
Investigation on Oil Physical States of Hybrid Shale Oil System: A Case Study on Cretaceous Second White Speckled Shale Formation from Highwood River Outcrop, Southern Alberta


Abstract
:1. Introduction
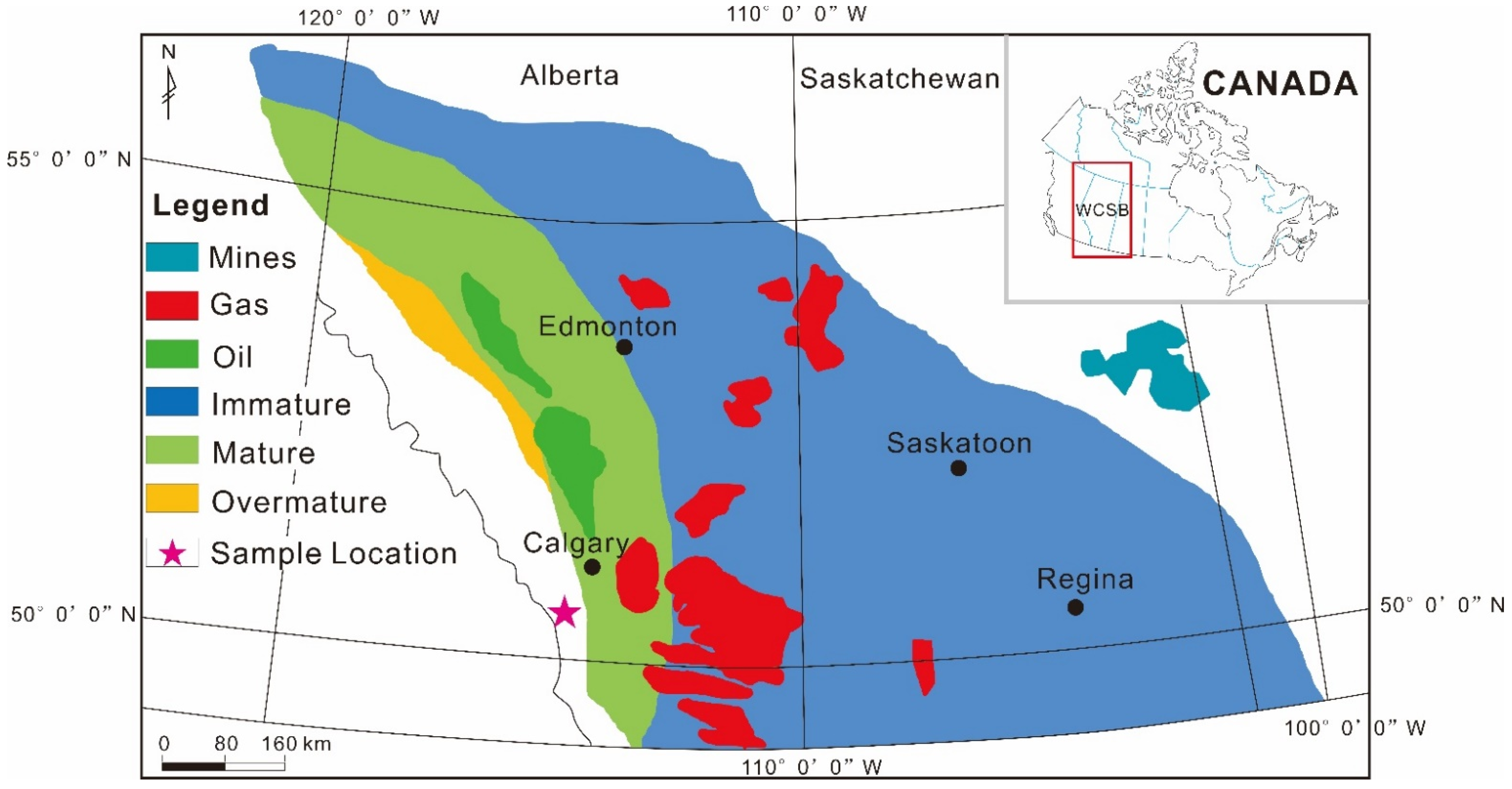
2. Geological Background
3. Samples and Methods
3.1. Samples
3.2. Analytical Methods
3.2.1. Rock-Eval Pyrolysis
3.2.2. Sequential Extraction
3.2.3. Gross Composition Separation (SARA)
3.2.4. Gas Chromatography–Mass Spectrometry (GC-MS)
4. Results
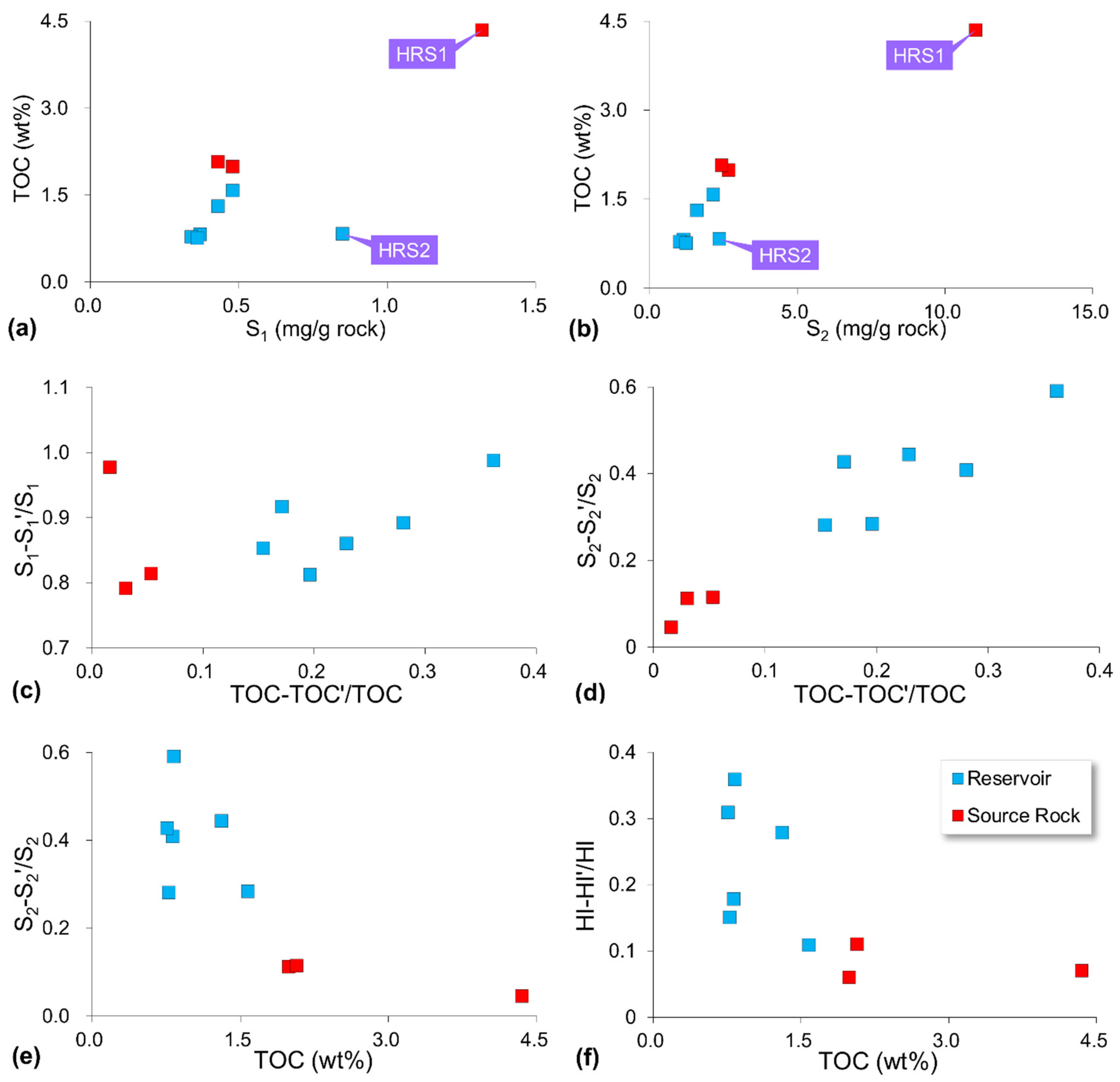
4.1. Rock-Eval Pyrolysis
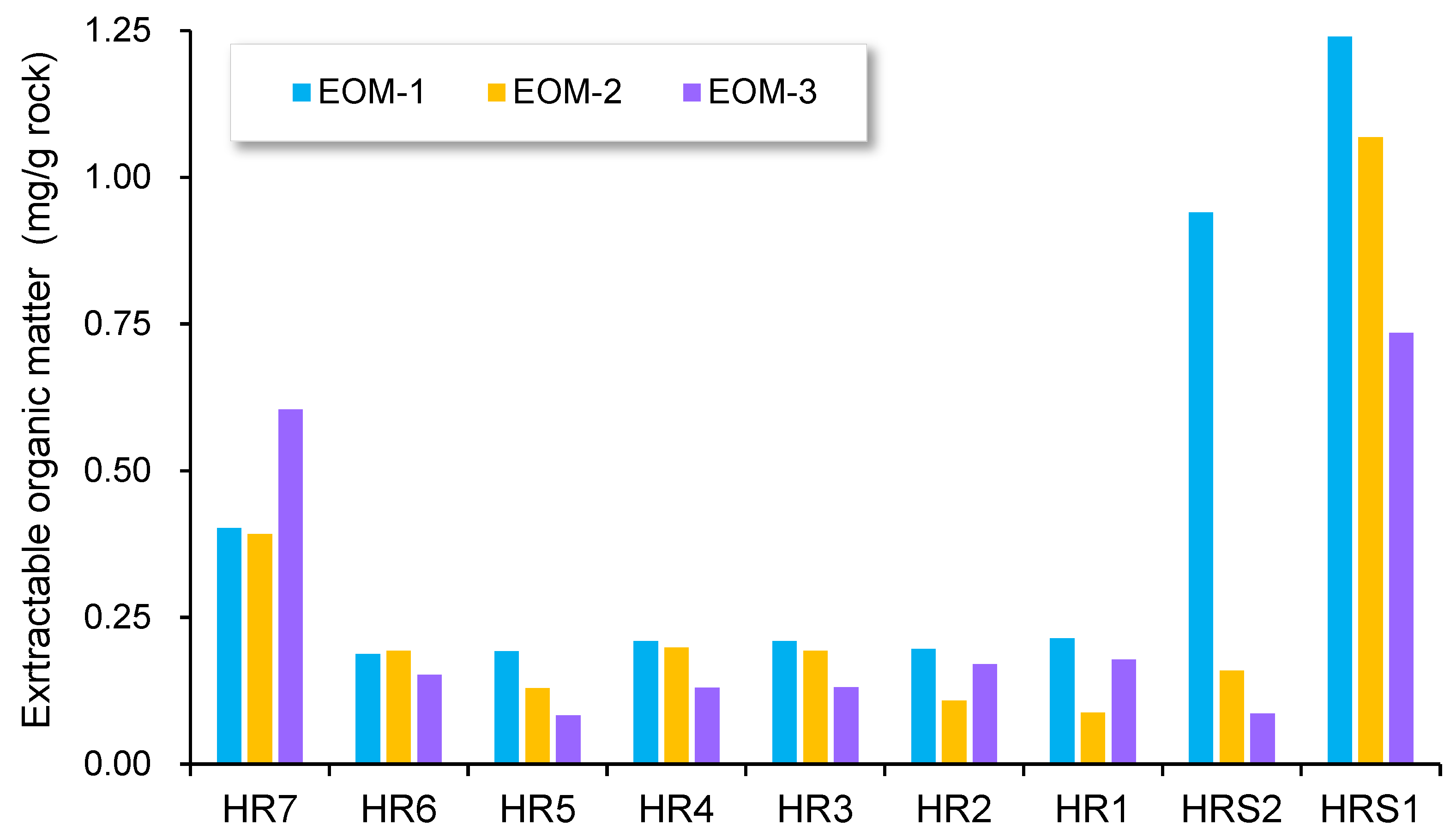
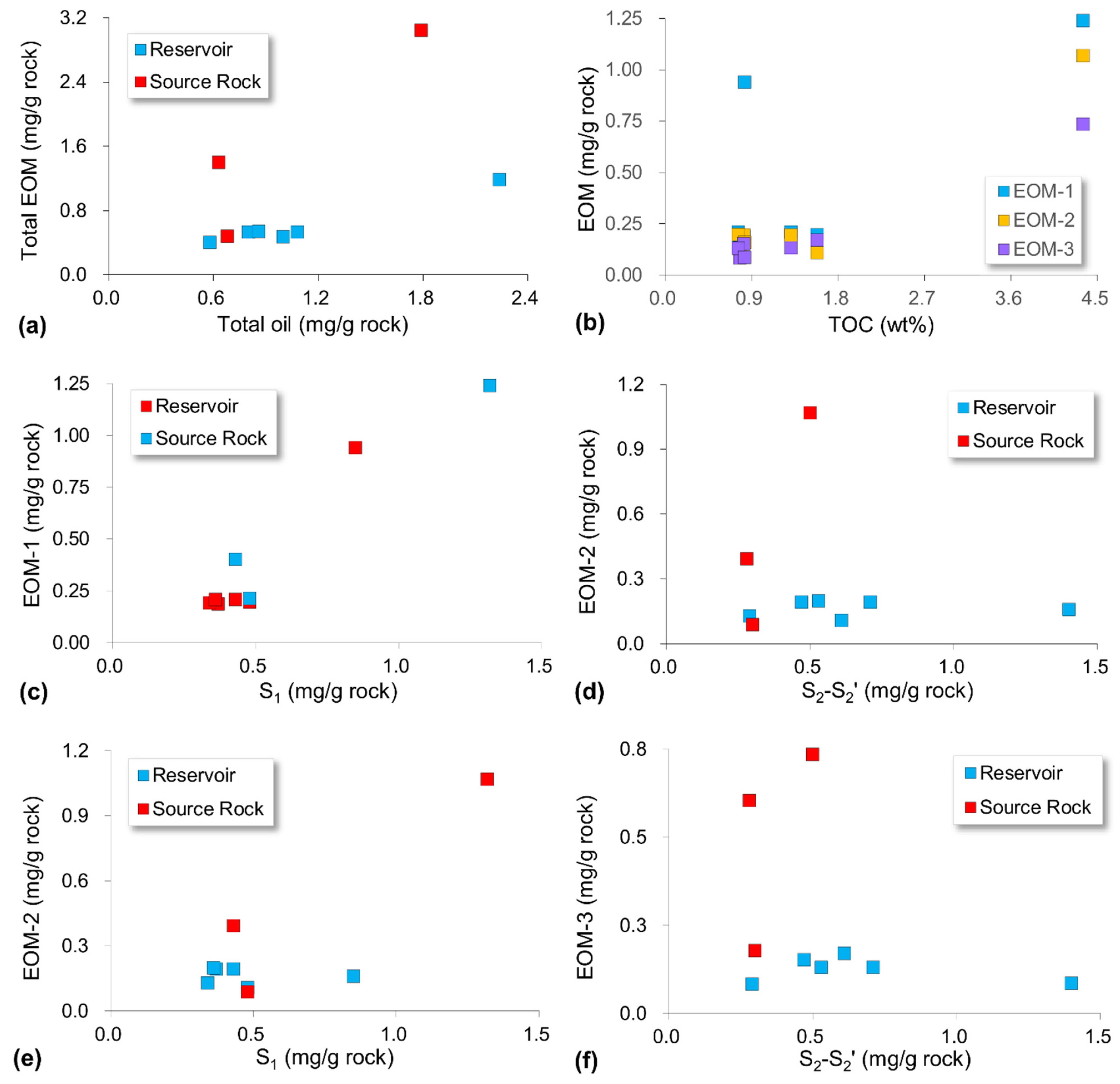
4.2. Sequential Extraction
4.3. SARA Compositions
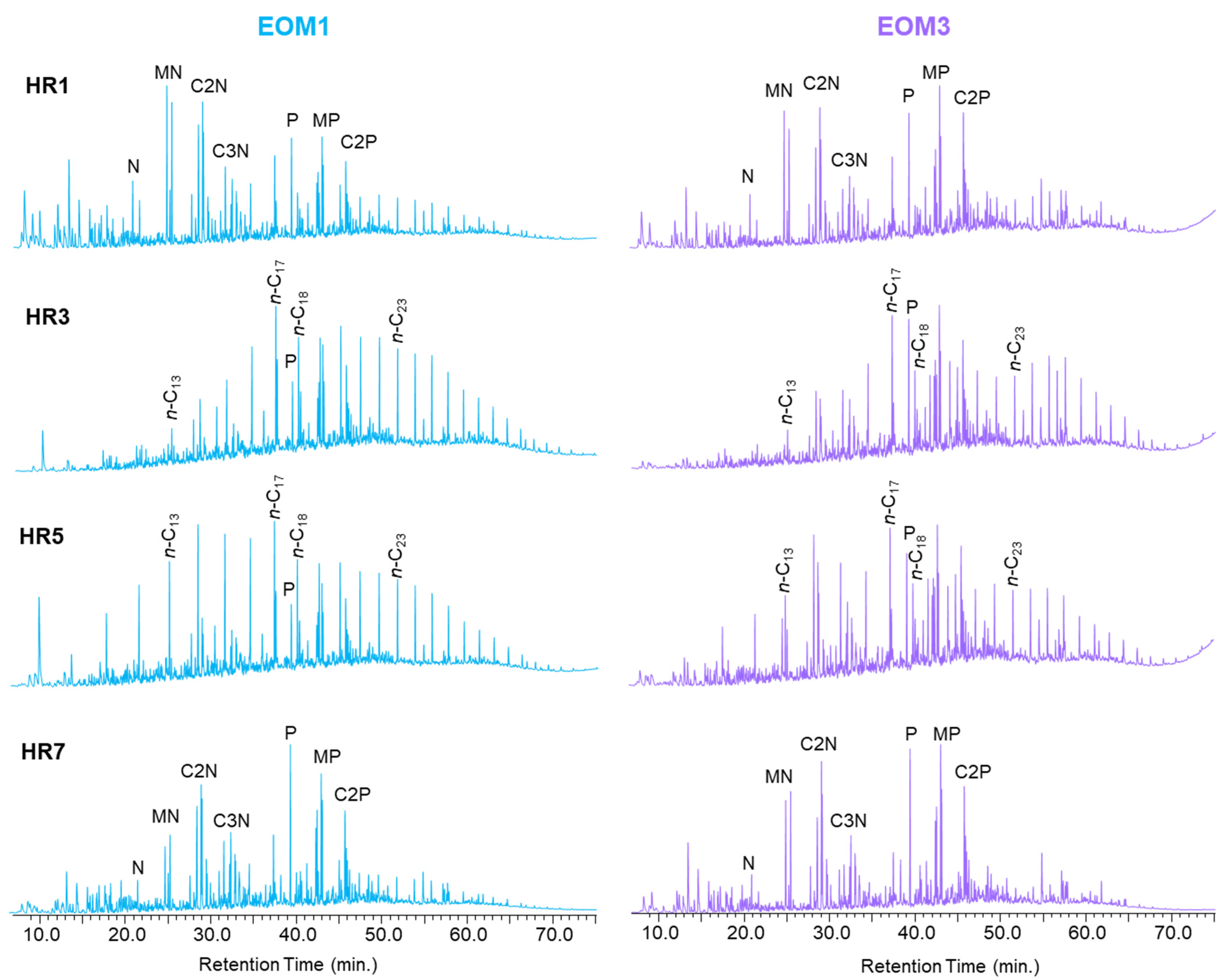
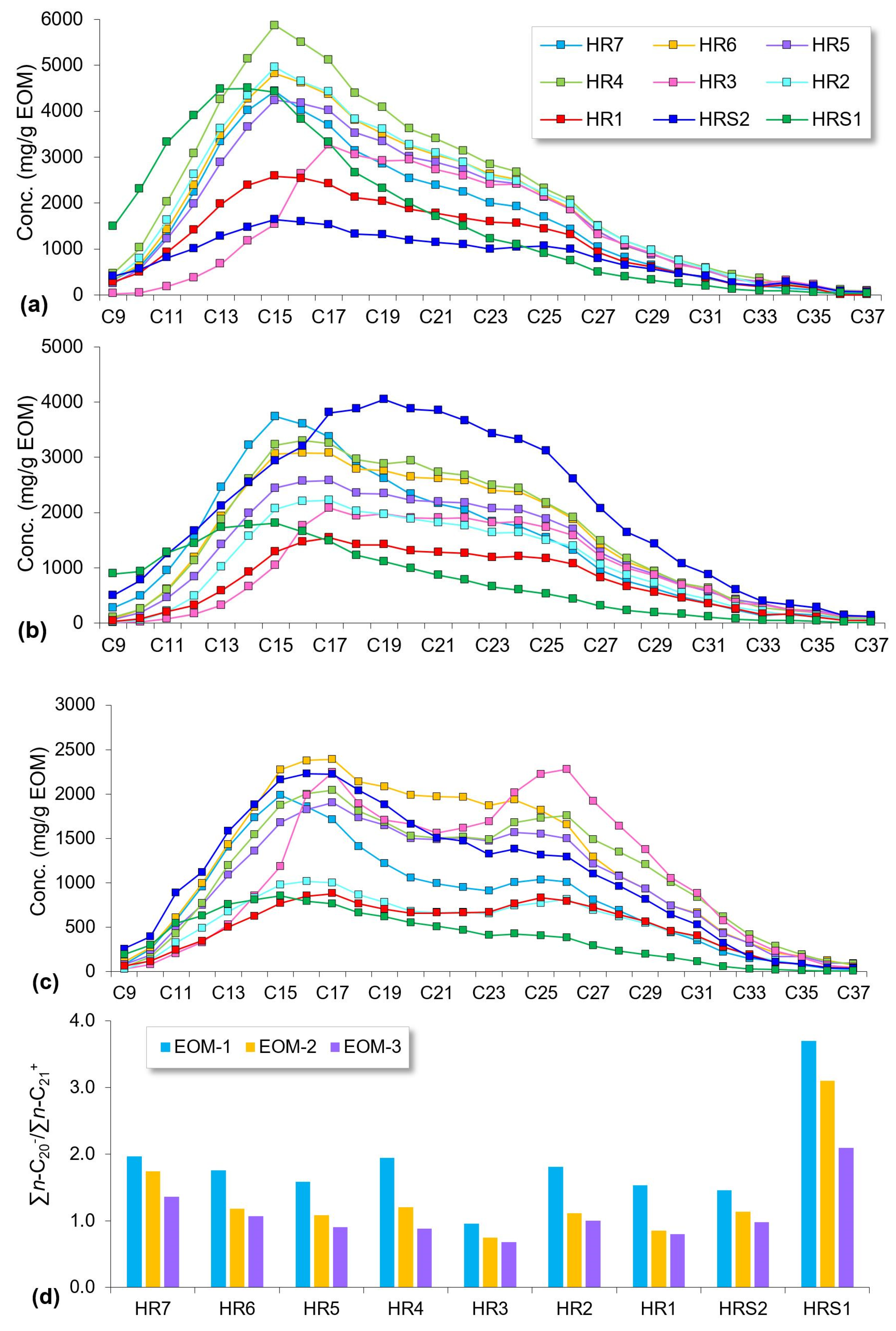
4.4. Molecular Compositions
5. Discussion
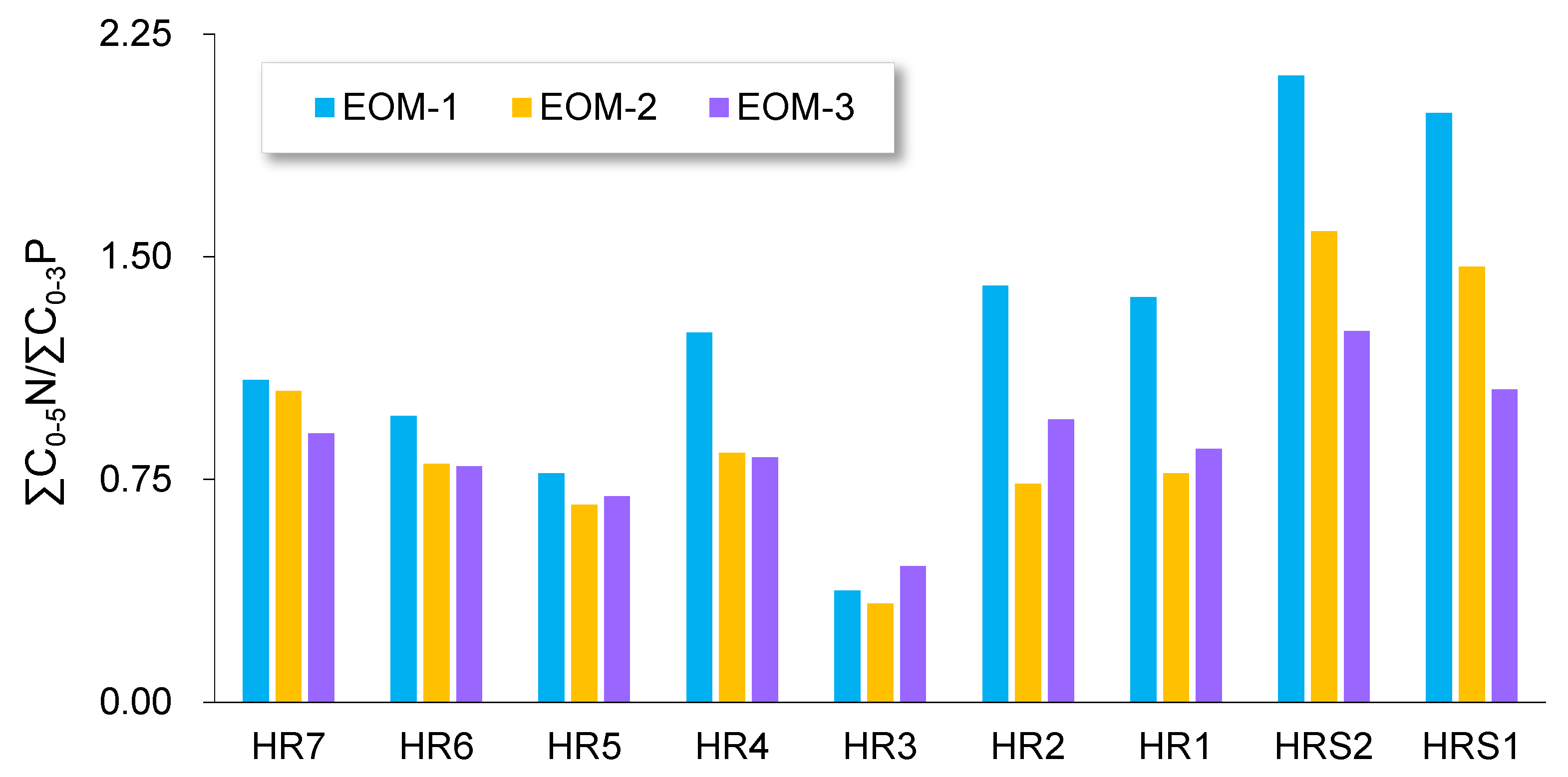
5.1. Oil Physical Status Illustrated by Sequential Extraction
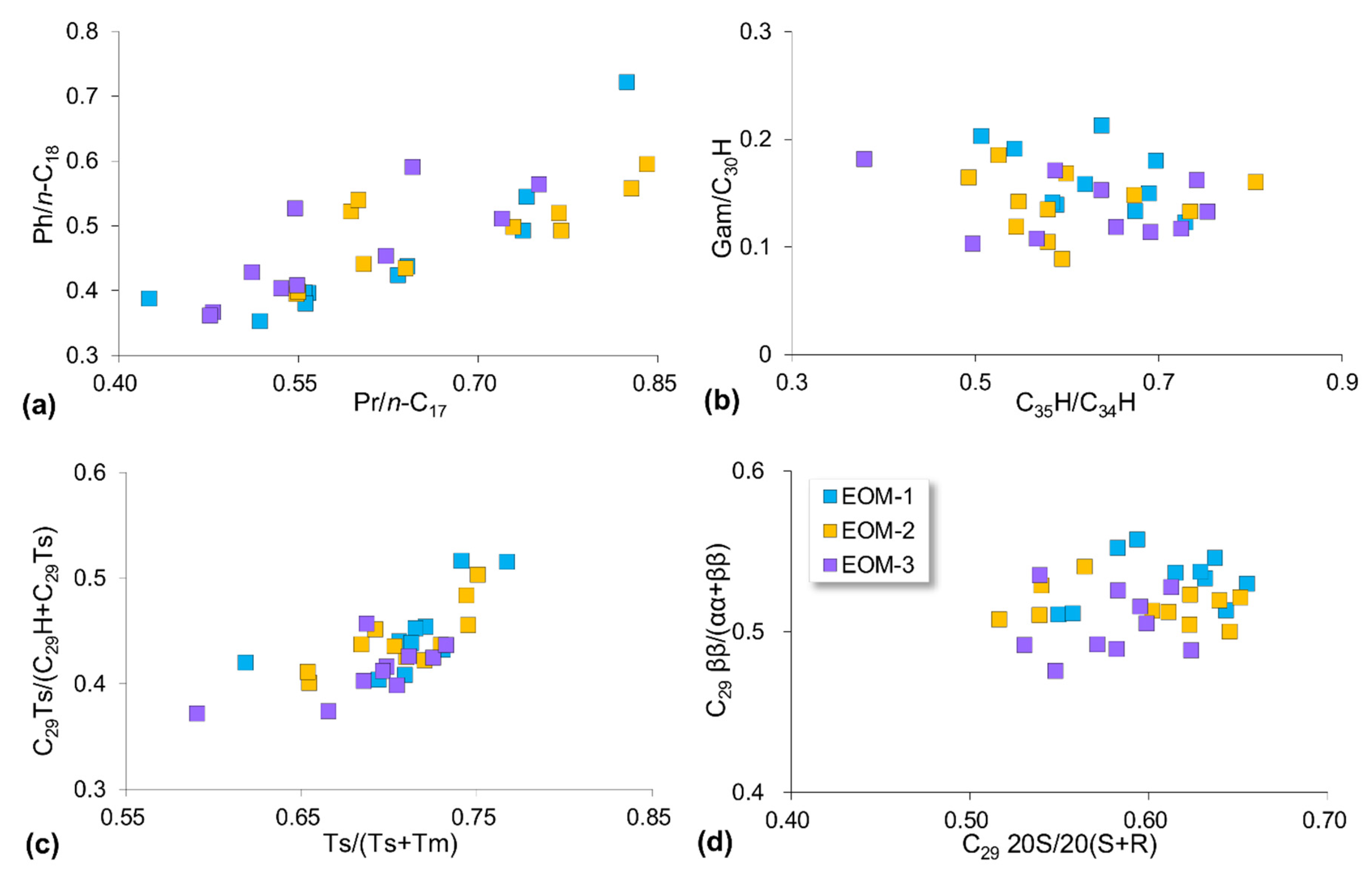
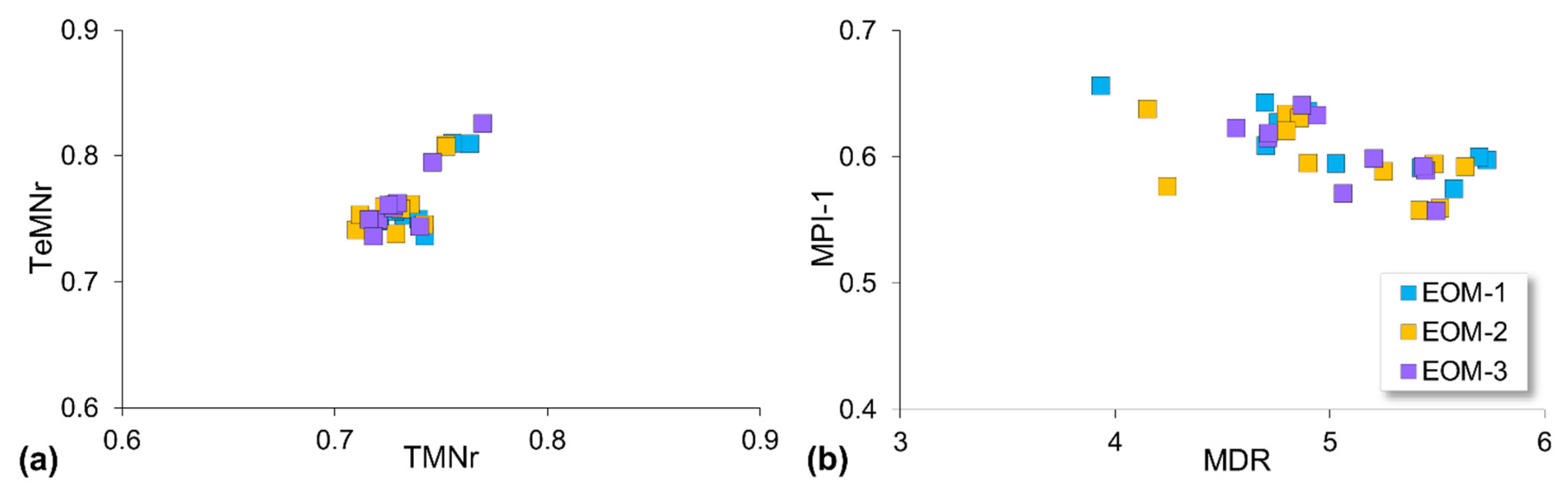
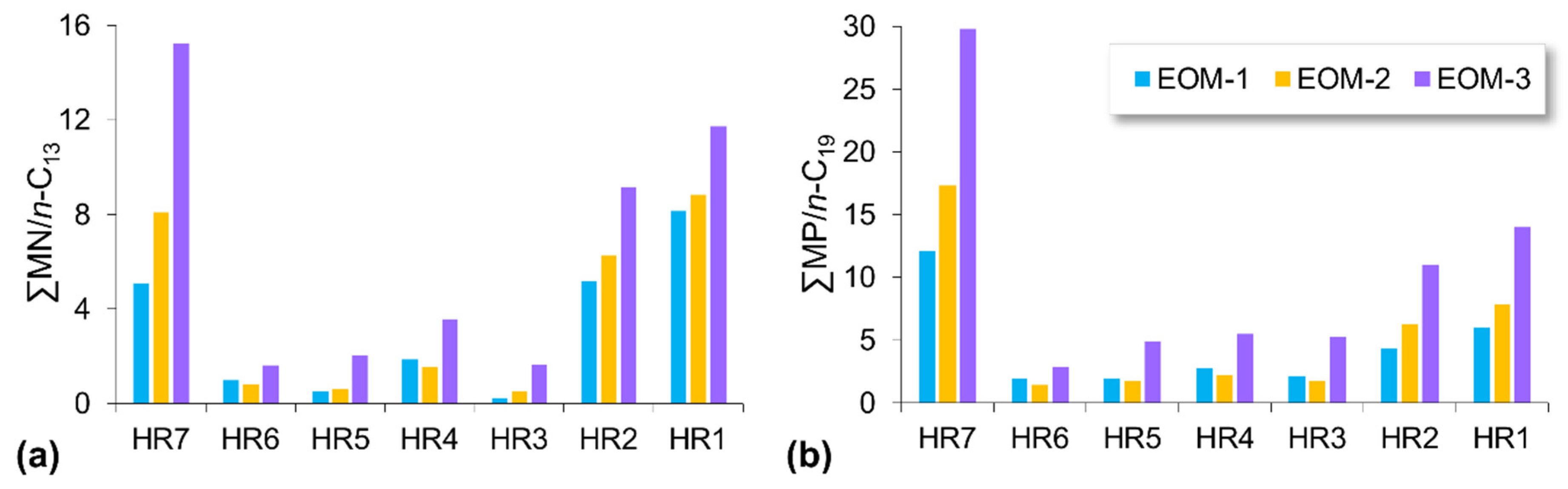
5.2. Compositional Fractionation during Sequential Extraction
5.3. Lithological Constraint on Oil Physical State and Exploration Potential
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonnenberg, S.A.; Pramudito, A. Petroleum geology of the giant Elm Coulee field, Williston Basin. Am. Assoc. Pet. Geol. Bull. 2009, 93, 1127–1153. [Google Scholar] [CrossRef]
- Clarkson, C.R.; Pedersen, P.K. Production Analysis of Western Canadian Unconventional Light Oil Plays. In Proceedings of the SPE Canadian Unconventional Resources Conference, Calgary, AB, Canada, 15–17 November 2011; p. 149005. [Google Scholar]
- Boak, J.; Kleinberg, R. Shale gas, tight oil, shale oil and hydraulic fracturing. In Future Energy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 67–95. [Google Scholar]
- Jarvie, D.M. Shale resource systems for oil and gas: Part 2—shale−oil resource systems. In Shale Reservoirs—Giant Resources for 21st Century; Beyer, J.A., Ed.; The American Association of Petroleum Geologists: Tulsa, OK, USA, 2012; Volume 97, pp. 89–119. [Google Scholar]
- Curiale, J.A.; Curtis, J.B. Organic geochemical applications to the exploration for source−rock reservoirs—A review. J. Unconv. Oil Gas Resour. 2016, 13, 1–31. [Google Scholar] [CrossRef]
