
Concomitant Calcium Channelopathies Involving CACNA1A and CACNA1F: A Case Report and Review of the Literature
 , ,
, , 
Abstract
:1. Introduction
1.1. Voltage-Gated Calcium Channels
1.2. SHM1 a CACNA1A-Related Phenotype
1.3. CACNA1F-Related Phenotype
1.4. Case Report
1.5. Objectives
2. Methods
2.1. Ethical Protocols
2.2. Systematic Review Procedure
2.3. Search Strategy
2.4. Criteria for Inclusion
2.5. Data Collection
3. Case Report
3.1. Clinical Findings
3.1.1. Developmental History
3.1.2. Neurological History
3.1.3. Medical History
3.2. Diagnostic Assessment
3.2.1. Genetic and Metabolic Testing
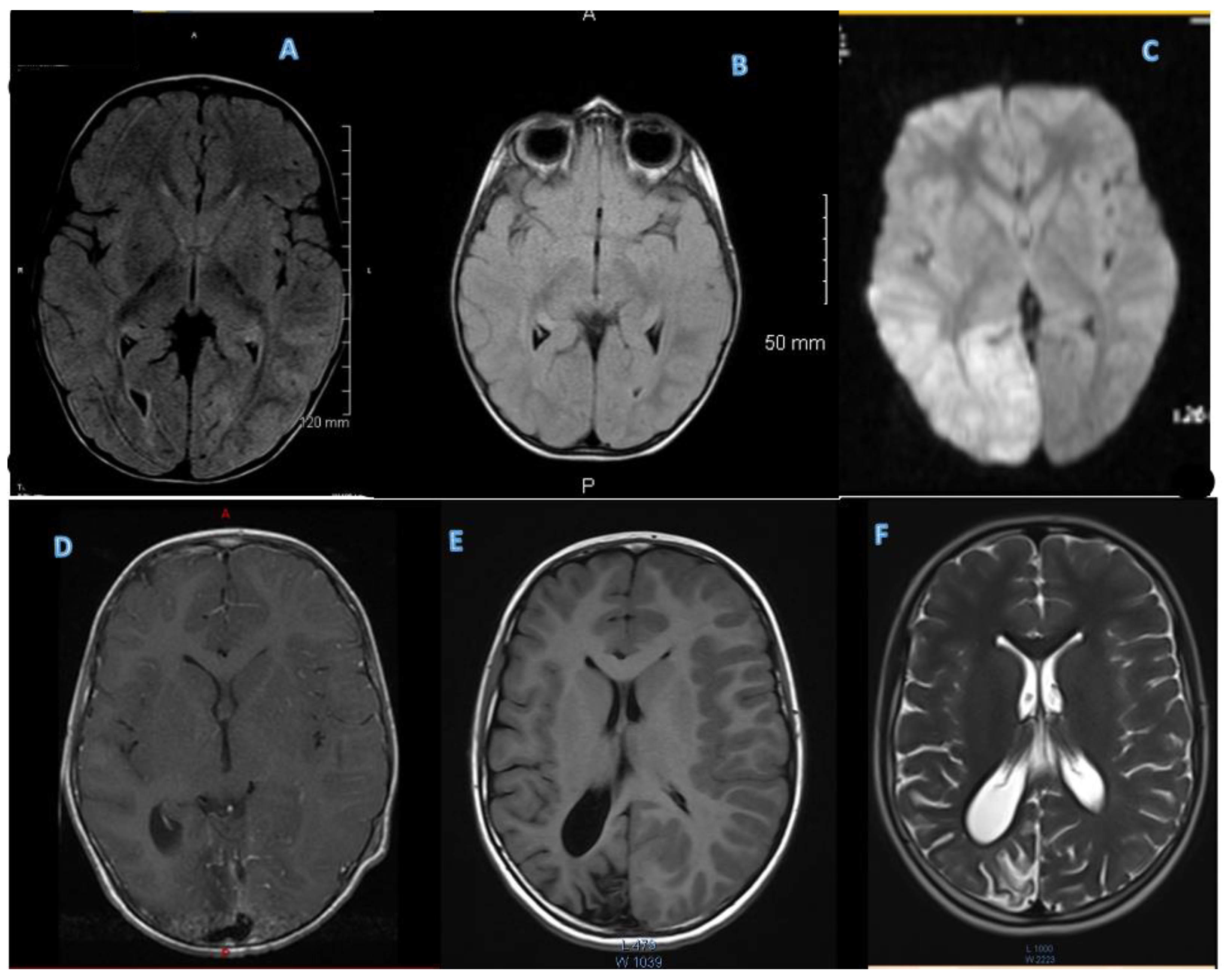
3.2.2. MRI
3.2.3. EEGs
3.2.4. Labs
3.2.5. Cardiac Evaluation
3.2.6. Ocular Assessment
3.2.7. Immune Analysis
3.3. Therapeutic Interventions and Follow-Up
4. A Review of the Literature
4.1. SHM1 a CACNA1A-Related Phenotype
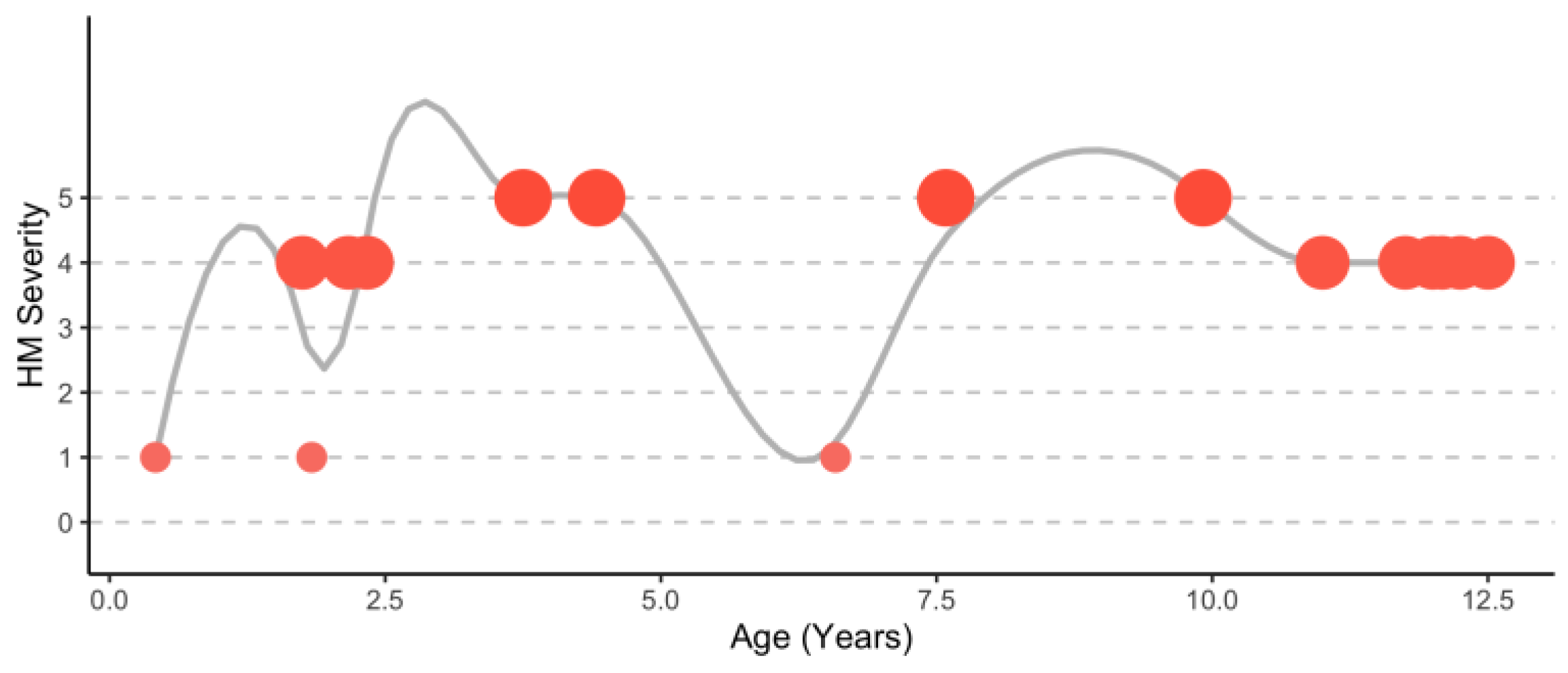
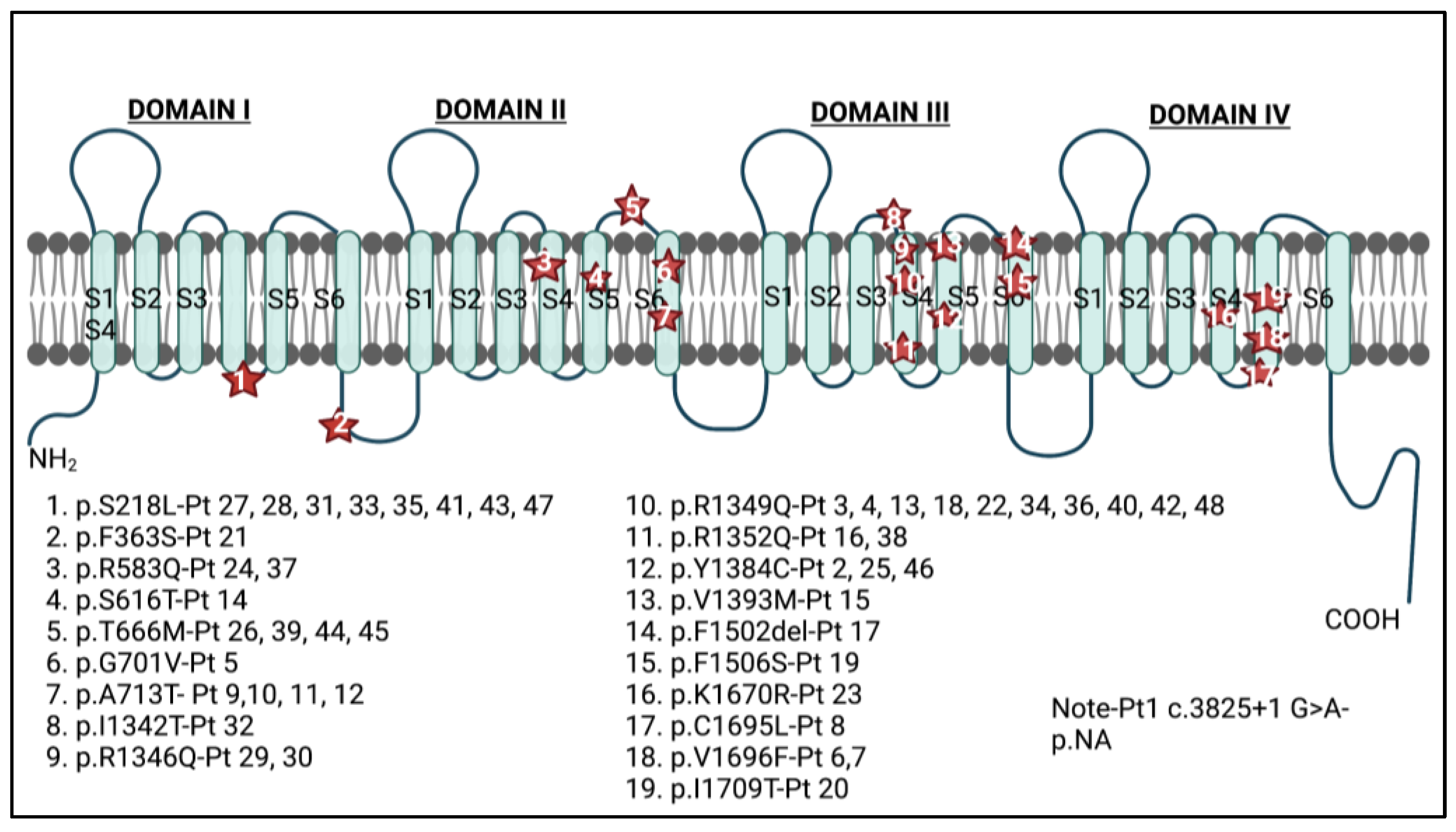
4.1.1. Demographic Information, Variant Location, and Variant Substitution
4.1.2. Hemiplegic Migraine Presentation during Attacks
4.1.3. Common Symptoms Outside of Attacks
4.1.4. Ataxia, Extra Pyramidal Movement Disorders, and Spasticity
4.1.5. Global Developmental Delay and Intellectual Disability
4.1.6. Brain Atrophy and Abnormal EEGs
4.1.7. Paroxysmal Events and Abnormal Eye Movements
4.1.8. Seizures
4.1.9. Other Findings Outside of Hemiplegic Attacks
4.2. A Scoping Review of CACNA1F-Related Phenotypes
4.2.1. Demographic Information, Variant Type, and Variant Location
4.2.2. Retinal Dystrophy Associated with CACNA1F
4.2.3. Other Ocular Manifestations
4.2.4. Neurocognitive Signs
4.2.5. Immune Symptoms
5. Discussion
5.1. Limitations
5.2. Clinical Implications
5.3. Future Research Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bidaud, I.; Mezghrani, A.; Swayne, L.A.; Monteil, A.; Lory, P. Voltage-gated calcium channels in genetic diseases. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Heyes, S.; Pratt, W.S.; Rees, E.; Dahimene, S.; Ferron, L.; Owen, M.; Dolphin, A. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 2015, 134, 36–54. [Google Scholar] [CrossRef]
- Carreno, O.; Corominas, R.; Serra, S.A.; Sintas, C.; Fernandez-Castllo, N.; Villa-Pueyo, M.; Toma, C.; Gene, G.; Pons, R.; Llaneza, M.; et al. Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: Clinical, genetic, and functional studies. Mol. Genet. Genom. Med. 2013, 1, 206–222. [Google Scholar] [CrossRef] [PubMed]
