In Silico Analysis of Hepatitis B Virus Occult Associated Mutations in Botswana Using a Novel Algorithm

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population
2.2. Determination of OBI-Associated Mutations
2.3. Ethical Considerations
2.4. In Silico Methods
2.5. Data Analysis
3. Results
OBI-Associated Mutations Phenotypic Results
Phyre2 Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Torbenson, M.; Thomas, D.L. Occult hepatitis B. Lancet Infect. Dis. 2002, 2, 479–486. [Google Scholar] [CrossRef]
- Raimondo, G.; Allain, J.P.; Brunetto, M.R.; Buendia, M.A.; Chen, D.S.; Colombo, M.; Craxi, A.; Donato, F.; Ferrari, C.; Gaeta, G.B.; et al. Statements from the taormina expert meeting on occult hepatitis B virus infection. J. Hepatol. 2008, 49, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Joller-Jemelka, H.I.; Grob, P.J.; Luthy, R.; Opravil, M. Frequent chronic hepatitis B virus infection in HIV-infected patients positive for antibody to hepatitis B core antigen only. Swiss HIV cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 1998, 17, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.; Rios, P.; Perez-Olmeda, M.; Soriano, V. Lack of ‘occult’ hepatitis B virus infection in HIV-infected patients. AIDS 2002, 16, 2099–2101. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Lee, C.K.; Wong, D.K.; Fung, J.; Hung, I.; Hsu, A.; But, D.Y.; Cheung, T.K.; Chan, P.; Yuen, J.C.; et al. Prevalence of occult hepatitis B infection in a highly endemic area for chronic hepatitis B: A study of a large blood donor population. Gut 2010, 59, 1389–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minuk, G.Y.; Sun, D.F.; Uhanova, J.; Zhang, M.; Caouette, S.; Nicolle, L.E.; Gutkin, A.; Doucette, K.; Martin, B.; Giulivi, A. Occult hepatitis B virus infection in a north american community-based population. J. Hepatol. 2005, 42, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-Melendez, G.; Panduro, A.; Fierro, N.A.; Roman, S. High prevalence of occult hepatitis B virus genotype h infection among children with clinical hepatitis in west Mexico. Memorias do Instituto Oswaldo Cruz 2014, 109, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.I.; Jensen, D.; Sarmiento, V.; Peirano, F.; Acuna, P.; Fuster, F.; Soto, S.; Ahumada, R.; Huilcaman, M.; Bruna, M.; et al. Presence of anti-HBc is associated to high rates of HBV resolved infection and low threshold for occult HBV infection in HIV patients with negative HBsAg in chile. J. Med. Virol. 2016, 88, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.G.; Makondo, E.; Martinson, N.A.; Kramvis, A. Hepatitis B virus infection in human immunodeficiency virus infected southern African adults: Occult or overt--that is the question. PLoS ONE 2012, 7, e45750. [Google Scholar] [CrossRef] [PubMed]
- Wester, C.W.; Bussmann, H.; Moyo, S.; Avalos, A.; Gaolathe, T.; Ndwapi, N.; Essex, M.; MacGregor, R.R.; Marlink, R.G. Serological evidence of HIV-associated infection among HIV-1-infected adults in Botswana. Clin. Infect. Dis. 2006, 43, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Davis, S.; Tolle, M.; Mabikwa, V.; Anabwani, G. Prevalence of hepatitis B and hepatitis C coinfections in an adult HIV centre population in Gaborone, Botswana. Am. J. Trop. Med. Hyg. 2011, 85, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.C.; Beloukas, A.; Malik, A.; Carlson, J.M.; Jooste, P.; Ogwu, A.; Shapiro, R.; Riddell, L.; Chen, F.; Luzzi, G.; et al. Prevalence and characteristics of hepatitis B virus (HBV) coinfection among HIV-positive women in south Africa and Botswana. PLoS ONE 2015, 10, e0134037. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Gaseitsiwe, S.; Moyo, S.; Thami, K.P.; Mohammed, T.; Setlhare, D.; Sebunya, T.K.; Powell, E.A.; Makhema, J.; Blackard, J.T.; et al. Slow CD4+ T-cell recovery in human immunodeficiency virus/hepatitis B virus-coinfected patients initiating truvada-based combination antiretroviral therapy in Botswana. Open Forum Infect. Dis. 2016, 3, ofw140. [Google Scholar] [CrossRef] [PubMed]
- Mbangiwa, T.; Kasvosve, I.; Anderson, M.; Thami, P.K.; Choga, W.T.; Needleman, A.; Phinius, B.B.; Moyo, S.; Leteane, M.; Leidner, J.; et al. Chronic and occult hepatitis B virus infection in pregnant women in Botswana. Genes 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Anderson, M.; Gyurova, I.; Ambroggio, L.; Moyo, S.; Sebunya, T.; Makhema, J.; Marlink, R.; Essex, M.; Musonda, R.; et al. High rates of occult hepatitis B virus infection in HIV-positive individuals initiating antiretroviral therapy in Botswana. Open Forum Infect. Dis. 2017, 4, ofx195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.L.; Li, X.; Li, J.; Zhang, Z.H. Genetic variation of occult hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 3531–3546. [Google Scholar] [CrossRef] [PubMed]
- Jilg, W.; Sieger, E.; Zachoval, R.; Schatzl, H. Individuals with antibodies against hepatitis B core antigen as the only serological marker for hepatitis B infection: High percentage of carriers of hepatitis B and C virus. J. Hepatol. 1995, 23, 14–20. [Google Scholar] [CrossRef]
- Campe, H.; Hillebrand, G.F.; Mairhofer, H.; Nitschko, H.; Jager, G. Undetected chronic hepatitis B virus infection of a vaccinated dialysis patient after liver transplantation. Nephrol. Dial. Transplant. 2005, 20, 1492–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, M.F.; Wong, D.K.; Lee, C.K.; Tanaka, Y.; Allain, J.P.; Fung, J.; Leung, J.; Lin, C.K.; Sugiyama, M.; Sugauchi, F.; et al. Transmissibility of hepatitis B virus (HBV) infection through blood transfusion from blood donors with occult HBV infection. Clin. Infect. Dis. 2011, 52, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Thiers, V.; Nakajima, E.; Kremsdorf, D.; Mack, D.; Schellekens, H.; Driss, F.; Goudeau, A.; Wands, J.; Sninsky, J.; Tiollais, P.; et al. Transmission of hepatitis B from hepatitis-B-seronegative subjects. Lancet 1988, 2, 1273–1276. [Google Scholar] [CrossRef]
- Ikeda, K.; Kobayashi, M.; Someya, T.; Saitoh, S.; Hosaka, T.; Akuta, N.; Suzuki, F.; Suzuki, Y.; Arase, Y.; Kumada, H. Occult hepatitis B virus infection increases hepatocellular carcinogenesis by eight times in patients with non-B, non-C liver cirrhosis: A cohort study. J. Viral Hepat. 2009, 16, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Kannangai, R.; Molmenti, E.; Arrazola, L.; Klein, A.; Choti, M.; Thomas, D.L.; Torbenson, M. Occult hepatitis B viral DNA in liver carcinomas from a region with a low prevalence of chronic hepatitis B infection. J. Viral Hepat. 