- Wu, T.; Pan, Z.; Liu, B.; Connell, L.D.; Sander, R.; Fu, X. Laboratory characterization of shale oil storage behavior: A comprehensive review. Energy Fuels 2021, 35, 7305–7318. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, H.; Li, Z.; Liu, M. Oil physical status in lacustrine shale reservoirs–A case study on Eocene Shahejie Formation shales, Dongying Depression, East China. Fuel 2019, 257, 116027. [Google Scholar] [CrossRef]
- Larter, S.; Huang, H.; Bennett, B.; Snowdon, L. What Don’t We Know about Self Sourced Oil Reservoirs: Challenges and Potential Solutions? In Proceedings of the SPE Canadian Unconventional Resources Conference, Calgary, AB, Canada, 30 October–1 November 2012; p. 162777. [Google Scholar]
- Raji, M.; Gröcke, D.R.; Greenwell, C.; Cornford, C. Pyrolysis, Porosity and Productivity in Unconventional Mudstone Reservoirs: ‘Free’ and ‘Adsorbed’ Oil. In Proceedings of the SPE/AAPG/SEG Unconventional Resources Technology Conference, San Antonio, TX, USA, 20–22 July 2015; p. 2172996. [Google Scholar]
- Zink, K.G.; Scheeder, G.; Stueck, H.L.; Biermann, S.; Blumenberg, M. Total shale oil inventory from an extended Rock-Eval approach on unextracted and extracted source rocks from Germany. Int. J. Coal Geol. 2016, 163, 186–194. [Google Scholar] [CrossRef]
- Abrams, M.A.; Gong, C.; Garnier, C.; Sephton, M.A. A new thermal extraction protocol to evaluate liquid rich unconventional oil in place and in−situ fluid chemistry. Mar. Pet. Geol. 2017, 88, 659–675. [Google Scholar] [CrossRef]
- Price, L.C.; Clayton, J.L. Extraction of whole versus ground source rocks-fundamental petroleum geochemical implications including oil−source rock correlation. Geochim. Cosmochim. Acta 1992, 56, 1213–1222. [Google Scholar] [CrossRef]
- Wilhelms, A.; Horstad, I.; Karlsen, D. Sequential extraction—A useful tool for reservoir geochemistry? Org. Geochem. 1996, 24, 1157–1172. [Google Scholar] [CrossRef]
- Schwark, L.; Stoddart, D.; Keuser, C.; Spitthoff, B.; Leythaeuser, D. A novel sequential extraction system for whole core plug extraction in a solvent flow through cell-application to extraction of residual petroleum from an intact pore−system in secondary migration studies. Org. Geochem. 1997, 26, 19–31. [Google Scholar] [CrossRef]