- García Segarra, N.; Gautschi, I.; Mittaz-Crettol, L.; Kallay Zetchi, C.; Al Qusairi, L.; Van Bemmeien, M.; Maeder, P.; Bonafe, L.; Schlid, L.; Roulet-Perez, E. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J. Neurol. Sci. 2014, 342, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wen, Y.; Zhang, Q.; Chen, Y.; Wang, J.; Shi, K.; Du, L.; Bao, X. CACNA1A Gene Variants in Eight Chinese Patients with a Wide Range of Phenotypes. Front. Pediatr. 2020, 8, 577544. [Google Scholar] [CrossRef] [PubMed]
- Blumkin, L.; Michelson, M.; Leshinsky-Silver, E.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J. Child Neurol. 2010, 25, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Curtain, R.P.; Smith, R.L.; Ovcaric, M.; Griffiths, L.R. Minor head trauma-induced sporadic hemiplegic migraine coma. Pediatr. Neurol. 2006, 34, 329–332. [Google Scholar] [CrossRef]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef]
- Gandini, M.A.; Souza, I.A.; Ferron, L.; Innes, A.M.; Zamponi, G.W. The de novo CACNA1A pathogenic variant Y1384C associated with hemiplegic migraine, early onset cerebellar atrophy and developmental delay leads to a loss of Cav2.1 channel function. Mol. Brain 2021, 14, 27. [Google Scholar] [CrossRef]
- Travaglini, L.; Nardella, M.; Bellacchio, E.; D’Amico, A.; Capuano, A.; Frusciante, R.; Di Capua, M.; Cusmai, R.; Barresi, S.; Morlino, S.; et al. Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur. J. Paediatr. Neurol. 2017, 21, 450–456. [Google Scholar] [CrossRef]
- Prontera, P.; Sarchielli, P.; Caproni, S.; Bedetti, C.; Cupini, L.M.; Calabresi, P.; Costa, C. Epilepsy in hemiplegic migraine: Genetic mutations and clinical implications. Cephalalgia 2018, 38, 361–373. [Google Scholar] [CrossRef]
- Stam, A.; Vanmolkot, K.; Kremer, H.; Ginjaar, I.B.; Frants, R.R.; Haan, J.; Ferrari, M.D.; Terwindt, G.M.; van den Maagdenberg, A.M. CACNA1A R1347Q: A frequent recurrent mutation in hemiplegic migraine. Clin. Genet. 2008, 74, 481–485. [Google Scholar] [CrossRef]
- Koschak, A.; Fernandez-Quintero, M.L.; Heigl, T.; Ruzza, M.; Seitter, H.; Zanetti, L. Cav1.4 dysfunction and congenital stationary night blindness type 2. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 1437–1454. [Google Scholar] [CrossRef] [PubMed]
- McRory, J.E.; Hamid, J.; Doering, C.J.; Garcia, E.; Parker, R.; Hamming, K.; Chen, L.; Hildebrand, M.; Beedle, A.; Feldcamp, L.; et al. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Mejecase, C.; Ali, S.M.A.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with angstrom land Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 171. [Google Scholar] [CrossRef]