2004, 11, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Nirei, K.; Tamura, A.; Nakamura, H.; Matsumura, H.; Oshiro, S.; Arakawa, Y.; Yamagami, H.; Tanaka, N.; Moriyama, M. Influence of occult hepatitis B virus coinfection on the incidence of fibrosis and hepatocellular carcinoma in chronic hepatitis C. Intervirology 2008, 51, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, G.; Pollicino, T.; Cacciola, I.; Caccamo, G.; Villari, D.; La Masa, T.; Restuccia, T.; Cucinotta, E.; Scisca, C.; Magazzu, D.; et al. Occult hepatitis B virus infection is associated with the development of hepatocellular carcinoma in chronic hepatitis C patients. Cancer 2006, 106, 1326–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollicino, T.; Squadrito, G.; Cerenzia, G.; Cacciola, I.; Raffa, G.; Craxi, A.; Farinati, F.; Missale, G.; Smedile, A.; Tiribelli, C.; et al. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004, 126, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Tamori, A.; Nishiguchi, S.; Kubo, S.; Enomoto, M.; Koh, N.; Takeda, T.; Shiomi, S.; Hirohashi, K.; Kinoshita, H.; Otani, S. Sequencing of human-viral DNA junctions in hepatocellular carcinoma from patients with HCV and occult HBV infection. J. Med. Virol. 2003, 69, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepat. Res. 2016, 2, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Thimme, R.; Blum, H.E. HBV life cycle and novel drug targets. Hepatol. Int. 2011, 5, 644–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.; Kramvis, A. Genotype d of hepatitis b virus and its subgenotypes: An update. Hepatol. Res. 2013, 43, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.O.; Alvarado-Mora, M.V.; Botelho, L.; Vieira, D.S.; Pinho, J.R.; Carrilho, F.J.; Honda, E.R.; Salcedo, J.M. Characterization of hepatitis B virus (HBV) genotypes in patients from Rondonia, Brazil. Virol. J. 2010, 7, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Yuan, Q.; Ge, S.X.; Wang, H.Y.; Zhang, Y.L.; Chen, Q.R.; Zhang, J.; Chen, P.J.; Xia, N.S. Molecular and phylogenetic analyses suggest an additional hepatitis B virus genotype “i”. PLoS ONE 2010, 5, e9297. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, Z.; Ling, C.; Zheng, W.; Zhu, C.; Carr, M.J.; Higgins, D.G. Hepatitis B virus subgenotyping: History, effects of recombination, misclassifications, and corrections. Infect. Genet. Evol. 2013, 16, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef] [PubMed]
- Ochwoto, M.; Kimotho, J.H.; Oyugi, J.; Okoth, F.; Kioko, H.; Mining, S.; Budambula, N.L.; Giles, E.; Andonov, A.; Songok, E.; et al. Hepatitis B infection is highly prevalent among patients presenting with jaundice in kenya. BMC Infect. Dis. 2016, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Gaseitsiwe, S.; Moyo, S.; Wessels, M.J.; Mohammed, T.; Sebunya, T.K.; Powell, E.A.; Makhema, J.; Blackard, J.T.; Marlink, R.; et al. Molecular characterisation of hepatitis B virus in HIV-1 subtype C infected patients in Botswana. BMC Infect. Dis. 2015, 15, 335. [Google Scholar] [CrossRef] [PubMed]
