
Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution



1
Key Laboratory of Germplasm Enhancement, Physiology and Ecology of Food Crops in Cold Region of Chinese Education Ministry, College of Agriculture, Northeast Agricultural University, Harbin 150030, China
2
Key Laboratory of Soybean Biology of Chinese Education Ministry, College of Agriculture, Northeast Agricultural University, Harbin 150030, China
*
Authors to whom correspondence should be addressed.
Pathogens 2022, 11(4), 419; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11040419
Submission received: 2 March 2022
/
Revised: 22 March 2022
/
Accepted: 28 March 2022
/
Published: 30 March 2022
(This article belongs to the Special Issue Evolution of Plant Viruses)
Abstract
:Cowpea mild mottle virus (CpMMV) is an economically significant virus that causes severe disease on several legume crops. Aside from recombination, other factors driving its rapid evolution are elusive. In this study, the synonymous codon pattern of CpMMV and factors shaping it were analyzed. Phylogeny and nucleotide composition analyses showed that isolates of different geography or hosts had very similar nucleotide compositions. Relative synonymous codon usage (RSCU) and neutrality analyses suggest that CpMMV prefers A/U-ending codons and natural selection is the dominative factor that affects its codon bias. Dinucleotide composition and codon adaptation analyses indicate that the codon pattern of CpMMV is mainly shaped by the requirement of escaping of host dinucleotide-associated antiviral responses and translational efficiency.
1. Introduction
Cowpea mild mottle virus (CpMMV), a whitefly (Bemisia tabaci)-transmitted virus of the genus Carlavirus within the family Betaflexviridae, has filamentous particles of about 650 nm in length and a diameter of about 13 nm [1]. The genome of CpMMV is composed of a positive-sense, single-stranded RNA (+ssRNA) that is capped at the 5′-end and polyadenylated at the 3′-end. The genome of CpMMV encodes a total number of six open reading frames (ORFs). The largest ORF on the 5′-termini encodes the viral RNA-dependent RNA polymerase (RdRp). ORF2-4 are partially overlapped and are composed of the so-called triple-gene-block (TGB) module, which encodes TGBp1, TGBp2, and TGBp3, respectively. ORF5 encodes the viral coat protein (CP), while ORF6 encodes a small cysteine-rich protein that may function as the pathogenicity factor and RNA silencing suppressor [2,3,4]. ORF1 is translated from genomic RNA, while ORF2-4 and ORF5-6 are translated from two subgenomic RNAs [5].
Although CMMV was thought to be only locally distributed when first found on cowpea (Vigna unguiculata) in Ghana [6], it has since been shown to have a worldwide distribution and a wide natural host range, e.g., tomato (Lycopersicon esculentum), groundnut (Arachis hypogaea), peanut grass (Arachis repens), soybean (Glycine max), French bean (Phaseolus vulgars), cowpea (Vigna unguiculata), urdbean (Vigna mungo Black gram), mungbean (Vigna radiata), Adzuki bean (Vigna angularis), Chia (Salvia hispanica), and papaya (Carica papaya) [7,8,9,10,11,12,13,14,15,16]. Recent outbreaks of CpMMV on soybean in Brazil and China further emphasize its economic importance [17,18]. Studies showed that CpMMV is abundant in genetic diversity, which may be responsible for its wide host range and pathogenicity on soybean [5,14,17]. Moreover, isolates of different origins showed distinct biological properties, suggesting their high genetic flexibility and adaptation [8,9,10]. Indeed, studies showed that recombination plays an important role in the adaptation and symptomology of CpMMV in soybean [18,19]; however, other genetic forces driving the evolution of CpMMV are still elusive.