- Leythaeuser, D.; Keuser, C.; Schwark, L. Molecular memory effects recording the accumulation history of petroleum reservoirs: A case study of the Heidrun Field, offshore Norway. Mar. Pet. Geol. 2007, 24, 199–220. [Google Scholar] [CrossRef]
- Mohnhoff, D.; Littke, R.; Krooss, B.M.; Weniger, P. Flow−through extraction of oil and gas shales under controlled stress using organic solvents: Implications for organic matter−related porosity and permeability changes with thermal maturity. Int. J. Coal Geol. 2016, 157, 84–99. [Google Scholar] [CrossRef]
- Qian, M.; Jiang, Q.; Li, M.; Li, Z.; Liu, P.; Ma, Y.; Cao, T. Quantitative characterization of extractable organic matter in lacustrine shale with different occurrences. Pet. Geol. Exp. 2017, 39, 278–286, (In Chinese with an English Abstract). [Google Scholar]
- Zhang, H.; Huang, H.; Li, Z.; Liu, M. Comparative study between sequential solvent−extraction and multiple isothermal stages pyrolysis: A case study on Eocene Shahejie Formation shales, Dongying Depression, East China. Fuel 2020, 263, 116591. [Google Scholar] [CrossRef]
- Behar, F.; Beaumont, V.; Penteado, H.D.B. Rock-Eval 6 technology: Performances and developments. Oil Gas Sci. Technol. 2001, 56, 111–134. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, M.; Qian, M.; Li, Z.; Li, Z.; Huang, Z.; Zhang, C.M.; Ma, Y. Quantitative characterization of shale oil in different occurrence states and its application. Pet. Geol. Experiment 2016, 38, 842–849, (In Chinese with an English Abstract). [Google Scholar]
- Romero−Sarmiento, M.F.; Pillot, D.; Letort, G.; Lamoureux−Var, V.; Beaumont, V.; Huc, A.Y.; Garcia, B. New Rock-Eval method for characterization of unconventional shale resource systems. Oil Gas Sci. Technol. 2016, 71, 1–9. [Google Scholar] [CrossRef]
- Peters, K.E. Guidelines for evaluating petroleum source rock using programmed pyrolysis. Am. Assoc. Pet. Geol. Bull. 1986, 70, 318–329. [Google Scholar]
