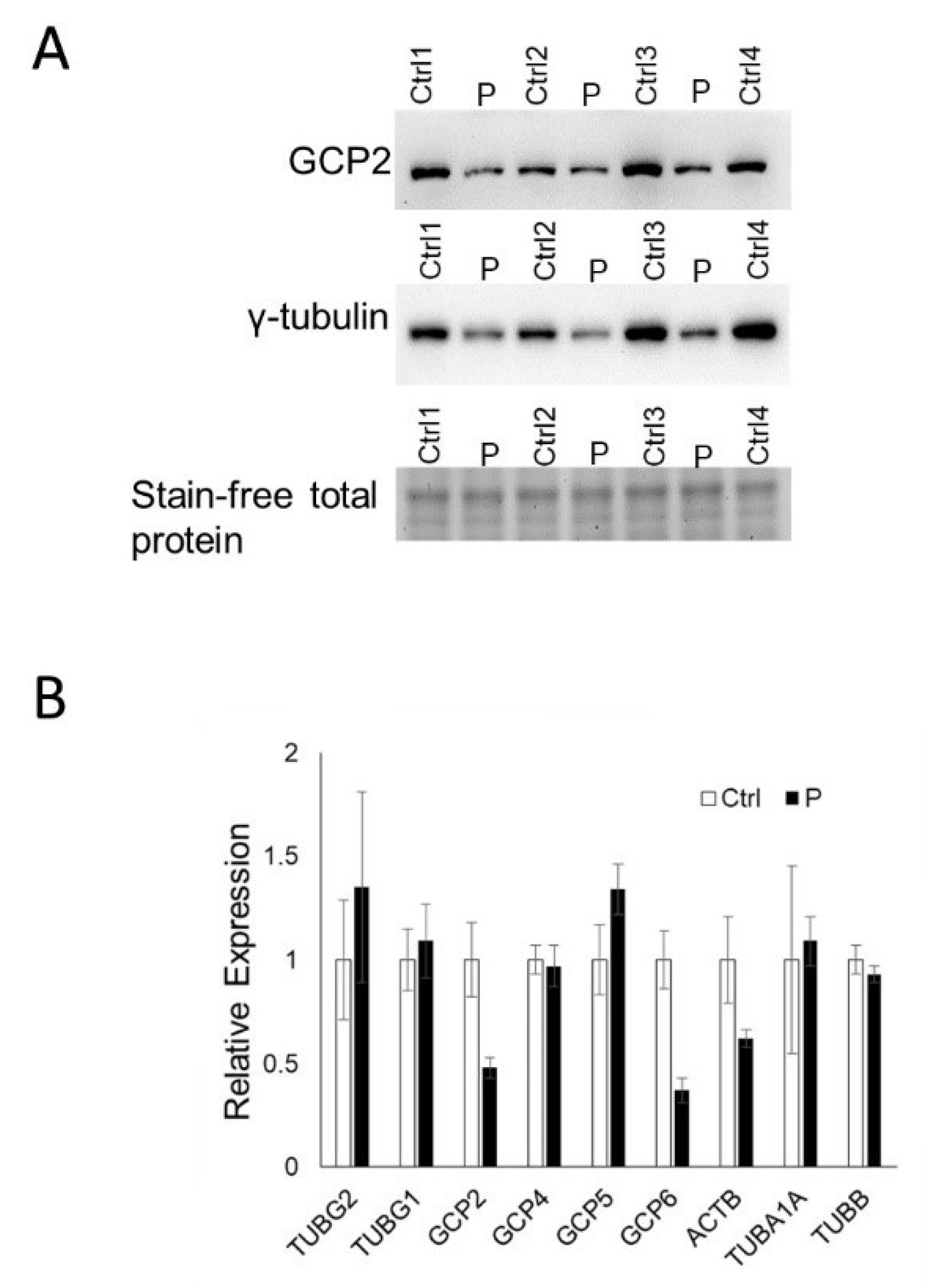
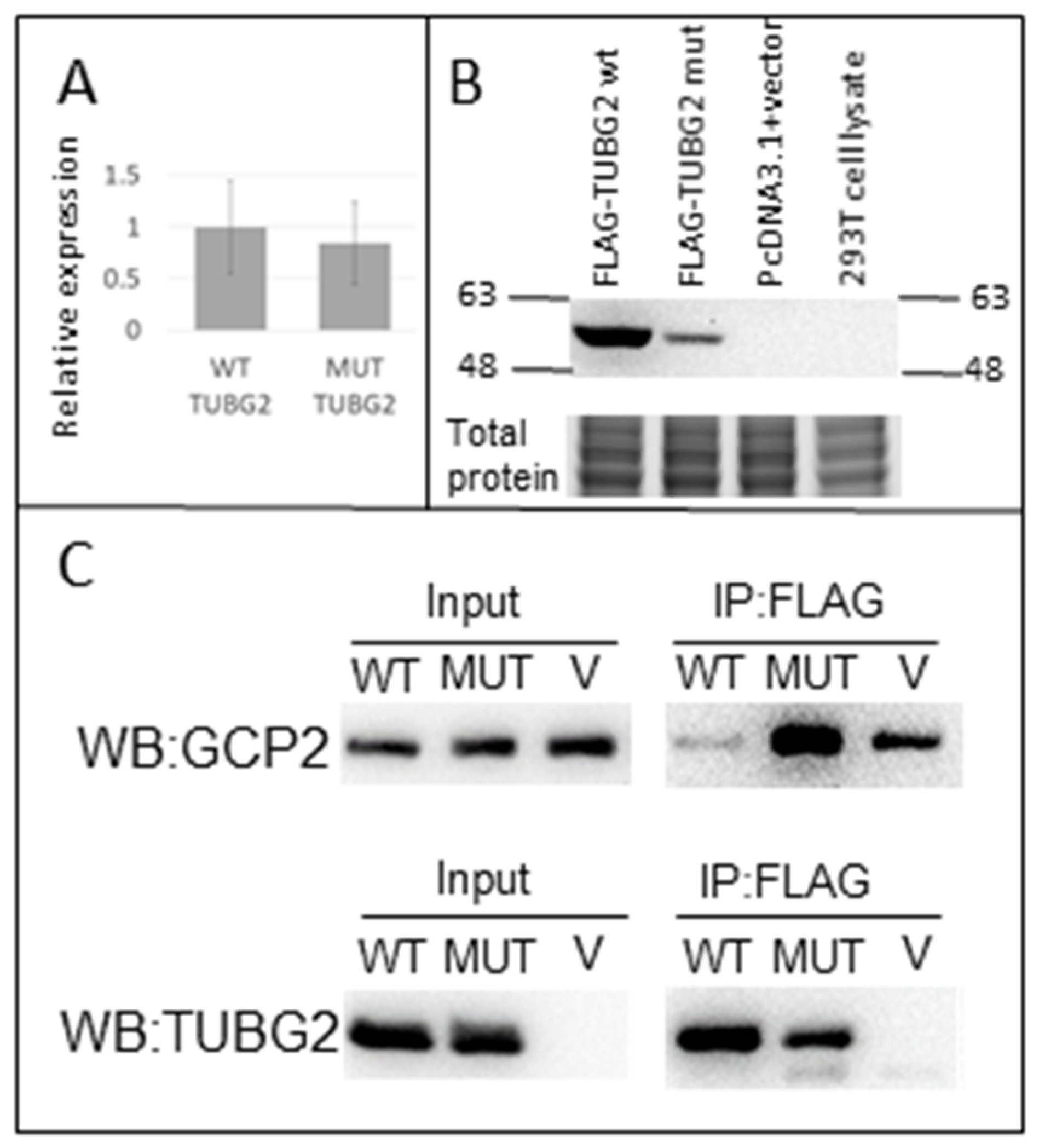
3.3. TUBG2 Expression and Function: Western Blot and RT-PCR
3.4. RT-PCR of TUBG1
3.5. RT-PCR of γ-Tubulin Complex Proteins
3.6. Co-Immunoprecipitation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stutterd, C.A.; Leventer, R.J. Polymicrogyria: A common and heterogeneous malformation of cortical development. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Leventer, R.J.; Jansen, A.; Pilz, D.T.; Stoodley, N.; Marini, C.; Dubeau, F.; Malone, J.; Mitchell, L.A.; Mandelstam, S.; Scheffer, I.E.; et al. Clinical and imaging heterogeneity of polymicrogyria: A study of 328 patients. Brain 2010, 133, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, C.; Ramgopal, S.; Fallil, Z.; Parulkar, I.; Alongi, R.; Knowlton, R.; Poduri, A.; EPGP Investigators. Polymicrogyria-associated epilepsy: A multicenter phenotypic study from the Epilepsy Phenome/Genome Project. Epilepsia 2013, 54, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkovich, A.J.; Linden, C.E. Congenital cytomegalovirus infection of the brain: Imaging analysis and embryologic considerations. AJNR Am. J. Neuroradiol. 1994, 15, 703–715. [Google Scholar] [PubMed]
- Dobyns, W.B.; Mirzaa, G.; Christian, S.L.; Petras, K.; Roseberry, J.; Clark, G.D.; Curry, C.J.; McDonald McGinn, D.; Medne, L.; Zackai, E.; et al. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am. J. Med. Genet. A 2008, 146A, 1637–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutterd, C.A.; Brock, S.; Stouffs, K.; Fanjul-Fernandez, M.; Lockhart, P.J.; McGillivray, G.; Mandelstam, S.; Pope, K.; Delatycki, M.B.; Jansen, A.; et al. Genetic heterogeneity of polymicrogyria: Study of 123 patients using deep sequencing. Brain Commun. 2020, 3, fcaa221. [Google Scholar] [CrossRef] [PubMed]