The genetic information is read by translational machinery in a triple-nucleotide manner from 5′ to 3′, each of which is called a genetic codon that is translated to a special amino acid or as a translation stop codon. Except for three stop codons, there are 61 codons for 20 natural amino acids, resulting in most amino acids being encoded by more than one codon. Codons for the same amino acid are known as synonymous codons. Synonymous codons are not used equally; instead, different species show different codon preferences. Even genes of the same species display differences in codon patterns. Codon usage bias can be affected by many factors, e.g., nucleotide composition, tRNA abundance, mutation bias, natural selection, gene length, and even expression level [20]. Viruses are obligate parasites that rely on host translation machinery for protein translation. As a result, viral codon patterns may be significantly affected by the hosts they are infecting. For instance, rubella virus (RUBV) and citrus tristeza virus (CTV) showed high codon adaptation to their respective hosts [21,22]. The increased nucleotide sequence data of CpMMV from different hosts in recent years allow us to gain detailed insight into its genetic properties, e.g., nucleotide composition, codon pattern, mutation bias, and the influence of host on these genetic properties. Our results showed that the host is an important factor driving the evolution of CpMMV by shaping its dinucleotide composition and codon pattern.
2. Materials and Methods
2.1. Data Availability
A total number of fifty-five full-length genome or the Coat protein (CP) sequences of CpMMV were retrieved from the National Center for Biotechnology Information (NCBI) GenBank nucleotide database. The database also includes the accession number, country of origin, isolate name, and host of each sequence—see Table 1. A total number of 122,218, 41,097, and 25,026 RefSeq mRNA sequences of soybean, cowpea, and papaya were downloaded from the NCBI GenBank nucleotide database.
2.2. Phylogenetic Analysis
A phylogenetic tree was constructed using the MEGA 11 software with the neighbor-joining (NJ) algorithm [23]. The nucleotide substitution model, mutation rate, and pattern were determined using the Model Selection function in MEGA 11. The bootstrapped confidence interval was based on 1000 replicates.
2.3. Calculation of the Relative Synonymous Codon Usage (RSCU)
RSCU is defined as the ratio of observed to expected codon frequency under equal codon usage without being affected by the amino acid compositions or the CDS sizes of different gene samples [24]. Synonymous codons with RSCU values lower than 1, equal to 1, and higher than 1 represent negative, no, and positive codon usage bias, respectively. Synonymous codons with RSCU values > 1.5 and <0.6 can be recognized as significantly overrepresented and underrepresented codons, respectively [25]. The RSCU of CpMMV was calculated with a homebuilt BioPython script. To calculate the RSCU values of host, open reading frames (ORFs) within mRNA sequences were extracted using a homemade BioPython script, which also removed any incomplete and erroneous sequences. The RSCU values of soybean, cowpea, and papaya were calculated by a BioPython script from a total number of 89,784, 41,014, and 25,430 full-length coding sequences, respectively. RSCU values of soybean leaf-specific and seed-specific genes were derived from [26], which were calculated from 58 and 21 highly expressed leaf-specific and seed-specific genes, respectively. All BioPython scripts are available upon request.
2.4. Nucleotide Composition Analysis
The ORFs in each CpMMV genome were retrieved manually and then were aligned by Clustal W 2.0 with default parameters [27]. Overall nucleotide composition (%A, %C, %U, and %G), nucleotide composition at the third codon position (%A3s, %C3s, %U3s, and %G3s), and mean frequencies of G + C nucleotide at the first, second, and third codon position (GC1s, GC2s, and GC3s) were calculated using the CodonW software version 1.4.2 with default parameters, which is available at http://codonw.sourceforge.net (accessed on 1 March 2022).
2.5. Analysis of Effective Number of Codons (ENC)
ENC is a simple and efficient method to quantify codon bias independent of gene length and amino acid composition [28]. The value of an ENC analysis ranges from 20 (when only one synonymous codon is used for the corresponding amino acid) to 61 (when all synonymous codons are used equally). The codon usage patterns were investigated by the NEC plot, a plot of NEC vs. GC3s at synonymous sites. In an ENC plot, the observed and expected ENC values are compared to determine the influence of structuring synonymous codon usage bias. An ENC plot is commonly used to determine the effect of G + C compositional constraints on codon usage bias. When the corresponding points fall near the expected curve, the mutation is the main force shaping codon usage. Alternatively, natural selection is the main force shaping codon usage when the corresponding points fall considerably below the expected curve.