- Said, Z.N. An overview of occult hepatitis B virus infection. World J. Gastroenterol. 2011, 17, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Mallet, V.; Vallet-Pichard, A.; Pol, S. The impact of human immunodeficiency virus on viral hepatitis. Liver Intern. 2011, 31, 135–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mphahlele, M.J.; Lukhwareni, A.; Burnett, R.J.; Moropeng, L.M.; Ngobeni, J.M. High risk of occult hepatitis B virus infection in HIV-positive patients from south africa. J. Clin. Virol. 2006, 35, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Arababadi, M.K.; Pourfathollah, A.A.; Jafarzadeh, A.; Hassanshahi, G.; Rezvani, M.E. Association of exon 9 but not intron 8 VDR polymorphisms with occult HBV infection in south-eastern Iranian patients. J. Gastroenterol. Hepatol. 2010, 25, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.A.; Gededzha, M.P.; Rentz, M.; Rakgole, N.J.; Selabe, S.G.; Seleise, T.A.; Mphahlele, M.J.; Blackard, J.T. Mutations associated with occult hepatitis B in HIV-positive south Africans. J. Med. Virol. 2015, 87, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Welge, J.A.; Shire, N.J.; Rouster, S.D.; Shata, M.T.; Sherman, K.E.; Blackard, J.T. Genomic variability associated with the presence of occult hepatitis B virus in HIV co-infected individuals. J. Viral Hepat. 2010, 17, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Welge, J.A.; Rouster, S.D.; Shata, M.T.; Sherman, K.E.; Blackard, J.T. Mutations associated with occult hepatitis B virus infection result in decreased surface antigen expression in vitro. J. Viral Hepat. 2012, 19, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, M.; Hannoun, C.; Kalinina, T.; Sommer, G.; Manegold, C.; Gunther, S. Functional analysis of hepatitis B virus reactivating in hepatitis B surface antigen-negative individuals. Hepatology 2005, 42, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Wang, Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J. Med. Virol. 2012, 84, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Jammeh, S.; Thomas, H.C.; Karayiannis, P. Replicative competence of the T131I, K141E, and G145R surface variants of hepatitis B virus. J. Infect. Dis. 2007, 196, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Yeh, C.T. Emergence of hepatitis B virus s gene mutants in patients experiencing hepatitis B surface antigen seroconversion after peginterferon therapy. Hepatology 2011, 54, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Yuan, Q.; Chen, P.J.; Zhang, Y.L.; Chen, C.R.; Zheng, Q.B.; Yeh, S.H.; Yu, H.; Xue, Y.; Chen, Y.X.; et al. Influence of mutations in hepatitis B virus surface protein on viral antigenicity and phenotype in occult HBV strains from blood donors. J. Hepatol. 2012, 57, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, T.; Iwanski, A.; Will, H.; Sterneck, M. Deficiency in virion secretion and decreased stability of the hepatitis B virus immune escape mutant G145R. Hepatology 2003, 38, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, V.; Tayal, R.; Nayak, B.; Acharya, S.K.; Panda, S.K. Occult hepatitis B virus infection in chronic liver disease: Full-length genome and analysis of mutant surface promoter. Gastroenterology 2004, 127, 1356–1371. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Kannangai, R.; Ray, S.C.; Thomas, D.L.; Torbenson, M. Comprehensive genetic and epigenetic analysis of occult hepatitis B from liver tissue samples. Clin. Infect. Dis. 2008, 46, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Teng, X.; Xu, W.Z.; Li, D.; Zhao, H.W.; Fu, L.J.; Zhang, F.M.; Gu, H.X. Molecular characterization and functional analysis of occult hepatitis B virus infection in Chinese patients infected with genotype c. J. Med. Virol. 2009, 81, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Gong, J.R.; Lee, S.A.; Kim, B.J. Discovery of a novel mutation (x8del) resulting in an 8-bp deletion in the hepatitis B virus x gene associated with occult infection in Korean vaccinated individuals. PLoS ONE 2015, 10, e0139551. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.A.; Kim, B.J. X region mutations of hepatitis b virus related to clinical severity. World J. Gastroenterol. 2016, 22, 5467–5478. [Google Scholar] [CrossRef] [PubMed]