- Parrini, E.; Conti, V.; Dobyns, W.B.; Guerrini, R. Genetic Basis of Brain Malformations. Mol. Syndromol. 2016, 7, 220–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137 Pt 6, 1676–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romaniello, R.; Arrigoni, F.; Fry, A.E.; Bassi, M.T.; Rees, M.I.; Borgatti, R.; Pilz, D.T.; Cushion, T.D. Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 2018, 61, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shen, K.M.; Zackai, E.H.; Bhoj, E.J. Clinical variability of TUBB-associated disorders: Diagnosis through reanalysis. Am. J. Med. Genet. Part A 2020, 182, 3035–3039. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.T.; Riggs, E.R.; Buchanan, A.H.; Ceyhan-Birsoy, O.; DiStefano, M.; Dwight, S.S.; Goldstein, J.; Ghosh, R.; Seifert, B.A.; Sneddon, T.P.; et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am. J. Hum. Genet. 2017, 100, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, S.; Stouffs, K.; Scalais, E.; D’Hooghe, M.; Keymolen, K.; Guerrini, R.; Dobyns, W.B.; Di Donato, N.; Jansen, A.C. Tubulinopathies continued: Refining the phenotypic spectrum associated with variants in TUBG1. Eur. J. Hum. Genet. 2018, 26, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.L.; Gilet, J.G.; Sulimenko, V.; Duchon, A.; Rudolf, G.; Runge, K.; Collins, S.C.; Asselin, L.; Broix, L.; Drouot, N.; et al. TUBG1 missense variants underlying cortical malformations disrupt neuronal locomotion and microtubule dynamics but not neurogenesis. Nat. Commun. 