- Galindo-Bocero, J.; Macías-Franco, S.; García-González, N.; Valles-Antuña, C.; Hernando Acero, I.; Rozas-Reyes, P. Diagnosis of an X-linked type 2 congenital stationary night blindness using electroretinography and CACNA1F sequencing. Arch. Soc. Española Oftalmol. 2020, 95, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef]
- Fenninger, F.; Jefferies, W.A. What’s Bred in the Bone: Calcium Channels in Lymphocytes. J. Immunol. 2019, 202, 1021–1030. [Google Scholar] [CrossRef]
- Badou, A.; Jha, M.K.; Matza, D.; Flavell, R.A. Emerging Roles of L-Type Voltage-Gated and Other Calcium Channels in T Lymphocytes. Front. Immunol. 2013, 4, 243. [Google Scholar] [CrossRef]
- Jha, A.; Singh, A.K.; Weissgerber, P.; Freichel, M.; Flockerzi, V.; Flavell, R.A.; Jha, M.K. Essential roles for Cavβ2 and Cav1 channels in thymocyte development and T cell homeostasis. Sci. Signal. 2015, 8, ra103. [Google Scholar] [CrossRef]
- Omilusik, K.; Priatel, J.J.; Chen, X.; Wang, Y.T.; Xu, H.; Choi, K.B.; Gopaul, R.; McIntyre-Smith, A.; Ten, H.; Tan, R.; et al. The CaV1.4 Calcium Channel Is a Critical Regulator of T Cell Receptor Signaling and Naive T Cell Homeostasis. Immunity 2011, 35, 349–360. [Google Scholar] [CrossRef]
- Kotturi, M.F.; Jefferies, W.A. Molecular characterization of L-type calcium channel splice variants expressed in human T lymphocytes. Mol. Immunol. 2005, 42, 1461–1474. [Google Scholar] [CrossRef]
- Ohmura, K.; Suzuki, Y.; Saito, Y.; Wada, T.; Goto, M.; Seto, S. Sporadic hemiplegic migraine presenting as acute encephalopathy. Brain Dev. 2012, 34, 691–695. [Google Scholar] [CrossRef]
- Sánchez-Albisua, I.; Schöning, M.; Jurkat-Rott, K.; Lerche, H. Possible effect of corticoids on hemiplegic attacks in severe hemiplegic migraine. Pediatr. Neurol. 2013, 49, 286–288. [Google Scholar] [CrossRef]
- Topakian, R.; Pischinger, B.; Stieglbauer, K.; Pichler, R. Rare clinical findings in a patient with sporadic hemiplegic migraine: FDG-PET provides diminished brain metabolism at 10-year follow-up. Cephalalgia 2014, 34, 392–396. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Camia, F.; Pisciotta, L.; Morana, G.; Schiaffino, M.C.; Renna, S.; Carrera, P.; Ferrari, M.; Baglietto, M.G.; Veneselli, E.; Siri, L.; et al. Combined early treatment in hemiplegic attacks related to CACNA1A encephalopathy with brain oedema: Blocking the cascade? Cephalalgia 2017, 37, 1202–1206. [Google Scholar] [CrossRef]
- Riant, F.; Ducros, A.; Ploton, C.; Barbance, C.; Depienne, C.; Tournier-Lasserve, E. De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology 2010, 75, 967–972. [Google Scholar] [CrossRef]
- Stubberud, A.; O’Connor, E.; Tronvik, E.; Houlden, H.; Matharu, M. R1352Q CACNA1A Variant in a Patient with Sporadic Hemiplegic Migraine, Ataxia, Seizures and Cerebral Oedema: A Case Report. Case Rep. Neurol. 2021, 13, 123–130. [Google Scholar] [CrossRef]