- Warner, N.; Locarnini, S. The antiviral drug selected hepatitis B virus rta181t/sw172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology 2008, 48, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Motta, J.S.; Mello, F.C.; Lago, B.V.; Perez, R.M.; Gomes, S.A.; Figueiredo, F.F. Occult hepatitis B virus infection and lamivudine-resistant mutations in isolates from renal patients undergoing hemodialysis. J. Gastroenterol. Hepatol. 2010, 25, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The psipred protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-tasser server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The swiss-model repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Svicher, V.; Cento, V.; Bernassola, M.; Neumann-Fraune, M.; Van Hemert, F.; Chen, M.; Salpini, R.; Liu, C.; Longo, R.; Visca, M.; et al. Novel Hbsag markers tightly correlate with occult HBV infection and strongly affect hbsag detection. Antivir. Res. 2012, 93, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, F.; Tsubota, A.; Hirokawa, T.; Kumada, H.; Yang, Z.; Tanaka, H. A unique amino acid substitution, t126i, in human genotype C of hepatitis B virus s gene and its possible influence on antigenic structural change. Gene 2006, 383, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.Y.; Wong, D.K.; Seto, W.K.; Zhang, A.Y.; Lee, C.K.; Lin, C.K.; Fung, J.; Lai, C.L.; Yuen, M.F. Sequence variations of full-length hepatitis B virus genomes in Chinese patients with HBsAg-negative hepatitis B infection. PLoS ONE 2014, 9, e99028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, E.A.; Boyce, C.L.; Gededzha, M.P.; Selabe, S.G.; Mphahlele, M.J.; Blackard, J.T. Functional analysis of ‘a’ determinant mutations associated with occult HBV in HIV-positive south africans. J. Gen. Virol. 2016, 97, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Williams, P.L.; Mayondi, G.K.; Leidner, J.; Holding, P.; Tepper, V.; Nichols, S.; Magetse, J.; Sakoi, M.; Moabi, K.; et al. Neurodevelopment of HIV-exposed and HIV-unexposed uninfected children at 24 months. Pediatrics 2017, 140, e20170988. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Choga, W.T.; Moyo, S.; Bell, T.G.; Mbangiwa, T.; Phinius, B.B.; Bhebhe, L.; Sebunya, T.K.; Lockman, S.; Marlink, R.; et al. Molecular characterization of near full-length genomes of hepatitis B virus isolated from HIV infected individuals in Botswana. Genes 2018, in press. [Google Scholar]