- Han, Y.; Mahlstedt, N.; Horsfield, B. The Barnett Shale: Compositional fractionation associated with intraformational petroleum migration, retention, and expulsion. Am. Assoc. Pet. Geol. Bull. 2015, 99, 2173–2202. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.; Lapierre, S. Integrating Solvent Extraction with Standard Pyrolysis to Better Quantify Thermal Maturity and Hydrocarbon Content in the Oil Window. In Proceedings of the SPE/AAPG/SEG Unconventional Resources Technology Conference, Denver, CO, USA, 25–27 August 2014; p. 1922397. [Google Scholar]
- Han, Y.; Horsfield, B.; Curry, D.J. Control of facies, maturation and primary migration on biomarkers in the Barnett Shale sequence in the Marathon 1 Mesquite well, Texas. Mar. Pet. Geol. 2017, 85, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Leythaeuser, D.; Schaefer, R.G.; Yukler, A. Role of diffusion in primary migration of hydrocarbons. Am. Assoc. Pet. Geol. Bull. 1982, 66, 408–429. [Google Scholar]
- Leythaeuser, D.; Schaefer, R.G.; Radke, M. Geochemical effects of primary migration of petroleum in Kimmeridge source rocks from Brae field area, North Sea. I: Gross composition and C15+−soluble organic matter and molecular composition of C15+−saturated hydrocarbons. Geochim. Cosmochim. Acta 1988, 52, 701–713. [Google Scholar] [CrossRef]
- Leythaeuser, D.; Radke, M.; Willsch, H. Geochemical effects of primary migration of petroleum in Kimmeridge source rocks from Brae field area, North Sea. II: Molecular composition of alkylated naphthalenes, phenanthrenes, benzo− and dibenzothiophenes. Geochim. Cosmochim. Acta 1988, 52, 2879–2891. [Google Scholar] [CrossRef]
- Lafargue, E.; Espitalié, J.; Jacobsen, T.; Eggen, S. Experimental simulation of hydrocarbon expulsion. Org. Geochem. 1990, 16, 121–131. [Google Scholar] [CrossRef]
- Sandvik, E.I.; Young, W.A.; Curry, D.J. Expulsion from hydrocarbon sources: The role of organic absorption. Org. Geochem. 1992, 19, 77–87. [Google Scholar] [CrossRef]
- Ritter, U. Fractionation of petroleum during expulsion from kerogen. J. Geochem. Explor. 2003, 78-79, 417–429. [Google Scholar] [CrossRef]
- Kelemen, S.R.; Walters, C.C.; Ertas, D.; Freund, H.; Curry, D.J. Petroleum Expulsion. Part 3. A model of chemically driven fractionation during expulsion of petroleum from kerogen. Energy Fuels 2006, 20, 309–319. [Google Scholar] [CrossRef]