2.6. Neutrality Plot Analysis
A neutrality plot was used to identify the effects of natural selection and mutation pressure on the codon pattern [29]. In a neutrality plot, the average GC content at GC12s was plotted against GC3s. A regression line was plotted between the GC3s-variable and the GC12s-variable. Unlike GC3s, GC1 and GC2 are subject to functional constraints, because a mutation at these positions usually leads to an amino acid change. As a result, the evolutionary speed of the mutation and natural selection pressure is expressed as the slope of the regression line, while the regression coefficient implies the mutation-selection equilibrium coefficient [30]. Theoretically, the mutation is recognized as the main force shaping the codon pattern when the slope of the regression line is close to 1, while natural selection is accepted as the main force when the slope of the regression line is close to 0.
2.7. Dinucleotide Odds Ratio
The dinucleotide odds ratio was calculated as described using a homebuilt BioPython script that is available upon request [31]. Based on the statistical theory, the dinucleotide can be recognized as significantly underrepresented if the ratio is equal or less than 0.78 and significantly overrepresented if the ratio is equal or higher than 1.23 [31].
3. Results
3.1. Phylogenetic Analysis
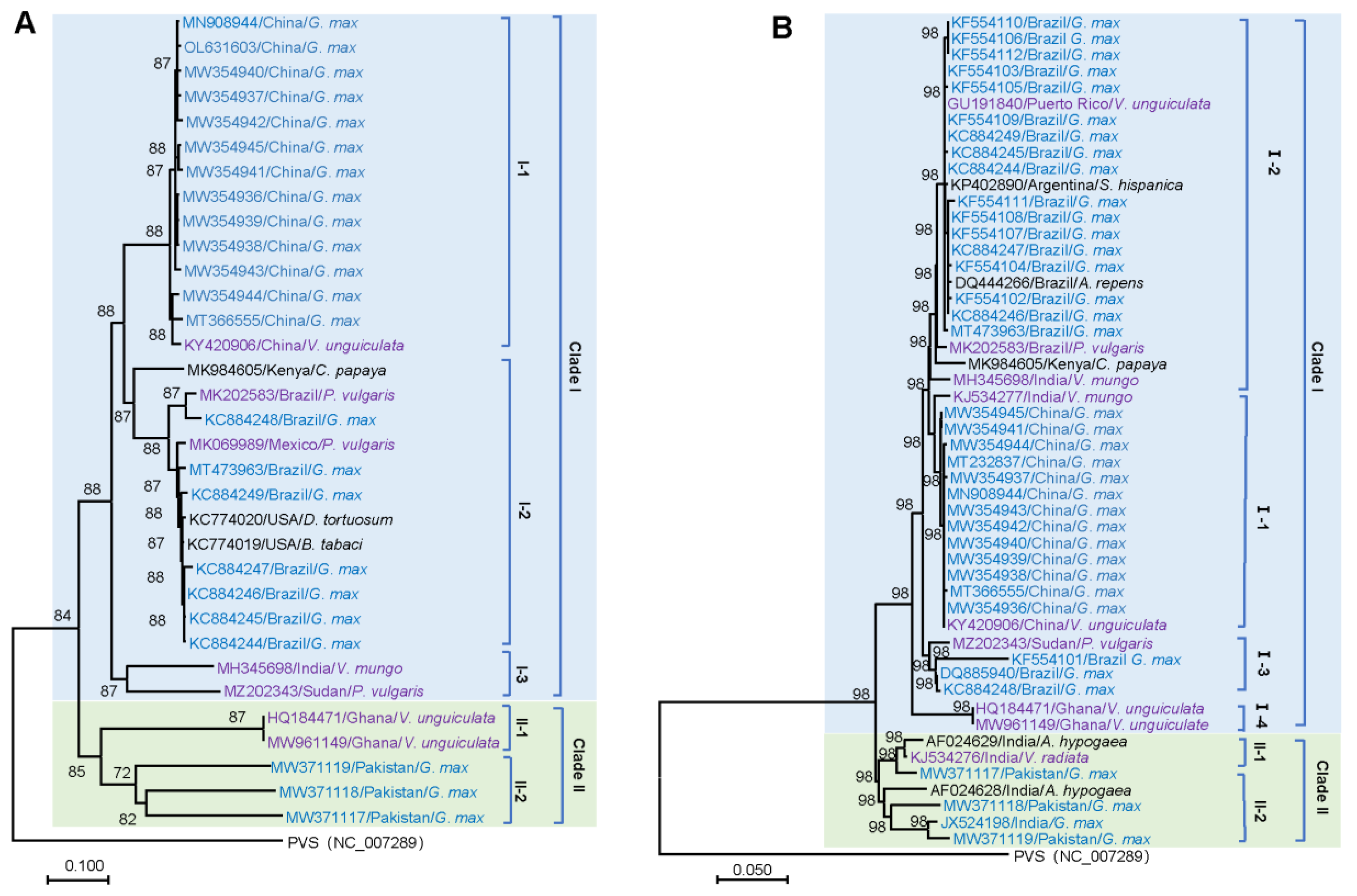
To gain insight into the phylogenetic relationship of CpMMV isolates, a neighbor-joining (NJ) phylogenetic tree was constructed using thirty-three full-length genomes of CpMMV (Table 1). Consistent with the previous study [32], the phylogenetic tree was apparently clustered into two major clades, namely Clade I and Clade II (Figure 1A). Clade I could be further separated into three subclades, namely subclades I-1, I-2, and I-3. Subclade I-1 contained thirteen isolates from China; subclade I-2 contained eleven isolates from America and a papaya isolate from Kenya (MK984605); subclade I-3 contained two isolates from India and Sudan, respectively. Clade II could be further divided into two subclades: subclade II-1 contained two isolates from Ghana; subclade II-2 contained three isolates from Pakistan. We further analyzed the phylogenetic relationship of the thirty-three CpMMV isolates based on hosts that were isolated. We found that isolates from soybean and cowpea were located in Clades I and II; isolates from other legumes and non-legume hosts were clustered in Clade I (Figure 1A).