- Drummond, A.J.; Rambaut, A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The clustal_x windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.G.; Yousif, M.; Kramvis, A. Bioinformatic curation and alignment of genotyped hepatitis B virus (HBV) sequence data from the Genbank public database. SpringerPlus 2016, 5, 1896. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.G.; Kramvis, A. Bioinformatics tools for small genomes, such as hepatitis B virus. Viruses 2015, 7, 781–797. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. The clinical implications of hepatitis B virus genotypes and HBeAg in pediatrics. Rev. Med. Virol. 2016, 26, 285–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkady, A.; Iijima, S.; Aboulfotuh, S.; Mostafa Ali, E.; Sayed, D.; Abdel-Aziz, N.M.; Ali, A.M.; Murakami, S.; Isogawa, M.; Tanaka, Y. Characteristics of escape mutations from occult hepatitis B virus infected patients with hematological malignancies in south Egypt. World J. Hepatol. 2017, 9, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.; Tang, X.; Lok, A.S.; Sureau, C.; Garcia, T.; Li, J.; Wands, J.; Tong, S. Impaired virion secretion by hepatitis B virus immune escape mutants and its rescue by wild-type envelope proteins or a second-site mutation. J. Virol. 2013, 87, 2352–2357. [Google Scholar] [CrossRef] [PubMed]
- Mello, F.C.; Martel, N.; Gomes, S.A.; Araujo, N.M. Expression of hepatitis B virus surface antigen containing y100c variant frequently detected in occult HBV infection. Hepat. Res. Treat. 2011, 2011, 695859. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Deng, W.; Deng, L.; Cao, L.; Qin, B.; Li, S.; Wang, Y.; Pei, R.; Yang, D.; Lu, M.; et al. Amino acid substitutions at positions 122 and 145 of hepatitis B virus surface antigen (HBsAg) determine the antigenicity and immunogenicity of HBsAg and influence in vivo HBsAg clearance. J. Virol. 2012, 86, 4658–4669. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, X.; Tian, Y.; Song, J.; Yang, D.; Roggendorf, M.; Lu, M.; Chen, X. Biological significance of amino acid substitutions in hepatitis B surface antigen (HBsAg) for glycosylation, secretion, antigenicity and immunogenicity of HBsAg and hepatitis B virus replication. J. Gen. Virol. 2010, 91, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.H.; Michailidis, E.; Ding, H.; Peng, Y.Q.; Su, M.Z.; Li, Y.; Liu, X.E.; Dao Thi, V.L.; Wu, X.F.; Schneider, W.M.; et al. Effects of amino acid substitutions in hepatitis B virus surface protein on virion secretion, antigenicity, HBsAg and viral DNA. J. Hepatol. 2017, 66, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddigh-Tonekaboni, S.; Waters, J.A.; Jeffers, S.; Gehrke, R.; Ofenloch, B.; Horsch, A.; Hess, G.; Thomas, H.C.; Karayiannis, P. Effect of variation in the common “a” determinant on the antigenicity of hepatitis B surface antigen. J. Med. Virol. 2000, 60, 113–121. [Google Scholar] [CrossRef]
- Zhou, H.; Gewaily, D.; Ahn, S.H.; Preskill, C.; Wang, Y.; Zong, L.; Zhang, J.; Han, K.H.; Wands, J.; Li, J.; et al. Sequence analysis and functional characterization of full-length hepatitis B virus genomes from korean cirrhotic patients with or without liver cancer. Virus Res. 2017, 235, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Qin, Y.; Guarnieri, M.; Garcia, T.; Kwei, K.; Mizokami, M.; Zhang, J.; Li, J.; Wands, J.R.; Tong, S. Impairment of hepatitis B virus virion secretion by single-amino-acid substitutions in the small envelope protein and rescue by a novel glycosylation site. J. Virol. 2010, 84, 12850–12861. [Google Scholar] [CrossRef] [PubMed]