- Humbertclaude, V.; Krams, B.; Nogue, E.; Nagot, N.; Annequin, D.; Tourniaire, B.; Tournier-Lasserve, E.; Riant, F.; Roubertie, A.; Episodic Syndrome Consortium. Benign paroxysmal torticollis, benign paroxysmal vertigo, and benign tonic upward gaze are not benign disorders. Dev. Med. Child Neurol. 2018, 60, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Vahedi, K.; Denier, C.; Ducros, A.; Bousson, V.; Levy, C.; Chabriat, H.; Haguenau, M.; Tournier-Lasserve, E.; Bousser, M.G. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology 2000, 55, 1040–1042. [Google Scholar] [CrossRef]
- de Vries, B.; Stam, A.H.; Beker, F.; van den Maagdenberg, A.M.; Vanmolkot, K.R.; Laan, L.; Ginjaar, I.B.; Frantis, R.R.; Lauffer, H.; Haan, J.; et al. CACNA1A mutation linking hemiplegic migraine and alternating hemiplegia of childhood. Cephalalgia 2008, 28, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, M.; Barth, M.; Gueden, S.; de Cepoy, P.D.; Aeby, A.; Vilain, C.; Hirsch, E.; Martin, A.D.S.; Portes, V.D.; Lesca, G.; et al. CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur. J. Paediatr. Neurol. 2021, 33, 75–85. [Google Scholar] [CrossRef]
- Humbertclaude, V.; Riant, F.; Krams, B.; Zimmerman, V.; Nagot, N.; Annequin, D.; Echienne, B.; Tournier-Lasserve, E.; Roubertie, A.; Episodic Syndrome Consortium. Cognitive impairment in children with CACNA1A mutations. Dev. Med. Child Neurol. 2020, 62, 330–337. [Google Scholar] [CrossRef]
- Gray, I.N.; Cristancho, A.G.; Licht, D.J.; Liu, G.T. Ocular Dipping in a Patient With Hemiplegic Migraine. J. Pediatr. Ophthalmol. Strabismus 2018, 55, e4–e6. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofori, A.; Fusi, L.; Gomitoni, A.; Grampa, G.; Bersano, A.; Lombardia GENS Collaborators. R583Q CACNA1A variant in SHM1 and ataxia: Case report and literature update. J. Headache Pain 2012, 13, 419–423. [Google Scholar] [CrossRef]
- Knierim, E.; Leisle, L.; Wagner, C.; Weschke, B.; Lucke, B.; Bohner, G.; Dreier, J.P.; Schuelke, M. Recurrent stroke due to a novel voltage sensor mutation in Cav2.1 responds to verapamil. Stroke 2011, 42, e14–e17. [Google Scholar] [CrossRef]
- Malpas, T.J.; Riant, F.; Tournier-Lasserve, E.; Vahedi, K.; Neville, B.G.R. Sporadic hemiplegic migraine and delayed cerebral oedema after minor head trauma: A novel de novo CACNA1A gene mutation. Dev. Med. Child Neurol. 2010, 52, 103–104. [Google Scholar] [CrossRef]