Since recombination is prevalent in CpMMV isolates [17,18,19], which may mask the real phylogenetic relationship of these isolates; therefore, we constructed a phylogenetic tree based on the CP gene, which has a low recombination ratio. A total number of 161 sequences of the CP gene were retrieved from the GenBank database. For clarity, sequences with higher than 99% nucleotide sequence similarities were removed. A phylogenetic tree was then constructed using the rest 51 sequences of the CP gene (Figure 1B). Consistent with the NJ tree based on full-length genomes, the 51 sequences of the CP gene were also clustered into two major Clades; however, the two isolates of subclade II-1 in the NJ tree constructed based on the full-length genome was comprised of the fourth subclade in Clade I. Geographic distribution analysis showed that isolates from India display a high degree of diversity since they were present in Clade I and Clade II. In this CP-based NJ tree, isolates from soybean were distributed in Clades I and II, isolates from cowpea were located in Clade I, and isolates from other legumes and non-legume hosts were found in Clades I and II (Figure 1B).
3.2. Nucleotide Composition Analysis
To gain insight into the genetic forces driving the evolution of CpMMV, we first analyzed the nucleotide composition of the full-length genome of the thirty-three CpMMV isolates (excluding the poly-A tail). Results showed that the overall nucleotide composition of CpMMV was T and U-rich; U and C were the most abundant and infrequent bases, respectively (Table 2). We further calculated the nucleotide composition in the protein-coding regions of the thirty-three CpMMV isolates. The GC1 position had the highest G + C content in the CpMMV coding sequence (0.41), while the GC2 position had the lowest G + C content (0.40). Further analysis of the third wobble codon position revealed a significantly higher abundance of U3 (0.41) and A3 (0.35) than G3 (0.28) and C3 (0.24) (Table 2). These results suggest that CpMMV displays a codon pattern that prefers A/U-ending codons; and, the codon pattern of CpMMV may be affected by compositional constraints. CpMMV had an overall effective number of codons (ENC) value of 52.23, suggesting a moderate but highly conserved codon pattern bias of CpMMV. The variations of nucleotide composition and ENC among isolates of Clade I and Clade II or of hosts were ignorable (Table 2), indicating that different isolates have similar mutation bias and natural selection.