- Velay, A.; Jeulin, H.; Eschlimann, M.; Malve, B.; Goehringer, F.; Bensenane, M.; Frippiat, J.P.; Abraham, P.; Ismail, A.M.; Murray, J.M.; et al. Characterization of hepatitis B virus surface antigen variability and impact on HBs antigen clearance under nucleos(t)ide analogue therapy. J. Viral Hepat. 2016, 23, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Candotti, D.; Allain, J.P. Specific amino acid substitutions in the s protein prevent its excretion in vitro and may contribute to occult hepatitis B virus infection. J. Virol. 2013, 87, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sullivan, P.F. Single nucleotide polymorphism genotyping: Biochemistry, protocol, cost and throughput. Pharmacogenom. J. 2003, 3, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Kala, D.; Dhiman, G.; Yadav, V.; Krokhotin, A.; Dokholyan, N.V. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in il8 gene. Sci. Rep. 2017, 7, 6525. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Chauhan, J.B. Computational analysis for the determination of deleterious nsSNPs in human MTHFD1 gene. Comput. Biol. Chem. 2017, 70, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Yachdav, G.; Rost, B. Snap predicts effect of mutations on protein function. Bioinformatics 2008, 24, 2397–2398. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Phenotype prediction for mucopolysaccharidosis type I by in silico analysis. Orphanet J. Rare Dis. 2017, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, C.E.; Kohlstedt, D.; Meisel, C.; Keller, K.; Becker, K.; Mackenroth, L.; Rump, A.; Schrock, E.; Wimberger, P.; Kast, K. BRCA1/2 missense mutations and the value of in-silico analyses. Eur. J. Med. Genet. 2017, 60, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, Y.; Zhang, Z.; Meng, Z.; Qin, L.; Lu, M.; Yang, D. The amino acid residues at positions 120 to 123 are crucial for the antigenicity of hepatitis B surface antigen. J. Clin. Microbiol. 2007, 45, 2971–2978. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Bertoletti, A.; Penna, A.; Cavalli, A.; Valli, A.; Missale, G.; Pilli, M.; Fowler, P.; Giuberti, T.; Chisari, F.V.; et al. Identification of immunodominant T cell epitopes of the hepatitis B virus nucleocapsid antigen. J. Clin. Investig. 1991, 88, 214–222. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variant | Genotype | PROVEAN Prediction # | PROVEAN Prediction * | SNAP2 Prediction | PolyPhen2 | References |
|---|---|---|---|---|---|---|
| sN146D, sC147Y, sY100C, sG130R, sC138Y, sP142S, sG145R, sG145E, sN146S, sC147R, sC149Y, sD144G | A | ✔ | ✔ | ✔ | ✔ | [81,82] |
| sS132P, sT114R, sT115A, sK141E | A | ✔ | ✔ | ✔ | [71,81] | |
| sC121A, sC124A, sQ129H, sC147A | A | ✔ | ✔ | ✔ | [81] | |
| sP120T | A | ✔ | ✔ | ✔ | [81] | |
| sP127S | A | ✔ | ✔ | [81] | ||
| sC149A | A | ✔ | ✔ | [81] | ||
| sC124R, sS136P, sC139R, sT140I, sD144A, sG145R, sG145A, sG145W, sG145I, sG145P, sG145N, sG145D, sK122I | B | ✔ | ✔ | ✔ | ✔ | [49] [83] [61] |
| sK141E, sK160N, sT123N | B | ✔ | ✔ | ✔ | [49,84] | |
| sQ129R | B | ✔ | [49] | |||
| sP120T | B | ✔ | ✔ | ✔ | [49] | |
| sA159G | B | ✔ | [84] | |||
| sG119R, sC124R, sC124Y, sS136P, sC139R, sK141E, sL21R, sT131I, sP142S | C | ✔ | ✔ | ✔ | ✔ | [49] [85] [86] |
| sG145R, sG145A | C | ✔ | ✔ | ✔ | [86] | |
| sL95W | C | ✔ | ✔ | ✔ | [85] | |
| sD144A | C | ✔ | ✔ | ✔ | [49] | |
| sT140I, sP120T, sE2G | C | ✔ | ✔ | [49] [85] | ||
| sL98V | C | ✔ | ✔ | [85] | ||
| sD99G | C | ✔ | ✔ | ✔ | [87] | |
| sQ129R | C | ✔ | [49] | |||
| sM133T | C | ✔ | ✔ | [88] | ||
| sI126S | C | [49] | ||||