- Zangaladze, A.; Asadi-Pooya, A.A.; Ashkenazi, A.; Sperling, M.R. Sporadic hemiplegic migraine and epilepsy associated with CACNA1A gene mutation. Epilepsy Behav. 2010, 17, 293–295. [Google Scholar] [CrossRef]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillion, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.; Tournier-Lasserve, E. The Clinical Spectrum of Familial Hemiplegic Migraine Associated with Mutations in a Neuronal Calcium Channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef]
- Debiais, S.; Hommet, C.; Bonnaud, I.; Barthez, M.A.; Rombaux, S.; Riant, F.; Autret, A. The FHM1 mutation S218L: A severe clinical phenotype? A case report and review of the literature. Cephalalgia 2009, 29, 1337–1339. [Google Scholar] [CrossRef]
- Boat, T.F.; Wu, J.T.; National Academies of Sciences, Engineering, and Medicine. Clinical Characteristics of Intellectual Disabilities. In Mental Disorders and Disabilities among Low-Income Children; National Academies Press: Washington, DC, USA, 2015. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK332877/ (accessed on 8 December 2022).
- Men, C.J.; Bujakowska, K.M.; Comander, J.; Place, E.; Bedoukian, E.C.; Zhu, X.; Leroy, B.P.; Fulton, A.B.; Pierce, E.A. The importance of genetic testing as demonstrated by two cases of CACNA1F-associated retinal generation misdiagnosed as LCA. Mol. Vis. 2017, 23, 695–706. [Google Scholar]
- Birtel, J.; Gliem, M.; Hess, K.; Birtel, T.; Holz, F.G.; Zechner, U.; Bolz, H.J.; Herrmann, P. Comprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies. Genes 2020, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Maconachie, G.D.E.; Sheth, V.; McLean, R.J.; Gottlob, I. Development and clinical utility of a novel diagnostic nystagmus gene panel using targeted next-generation sequencing. Eur. J. Hum. Genet. 2017, 25, 725–734. [Google Scholar] [CrossRef]
- Kim, H.M.; Joo, K.; Han, J.; Woo, S.J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes 2021, 12, 789. [Google Scholar] [CrossRef]
- Jacobi, F.K.; Hamel, C.P.; Arnaud, B.; Blin, N.; Broghammer, M.; Jacobi, P.C.; Apfelstedt-Sylla, E.; Pusch, C.M. A novel CACNA1F mutation in a french family with the incomplete type of X-linked congenital stationary night blindness. Am. J. Ophthalmol. 2003, 135, 733–736. [Google Scholar] [CrossRef]
- Zito, I.; Allen, L.E.; Patel, R.J.; Meindl, A.; Bradshaw, K.; Yates, J.R.; Bird, A.C.; Erskine, L.; Cheetham, M.E.; Webster, A.R.; et al. Mutations in the CACNA1F and NYX genes in British CSNBX families. Hum. Mutat. 2003, 21, 169. [Google Scholar] [CrossRef]
- Vincent, A.; Wright, T.; Day, M.A.; Westall, C.A.; Héon, E. A novel p.Gly603Arg mutation in CACNA1F causes Åland island eye disease and incomplete congenital stationary night blindness phenotypes in a family. Mol. Vis. 2011, 17, 3262–3270. [Google Scholar]