- Eseme, E.; Littke, R.; Krooss, B.M.; Schwaubauer, J. Experimental investigation of the compositional variation of petroleum during primary migration. Org. Geochem. 2007, 38, 1373–1397. [Google Scholar] [CrossRef]
- Bloch, J.D.; Schroder−Adams, C.J.; Leckie, D.A.; Craig, J.; McIntyre, D.J. Sedimentology, Micropaleontology, Geochemistry and Hydrocarbon Potential of Shale from the Cretaceous Lower Colorado Group in Western Canada; Geological Survey of Canada Offices: Ottawa, ON, Canada, 1999. [Google Scholar]
- Schröder−Adams, C.J.; Leckie, D.A.; Bloch, J.; Craig, J.; McIntyre, D.J.; Adams, P.J. Paleoenvironmental changes in the Cretaceous (Albian to Turonian) Colorado Group of western Canada: Microfossil, sedimentological and geochemical evidence. Cretac. Res. 1996, 17, 311–365. [Google Scholar] [CrossRef]
- Furmann, A.; Mastalerz, M.; Brassell, S.C.; Pedersen, P.K.; Zajac, N.A.; Schimmelmann, A. Organic matter geochemistry and petrography of Late Cretaceous (Cenomanian−Turonian) organic−rich shales from the Belle Fourche and Second White Specks formations, west−central Alberta, Canada. Org. Geochem. 2015, 85, 102–120. [Google Scholar] [CrossRef] [Green Version]
- Synnott, D.P.; Dewing, K.; Sanei, H.; Pedersen, P.K.; Ardakani, O.H. Influence of refractory organic matter on source rock hydrocarbon potential: A case study from the Second White Specks and Belle Fourche formations, Alberta, Canada. Mar. Pet. Geol. 2017, 85, 220–232. [Google Scholar] [CrossRef]
- Huang, H.; di Primio, R.; Pedersen, J.H.; Silva, R.; Algeer, R.; Ma, J.; Larter, S. On the determination of oil charge history and the practical application of molecular maturity markers. Mar. Pet. Geol. 2022, 139, 105586. [Google Scholar] [CrossRef]
- Mossop, G.; Shetsen, I. (co−compilers); Geological Atlas of the Western Canada Sedimentary Basin; Alberta Geological Survey: Edmonton, AB, Canada, 1994. [Google Scholar]
- Chen, Z.; Osadetz, K.G. An assessment of tight oil resource potential in Upper Cretaceous Cardium formation, Western Canada sedimentary basin. Pet. Explor. Dev. 2013, 40, 344–353. [Google Scholar] [CrossRef]