3.3. Natural Selection Is the Major Force Influencing the Codon Usage of CpMMV
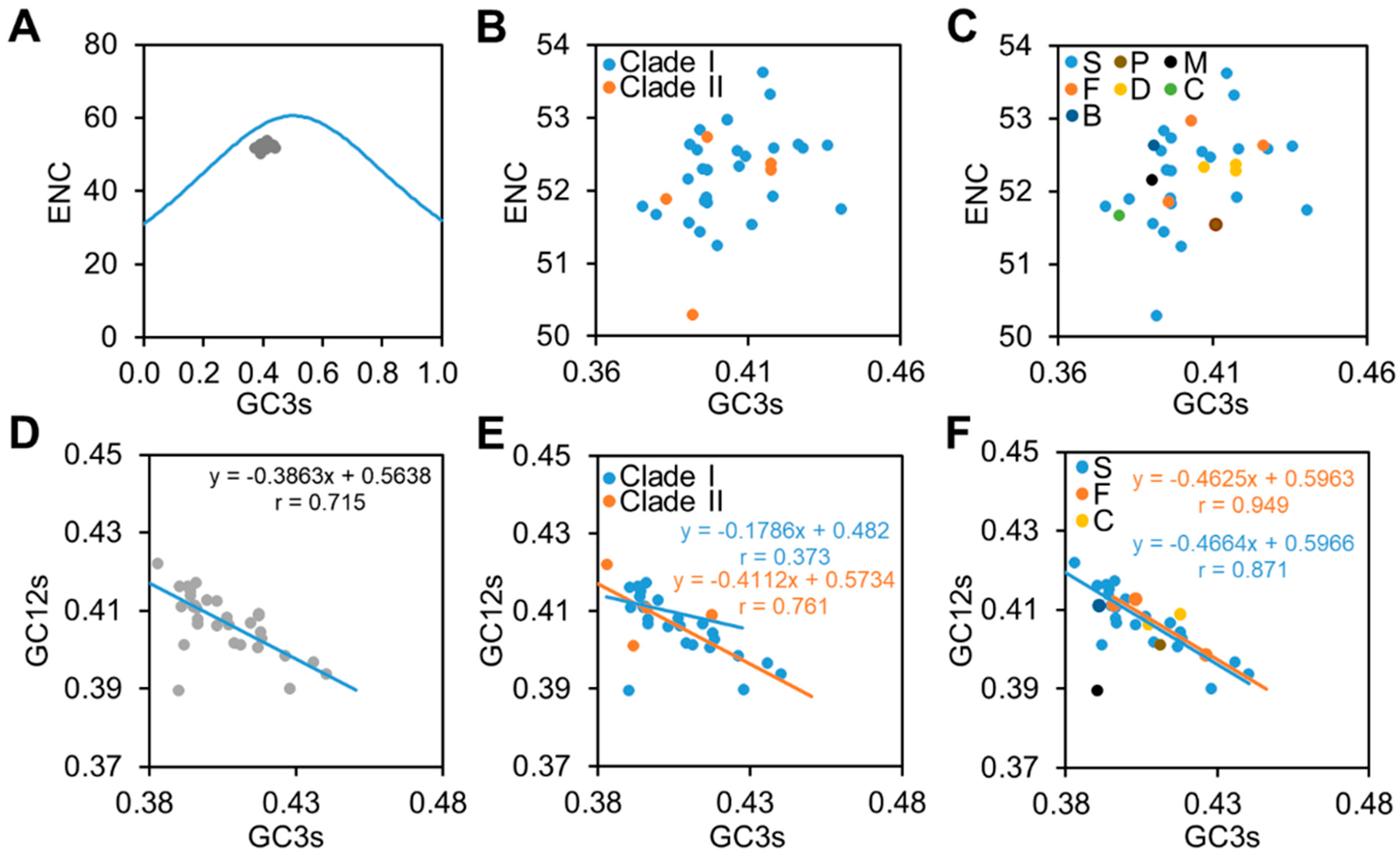
To evaluate the forces shaping the codon pattern patterns of CpMMV, an ENC plot and neutrality analysis were carried out. In the ENC plot, all CpMMV isolates were clustered together and located below the expected curve, suggesting that the codon pattern of CpMMV is subjected to GC compositional constraints (Figure 2A). A detailed survey showed that isolates of Clade I and Clade II of different hosts were distributed in the same area (Figure 2B,C), further confirming that different CpMMV isolates are subjected to similar GC compositional constraints. In the neutrality analysis (Figure 2D), average values of GC3s were negatively correlated with the average values of GC12s (r = 0.715; p < 0.01); however, the slope of the regression line was 0.3863, which suggests that mutation pressure accounts for approximately 38.63% of variations and natural selection accounts for 61.37% of variations. Neutrality analysis was also performed according to their phylogenetic relationships and hosts. Results showed that the average values of GC3s were also negatively correlated with the average values of GC12s (Figure 2E,F) and slopes values were closer to 0 than to 1. Thus, natural selection instead of mutation pressure plays a dominant role in shaping the codon pattern of CpMMV.
3.4. RSCU Patterns of CpMMV
To gain insight into the codon pattern of CpMMV, we calculated the overall RSCU values of the thirty-three CpMMV isolates (Table 3). Among the 59 synonymous codons, 9 and 12 were identified as significantly preferred codons (RSCU value > 1.5) and unpreferred codons (RSCU value < 0.6), respectively. An analysis of the base composition at the third codon showed that 7 out of 9 significant codons and 24 out of 27 preferred codons (RSCU value > 1.0) were A or U-ending, while 9 out the 12 significantly unpreferred codons were G or C-ending, suggesting that CpMMV prefers A/U-ending codons.