- Allen, L.E. Genotype-phenotype correlation in British families with X linked congenital stationary night blindness. Br. J. Ophthalmol. 2003, 87, 1413–1420. [Google Scholar] [CrossRef]
- Kimchi, A.; Meiner, V.; Silverstein, S.; Macarov, M.; Mor-Shaked, H.; Blumenfeld, A.; Audo, I.; Zeitz, C.; Mechoulam, H.; Banin, E.; et al. An Ashkenazi Jewish founder mutation in CACNA1F causes retinal phenotype in both hemizygous males and heterozygous female carriers. Ophthalmic Genet. 2019, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, S.J.; Thomas, M.G.; Jung, J.H.; Oh, E.H.; Shin, J.H.; Cho, J.W.; Kim, H.S.; Park, J.Y.; Choi, S.Y.; et al. Diagnostic yield of targeted next-generation sequencing in infantile nystagmus syndrome. Ophthalmic Genet. 2021, 42, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Pasutto, F.; Ekici, A.; Reis, A.; Kremers, J.; Huchzermeyer, C. Novel truncating mutation in CACNA1F in a young male patient diagnosed with optic atrophy. Ophthalmic Genet. 2018, 39, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guan, L.; Xiao, X.; Zhang, J.; Li, S.; Jiang, H.; Jia, X.; Yang, J.; Guo, X.; Yin, Y.; et al. Mutation analysis in 129 genes associated with other forms of retinal dystrophy in 157 families with retinitis pigmentosa based on exome sequencing. Mol. Vis. 2015, 21, 477–486. [Google Scholar] [PubMed]
- Kessi, M.; Chen, B.; Peng, J.; Yan, F.; Yang, L.; Yin, F. Calcium channelopathies and intellectual disability: A systematic review. Orphanet J. Rare Dis. 2021, 16, 219. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Channel Type | Gene Name | Protein | Chromosome | Associated Condition-OMIM# | Primary Tissue Expression |
|---|---|---|---|---|---|
| L-type | CACNA1S | Cav1.1 | 1q31-32 | HOKPP1 1-170400; MSH5 2-601887; TTPP1 3-188580 | Muscle Skeletal |
| CACNA1C | Cav1.2 | 12p13.3 | TS 4-601005; NEDHLSS 5-620029; LQT8 6-618447; BRGDA3 7-611875 | GI, GU, Heart, Brain | |
| CACNA1D | Cav1.3 | 3p14.3 | SANDD 8-614896; PASNA 9-615474 | Lung, Adrenal, Pituitary | |
| CACNA1F | Cav1.4 | Xp11.23 | CORDX3 10-300476; CSNB2A 11-300071; AIED 12-300600 | Retina, GI, Lymph, Spleen | |
| P/Q type | CACNA1A | Cav2.1 | 19p13.1 | EA2 13-108500; SCA6 14-183086; FHM1 15-141500; DEE42 16-617106 | Brain-Cerebellum, Stomach |
| N-type | CACNA1B | Cav2.2 | 9p34 | KLEFS1 17-610253; NEDNAH 18-618497; DYT23 19-614860 | Brain-Cerebellum, Cortex |
| R-type | CACNA1E | Cav2.3 | 1q25031 | DEE69 20-618285 | Brain (throughout) |