2019, 10, 2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Yuba-Kubo, A.; Kubo, A.; Hata, M.; Tsukita, S. Gene knockout analysis of two gamma-tubulin isoforms in mice. Dev. Biol. 2005, 282, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, N.; Timms, A.E.; Aldinger, K.A.; Mirzaa, G.M.; Bennett, J.T.; Collins, S.; Olds, C.; Mei, D.; Chiari, S.; Carvill, G.; et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet. Med. 2018, 11, 1354–1364. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thulasirajah, S.; Wang, X.; Sell, E.; Dávila, J.; Dyment, D.A.; Kernohan, K.D. A De Novo Missense Variant in TUBG2 in a Child with Global Developmental Delay, Microcephaly, Refractory Epilepsy and Perisylvian Polymicrogyria. Genes 2023, 14, 108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010108
Thulasirajah S, Wang X, Sell E, Dávila J, Dyment DA, Kernohan KD. A De Novo Missense Variant in TUBG2 in a Child with Global Developmental Delay, Microcephaly, Refractory Epilepsy and Perisylvian Polymicrogyria. Genes. 2023; 14(1):108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010108
Chicago/Turabian StyleThulasirajah, Salini, Xueqi Wang, Erick Sell, Jorge Dávila, David A. Dyment, and Kristin D. Kernohan. 2023. "A De Novo Missense Variant in TUBG2 in a Child with Global Developmental Delay, Microcephaly, Refractory Epilepsy and Perisylvian Polymicrogyria" Genes 14, no. 1: 108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010108