| sT116N | D | ✔ | ✔ | ✔ | [66] | |
| sP120T | D | ✔ | ✔ | ✔ | [80] | |
| sR122P, sG145R | D | ✔ | ✔ | ✔ | [66] [85] | |
| sT125M | D | ✔ | ✔ | ✔ | ✔ | [89] |
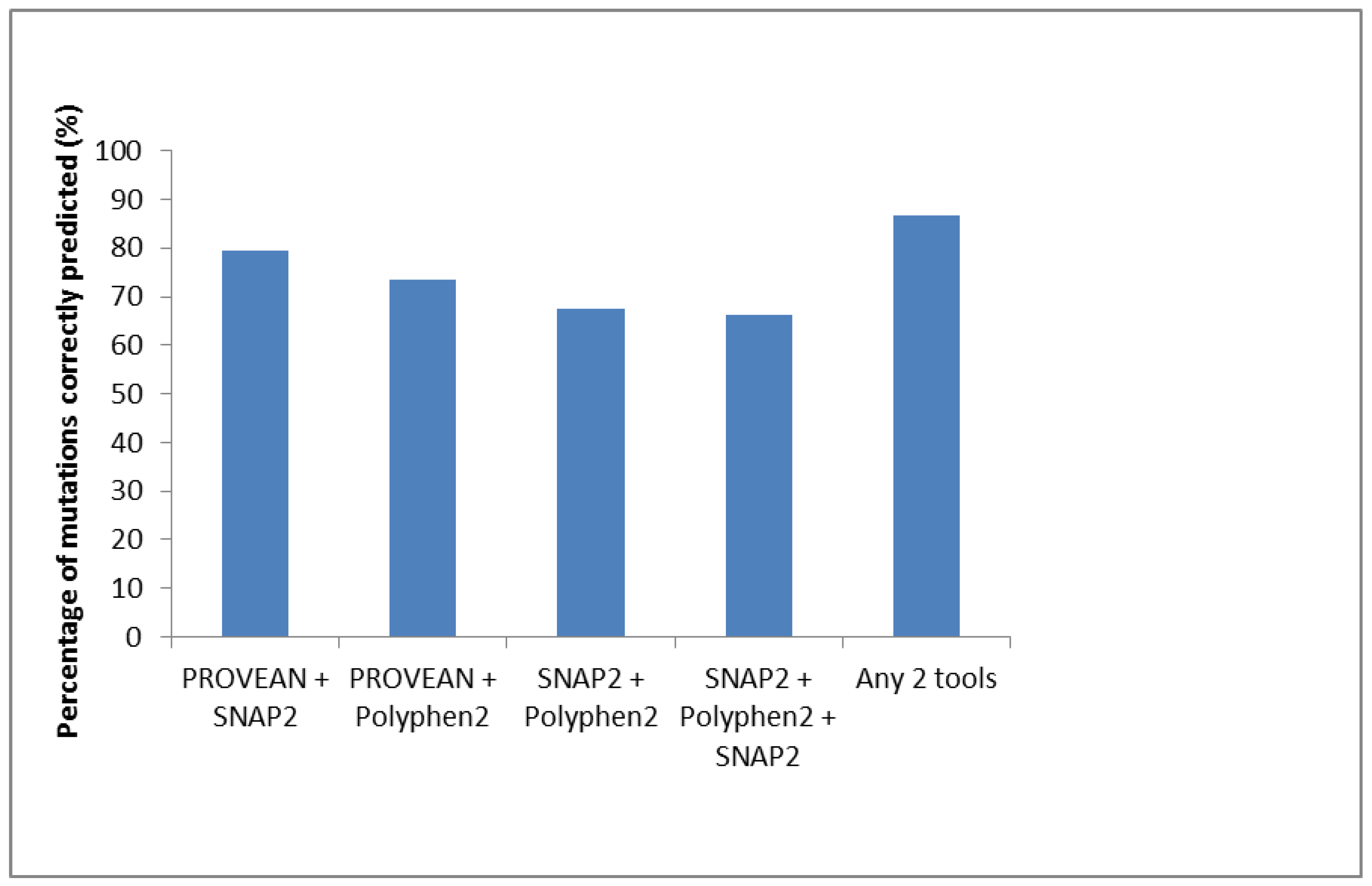
| Total (Effect) | 52 (76.5%) | 65 (95.6%) | 55(80.1%) | 52 (76.5%) | ||
| Total (Neutral) | 16 | 3 | 13 | 16 |
| Variant | Region | Genotype | PROVEAN Prediction # | PROVEAN Prediction * | SNAP2 | PolyPhen2 | Reference |
|---|---|---|---|---|---|---|---|
| sM103I | S | A | ✔ | ✔ | [44] | ||
| sK122R | S | A | [44] | ||||
| tpP100S | TP | A | ✔ | ✔ | ✔ | [45] | |
| sP111S, sS154P, sK122P, sK122W | S | B | ✔ | ✔ | ✔ | ✔ | [83,90] |
| sG112E, sG119E, sW165R, sK122G, sK122L, sK122D | S | B | ✔ | ✔ | ✔ | [83,90] | |
| sQ129R | S | B | ✔ | [90] | |||
| sI150T | S | B | ✔ | ✔ | ✔ | [90] | |
| sK122M, sK122H | S | B | ✔ | ✔ | ✔ | [83] | |
| sK122Q | S | B | [83] | ||||
| sK122T, sK122E, sK122N | S | B | ✔ | ✔ | [83] | ||
| tpQ177H | TP | B | ✔ | [45] | |||
| tpR27L | TP | B | ✔ | ✔ | [45] | ||
| sC121Y | S | C | ✔ | ✔ | ✔ | ✔ | [87] |
| sR24K, sT47A, sT47K, sI126S, sF134Y | S | C | [85,86,87] | ||||
| sQ101R | S | D | ✔ | [66] | |||
| sS167L | S | D | ✔ | ✔ | ✔ | [66] | |
| sS143L | S | D | ✔ | ✔ | [66] | ||
| total | 18 (56.3%) | 8 (25%) | 11 (34.4%) | 23 (71.9%) |
| Variant | ORF | Genotype | Final Result |
|---|---|---|---|
| sL97P, sT114I, sC124Y *, sN131K *, sP217L * | S | A1 | Deleterious |
| sQ129H * | S | D3 | Deleterious |
| PreS1S78N | PreS1 | D3 | Deleterious |
| PreS2F22P, PreS2F22H | PreS2 | D3 | Deleterious |
| xS11A, xV15I | X | A1 | Neutral |
| xS31A, xS101P, xL116V | X | D3 | Neutral |
| xP11S, xQ87L | X | D3 | Deleterious |
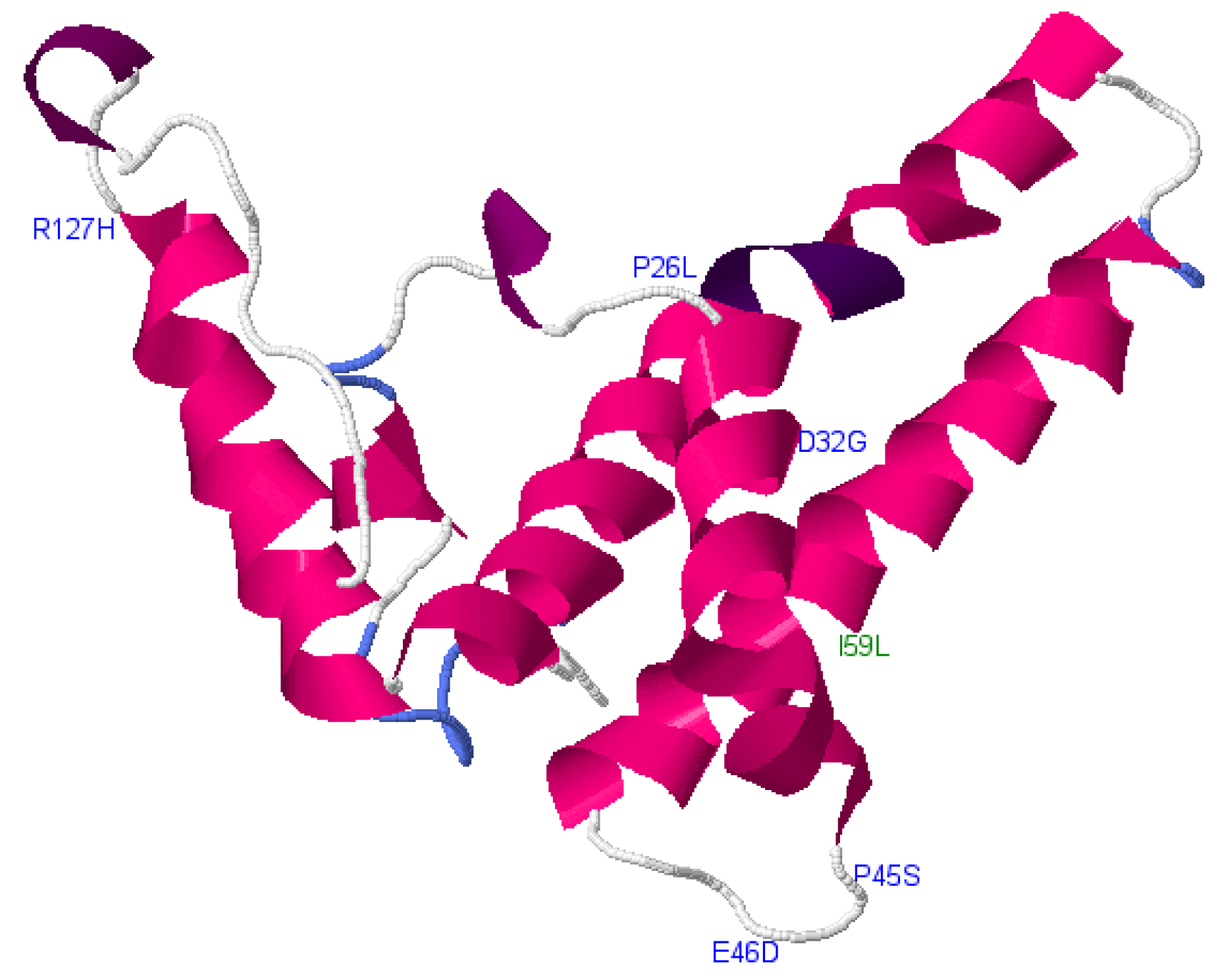
| cS26P, cD32G, cP45S, cE46D, cR127H | Core | A1 | Deleterious |
| cI59L | Core | A1 | Neutral |
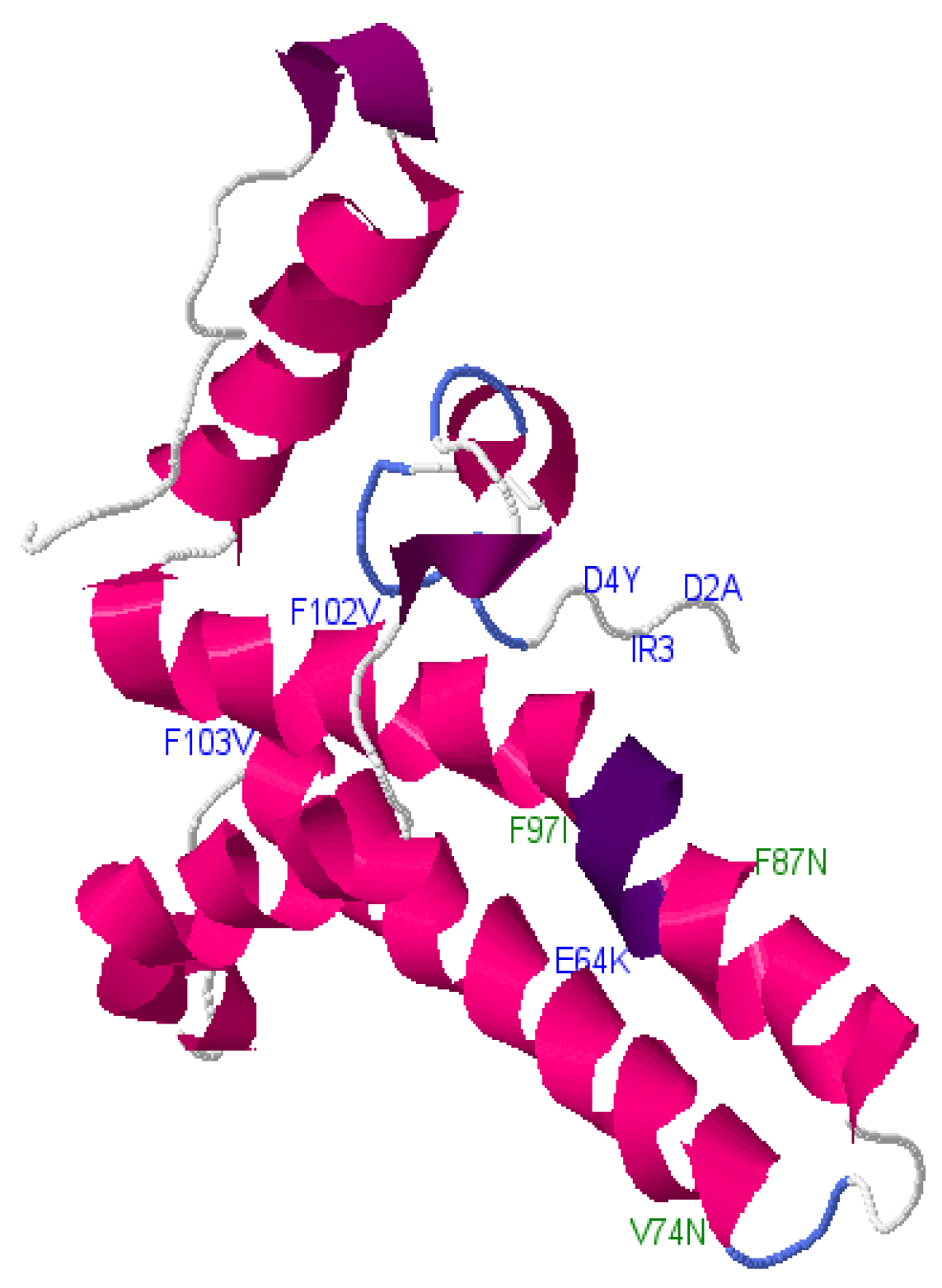
| cV74N, cS87N, cF97I | Core | D3 | Neutral |
| cD2A, D4Y, cI3R, cE64K, cW102V, cF103V | Core | D3 | Deleterious |
| tpN120Y, tpK155R | Pol-TP | A1 | Deleterious |
| spS91T, spS133G | Pol-Spacer | A1 | Neutral |
| rtL140I, rtA329T | Pol-RT | A1 | Neutral |
| rtT225A | Pol-RT | A1 | Deleterious |
| rhI81M | Pol-RH | A1 | Neutral |
| spW64R, spP103S | Pol-Spacer | D3 | Neutral |
| rtY257F | Pol-RT | D3 | Neutral |
| rtT128I | Pol-RT | D3 | Deleterious |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, M.; Choga, W.T.; Moyo, S.; Bell, T.G.; Mbangiwa, T.; Phinius, B.B.; Bhebhe, L.; Sebunya, T.K.; Makhema, J.; Marlink, R.; et al. In Silico Analysis of Hepatitis B Virus Occult Associated Mutations in Botswana Using a Novel Algorithm. Genes 2018, 9, 420. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090420
Anderson M, Choga WT, Moyo S, Bell TG, Mbangiwa T, Phinius BB, Bhebhe L, Sebunya TK, Makhema J, Marlink R, et al. In Silico Analysis of Hepatitis B Virus Occult Associated Mutations in Botswana Using a Novel Algorithm. Genes. 2018; 9(9):420. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090420
Chicago/Turabian StyleAnderson, Motswedi, Wonderful T. Choga, Sikhulile Moyo, Trevor Graham Bell, Tshepiso Mbangiwa, Bonolo B. Phinius, Lynette Bhebhe, Theresa K. Sebunya, Joseph Makhema, Richard Marlink, and et al. 2018. "In Silico Analysis of Hepatitis B Virus Occult Associated Mutations in Botswana Using a Novel Algorithm" Genes 9, no. 9: 420. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090420