| T-type | CACNA1G | Cav3.1 | 17q22 | SCA42 21-616795; SCA42ND 22-618087 | Brain, Endometrium |
| CACNA1H | Cav3.2 | 16p13.3 | HALD4 23-617027; ECA6 24-611942 [Disputed gene-disease relationship] | GU, GI | |
| CACNA1I | Cav3.3 | 22q13 | NEDSIS 25-620114 | Brain, Thyroid |
| Possible triggers | High alt. | X | |||||||||||||||
| Airborne allergen | X | X | X | X | X | X | X | X | X | ||||||||
| Head trauma | X | ||||||||||||||||
| Vitamins | X | ||||||||||||||||
| Food-diet | X | X | X | X | X | ||||||||||||
| Symptoms | Abn. MRI | X | X | X | X | X | |||||||||||
| Abn. EEG | X | X | X | X | X | ||||||||||||
| Abn. Labs | X | X | X | X | X | X | X | ||||||||||
| Transient blindness | X | X | X | X | X | X | |||||||||||
| Encephalopathy | X | X | X | X | X | X | X | X | X | ||||||||
| Hemiplegia | X | X | X | X | X | X | X | X | X | X | |||||||
| Fever | X | X | X | X | X | X | X | X | X | X | X | ||||||
| Status Epilepticus | X | X | X | ||||||||||||||
| Seizure | X | X | X | X | X | ||||||||||||
| Tachycardia | X | X | X | X | X | X | X | X | |||||||||
| Apnea | X | X | X | ||||||||||||||
| Dyskinesia | X | X | X | X | X | ||||||||||||
| Coma | X | X | X | X | X | X | |||||||||||
| Lethargy | X | X | X | X | X | X | X | X | X | X | X | ||||||
| Vomiting | X | X | X | X | X | X | X | X | X | X | X | ||||||
| Duration | <20 s | 5 days | <24 h | 4 days | 5 days | 5 days | 11 days | <6 h | 16 days | 6 days | 4 days | 3 days | 5 days | 5 days | 3 days | 2 days | |
| Age | 5 M | 1 Y 9 M | 1 Y 10 M | 2 Y 2 M | 2 Y 4 M | 3 Y 9 M | 4 Y 5 M | 6 Y 7 M | 7 Y 7 M | 9 Y 11 M | 11 Y | 11 Y 9 M | 12 Y | 12 Y 1 M | 12 Y 3 M | 12 Y 6 M | |
| Episode | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| Symptoms During Hemiplegic Attacks | N (%) |
|---|---|
| Migraine with aura | 33/33 (100%) |
| Hemiplegia | 33/33 (100%) |
| Coma/altered consciousness | 25/33 (75.8%) |
| Seizure (including status epilepticus) | 25/33 (75.8%) |
| Cerebral edema | 15/33 (45.5%) |
| EEG slowing or spike waves | 12/33 (36.4%) |
| Eye abnormalities (including transient blindness) | 11/33 (33.3%) |
| Encephalopathy | 11/33 (33.3%) |
| Dystonic storms | 4/33 (12.1%) |
| Lethargy | 3/33 (9.1%) |
| Fever (noninfectious) | 17/33 (51.5%) |
| Vomiting/nausea | 13/33 (39.4%) |
| Apnea | 4/33 (12.1%) |
| Other symptoms | |
| Speech difficulties | 7/33 (21.2%) |
| Photophobia | 4/33 (12.1%) |
| Phonophobia | 2/33 (6.1%) |
| Abnormal CSF (including high IL-6) | 2/33 (6.1%) |
| Symptoms Outside of Hemiplegic Attacks | No. (%) |
|---|---|
| Movement disturbance | 37/46 (80.4%) |
| Ataxia | 35/46 (76.1%) |
| Dystonia | 29/46 (63.0%) |
| Hypotonia | 24/46 (52.2%) |