- Aviles, M.A.; Ardakani, O.H.; Cheadle, B.A.; Sanei, H. Organic petrography and geochemical characterization of the Upper Cretaceous Second White Specks and Upper Belle Fourche alloformations, west−central Alberta: Analysis of local maturity anomalies. Int. J. Coal Geol. 2019, 203, 60–73. [Google Scholar] [CrossRef]
- Creaney, S.; Allan, J. Hydrocarbon generation and migration in the Western Canada sedimentary basin. Geol. Soc. Lond. Spec. Publ. 1990, 50, 189–202. [Google Scholar] [CrossRef]
- Komaromi, B.A.; Pedersen, P.K. Comparative Fracture Analysis and Characterization of the Upper Cretaceous Jumping Pound Sandstone and Second White Specks Formation, Southwestern Alberta. In Proceedings of the GeoConvention, Calgary, AB, Canada, 12–16 May 2014. [Google Scholar]
- Delvaux, D.; Martin, H.; Leplat, P.; Paulet, J. Comparative Rock-Eval pyrolysis as an improved tool for sedimentary organic matter analysis. Org. Geochem. 1990, 16, 1221–1229. [Google Scholar] [CrossRef]
- Lafargue, W.; Espitalié, J.; Broks, T.M.; Nyland, B. Experimental simulation of primary migration. Org. Geochem. 1994, 22, 575–586. [Google Scholar] [CrossRef]
- Sinninghe Damsté, J.S.; Kenig, F.; Koopmans, M.P.; Koster, J.; Schouten, S.; Hayes, J.M.; de Leeuw, J.W. Evidence for gammacerane as an indicator of water column stratification. Geochim. Cosmochim. Acta 1995, 59, 1895–1900. [Google Scholar] [CrossRef] [Green Version]
- Huang, H. The effect of biodegradation on gammacerane in crude oils. Biodegradation 2017, 28, 313–326. [Google Scholar] [CrossRef]
- Peters, K.E.; Moldowan, J.M. Effects of source, thermal maturity and biodegradation on the distribution and isomerization of homohopanes in petroleum. Org. Geochem. 1991, 17, 47–61. [Google Scholar] [CrossRef]
- Peters, K.E.; Walters, C.C.; Moldowan, J.M. The Biomarker Guide; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Van Aarssen, B.G.; Bastow, T.P.; Alexander, R.; Kagi, R.I. Distributions of methylated naphthalenes in crude oils: Indicators of maturity, biodegradation and mixing. Org. Geochem. 1999, 30, 1213–1227. [Google Scholar] [CrossRef]
- Radke, M.; Willsch, H.; Leythaeuser, D.; Teichmüller, M. Aromatic components of coal: Relation of distribution pattern to rank. Geochim. Cosmochim. Acta 1982, 46, 1831–1848. [Google Scholar] [CrossRef]