We further analyzed the RSCU of CpMMV based on their phylogenetic relationship. The average RSCU values of Clades I and II were very close to that of overall RSCU values (Table S1). Especially, the significantly preferred and unpreferred codons of Clades I and II were identical to that of the overall RSCU values (Figure 3A; Table S1). The RSCU values of the thirty-three CpMMV isolates according to their hosts were also calculated (Table S1). Noticeably, RSCU values of several synonymous codons, e.g., CCU, CCU, UGU, UGC, UUA, and UUG, showed high variations (Figure 3B). Specifically, CCA had the highest range (R = 0.734), followed by CCU (R = 0.689), UGU/UGC (R = 0.612), and UUA (R = 0.528) (Figure 3C). The extremum RSCU values were observed between soybean, cowpea, mungbean, and papaya-infecting CpMMV isolates, indicating that the codon pattern of CpMMV may be strongly affected by these hosts.
3.5. Dinucleotide Frequency Affects the Codon Pattern of CpMMV
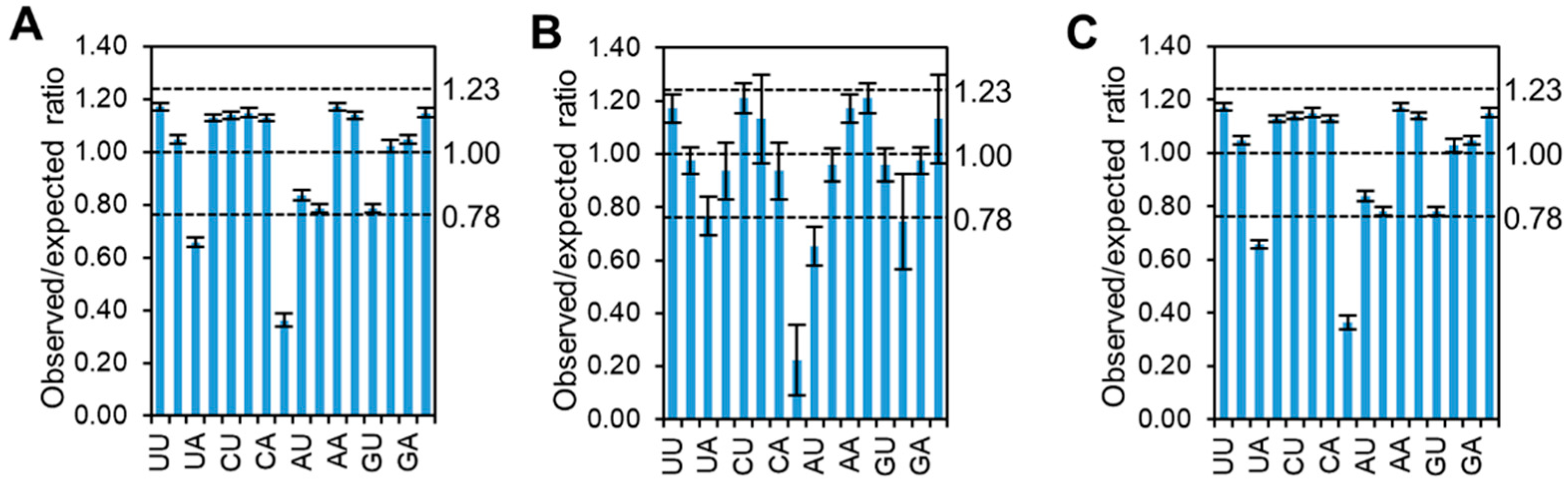
Since the codon pattern of CpMMV is subject to GC compositional constraint and previous studies showed that several pairs of dinucleotides in RNA viruses were strongly subjected to host selection [31,33], the dinucleotide frequency of CpMMV was analyzed to gain insight into the constraint of dinucleotide bias on the codon pattern. Results showed that CpG and UpA were significantly underrepresented (observed/expected ratio ≤ 0.78) in both the coding region and untranslated regions (UTRs), while no dinucleotide pairs were found to be significantly overrepresented (observed/expected ratio ≥ 1.23) (Figure 4), suggesting that the codon pattern of CpMMV may be affected by CpG and UpA dinucleotide bias. A direct comparison of the RSCU values of codons with an underrepresented dinucleotide showed that the codon pattern is highly affected by the dinucleotide frequency. For instance, four