| Myoclonus | 6/46 (13.0%) |
| Pyramidal signs | 6/46 (13.0%) |
| Tremor | 11/46 (23.9%) |
| Dyskinesia | 4/46 (8.7%) |
| Global developmental delay | 29/46 (63.0%) |
| Speech disturbance | 13/46 (28.3%) |
| Intellectual disability | 30/46 (65.2%) |
| Severe-IQ (20–35) | 7/30 (23.4%) |
| Moderate-IQ (36–49) | 4/30 (13.3%) |
| Mild-IQ (50–69) | 9/30 (30.0%) |
| Undefined | 10/30 (33.3%) |
| Brain atrophy | 28/37 (75.7%) |
| Progressive | 9/28 (32.1%) |
| Paroxysmal actions | 9/46 (19.6%) |
| Abnormal eye movements | 22/46 (47.8%) |
| Seizures | 11/46 (23.9%) |
| Clinical Features | Case Study | SHMI (CACNA1A) |
|---|---|---|
| Symptoms During Hemiplegic Attacks | ||
| Migraine with aura | + | + * |
| Hemiplegia | + | + * |
| Neurologic manifestations | ||
| Coma/altered consciousness | + | + * |
| Seizure (including status epilepticus) | + | + |
| Cerebral edema | + | + * |
| EEG slowing or spike waves | + | + |
| Eye abnormalities | + | + |
| Encephalopathy | + | + |
| Dystonic storms | + | + |
| Lethargy | + | + * |
| Autonomic manifestations | ||
| Fever (noninfectious) | + | + * |
| Vomiting/nausea | + | + |
| Apnea | + | + |
| Other Symptoms | ||
| Speech difficulties | + | + * |
| Photophobia | + | + |
| Phonophobia | + | |
| Abnormal CSF | + | + |
| Symptoms Outside of Hemiplegic Attacks | ||
| Movement disorders | ||
| Ataxia | + | + |
| Dystonia | + | + |
| Pyramidal signs | + | + |
| Tremor | + | + * |
| Dyskinesia | + | + |
| Global developmental delay | + | + |
| Verbal conditions | + | + |
| Intellectual disability | + | + |
| Brain atrophy | + | + * |
| Paroxysmal actions | + | + |
| Abnormal eye movements | + | + * |
| Seizures | + * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaare, D.; Sarasua, S.M.; Lusk, L.; Parthasarathy, S.; Wang, L.; Helbig, I.; Boccuto, L. Concomitant Calcium Channelopathies Involving CACNA1A and CACNA1F: A Case Report and Review of the Literature. Genes 2023, 14, 400. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020400
Schaare D, Sarasua SM, Lusk L, Parthasarathy S, Wang L, Helbig I, Boccuto L. Concomitant Calcium Channelopathies Involving CACNA1A and CACNA1F: A Case Report and Review of the Literature. Genes. 2023; 14(2):400. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020400
Chicago/Turabian StyleSchaare, Donna, Sara M. Sarasua, Laina Lusk, Shridhar Parthasarathy, Liangjiang Wang, Ingo Helbig, and Luigi Boccuto. 2023. "Concomitant Calcium Channelopathies Involving CACNA1A and CACNA1F: A Case Report and Review of the Literature" Genes 14, no. 2: 400. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020400