- Radke, M.; Welte, D.H.; Willsch, H. Maturity parameters based on aromatic hydrocarbons: Influence of the organic matter type. Org. Geochem. 1986, 10, 51–63. [Google Scholar] [CrossRef]
- Li, S.; Li, S.; Guo, R.; Zhou, X.; Wang, Y.; Chen, J.; Zhang, J.; Hao, L.; Ma, X.; Qiu, J. Occurrence state of soluble organic matter in shale oil reservoirs from the Upper Triassic Yanchang Formation in the Ordos Basin, China: Insights from multipolarity sequential extraction. Nat. Resour. Res. 2021, 30, 4379–4402. [Google Scholar] [CrossRef]
- Sajgo, C.; Maxwell, J.R. Evaluation of fractionation effects during the early stages of primary migration. Org. Geochem. 1983, 5, 65–73. [Google Scholar] [CrossRef]
- Pepper, A.S.; Corvi, P.J. Simple kinetic models of petroleum formation. Part I: Oil and gas generation from kerogen. Mar. Pet. Geol. 1995, 12, 291–319. [Google Scholar] [CrossRef]
- Larter, S. Some pragmatic perspectives in source rock geochemistry. Mar. Pet. Geol. 1988, 5, 194–204. [Google Scholar] [CrossRef]
- Stainforth, J.G.; Reinders, J.E.A. Primary migration of hydrocarbons by diffusion through organic matter networks, and its effect on oil and gas generation. Org. Geochem. 1990, 16, 61–74. [Google Scholar] [CrossRef]
- Mackenzie, A.S.; Disko, U.; Rullkötter, J. Determination of hydrocarbon distributions in oils and sediment extracts by gas chromatography—high resolution mass spectrometry. Org. Geochem. 1983, 5, 57–63. [Google Scholar] [CrossRef]
- Li, Z.; Huang, H.; Zhang, S. The effect of biodegradation on bound biomarkers released from intermediate−temperature gold−tube pyrolysis of severely biodegraded Athabasca bitumen. Fuel 2020, 263, 116669. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Height | S1 | S2 | PC | TOC | Tmax | PI | HI | OSI |
|---|---|---|---|---|---|---|---|---|---|
| (m) | (mg/g Rock) | (mg/g Rock) | (%) | (%) | (°C) | (mg/g TOC) | (mg/g TOC) | ||
| HR7 | 58.5 | 0.43 | 2.44 | 0.24 | 2.07 | 450 | 0.15 | 118 | 20.77 |
| HR6 | 54 | 0.37 | 1.15 | 0.13 | 0.82 | 448 | 0.24 | 140 | 45.12 |
| HR5 | 51 | 0.34 | 1.03 | 0.11 | 0.78 | 447 | 0.25 | 132 | 43.59 |
| HR4 | 47 | 0.36 | 1.24 | 0.13 | 0.76 | 445 | 0.23 | 163 | 47.37 |
| HR3 | 44 | 0.43 | 1.6 | 0.17 | 1.31 | 452 | 0.21 | 122 | 32.82 |
| HR2 | 42 | 0.48 | 2.15 | 0.22 | 1.58 | 452 | 0.18 | 136 | 30.38 |
| HR1 | 25.1 | 0.48 | 2.68 | 0.26 | 1.99 | 452 | 0.15 | 135 | 24.12 |
| HRS2 | 17.05 | 0.85 | 2.37 | 0.27 | 0.83 | 439 | 0.27 | 286 | 102.41 |
| HRS1 | 17 | 1.32 | 11.04 | 1.03 | 4.35 | 443 | 0.11 | 254 | 30.34 |
| HR7’ * | 58.5 | 0.08 | 2.16 | 0.19 | 1.96 | 448 | 0.04 | 110 | 4.08 |
| HR6’ | 54 | 0.04 | 0.68 | 0.06 | 0.59 | 447 | 0.06 | 115 | 6.78 |
| HR5’ | 51 | 0.05 | 0.74 | 0.07 | 0.66 | 446 | 0.06 | 112 | 7.58 |
| HR4’ | 47 | 0.03 | 0.71 | 0.06 | 0.63 | 447 | 0.04 | 113 | 4.76 |
| HR3’ | 44 | 0.06 | 0.89 | 0.08 | 1.01 | 452 | 0.06 | 88 | 5.94 |
| HR2’ | 42 | 0.09 | 1.54 | 0.14 | 1.27 | 447 | 0.06 | 121 | 7.09 |
| HR1’ | 25.1 | 0.1 | 2.38 | 0.21 | 1.93 | 448 | 0.04 | 123 | 5.18 |
| HRS2’ | 17.05 | 0.01 | 0.57 | 0.05 | 0.53 | 448 | 0.02 | 108 | 1.89 |
| HRS1’ | 17 | 0.03 | 11.14 | 0.93 | 4.28 | 450 | 0 | 260 | 0.7 |
| Sample | Fraction | %Sat | %Aro | %Resin | %Asph |
|---|---|---|---|---|---|
| HR1 | EOM-1 | 47.7 | 25.6 | 5.8 | 20.9 |
| EOM-3 | 42.9 | 19.4 | 6.3 | 31.4 | |
| HR3 | EOM-1 | 29.1 | 23.6 | 14.5 | 32.7 |
| EOM-3 | 27.2 | 20.5 | 11.5 | 40.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Huang, H.; Yin, M. Investigation on Oil Physical States of Hybrid Shale Oil System: A Case Study on Cretaceous Second White Speckled Shale Formation from Highwood River Outcrop, Southern Alberta. Minerals 2022, 12, 802. https://0-doi-org.brum.beds.ac.uk/10.3390/min12070802
Zhang H, Huang H, Yin M. Investigation on Oil Physical States of Hybrid Shale Oil System: A Case Study on Cretaceous Second White Speckled Shale Formation from Highwood River Outcrop, Southern Alberta. Minerals. 2022; 12(7):802. https://0-doi-org.brum.beds.ac.uk/10.3390/min12070802
Chicago/Turabian StyleZhang, Hong, Haiping Huang, and Mengsha Yin. 2022. "Investigation on Oil Physical States of Hybrid Shale Oil System: A Case Study on Cretaceous Second White Speckled Shale Formation from Highwood River Outcrop, Southern Alberta" Minerals 12, no. 7: 802. https://0-doi-org.brum.beds.ac.uk/10.3390/min12070802