of the six synonymous Arg containing CpG on the first and second codon position (CGN; N indicates A, U, G, or C) were underrepresented (Table 2). Codons containing CpG on the second and third codon position (ACG (coding Thr), UCG (coding Ser), GCG (coding Ala), and CCG (coding Pro)) were also significantly unpreferred (Table 3). Although UpA is contained in two stop codons, we still could find the RSCU values of three out of the four codons containing UpA on the second and third codon position (AUA (coding Ile), GUA (coding Val), and CUA (coding Leu)) were lower than expected (Table 2).
3.6. Codon Adaptation Analysis
To further gain insight into the impact of the host on CpMMV codon pattern, the overall codon pattern of soybean, cowpea, and papaya were calculated using all available nuclear genes in the GenBank nucleotide database (mungbean was not included due to data availability; Table S2). Corresponding analyses were then performed between RSCU values of CpMMV isolates from soybean (CpMMV-soybean), CpMMV isolates from cowpea (CpMMV-cowpea), or CpMMV isolates from papaya (CpMMV-papaya) and that of soybean, cowpea, and papaya. As expected, the codon pattern of CpMMV-cowpea was most adapted to cowpea according to the correlation coefficient (Figure 5A–C). Interestingly, the codon patterns of CpMMV-papaya and CpMMV-soybean also displayed the highest adaptation to that of cowpea but not papaya or soybean (Figure 5D–I), indicating that cowpea might be the original host of CpMMV. Compared to soybean, the codon pattern of CpMMV-papaya was more similar to that of papaya (Figure 5D,E), suggesting that the infection of papaya causes the readaptation of the codon pattern from cowpea to papaya. The codon pattern of CpMMV-soybean displayed a similar correlation coefficient to the codon pattern of soybean and papaya (Figure 5G–I), implying that papaya and soybean have similar codon usage or CpMMV-soybean possibly originated from papaya.
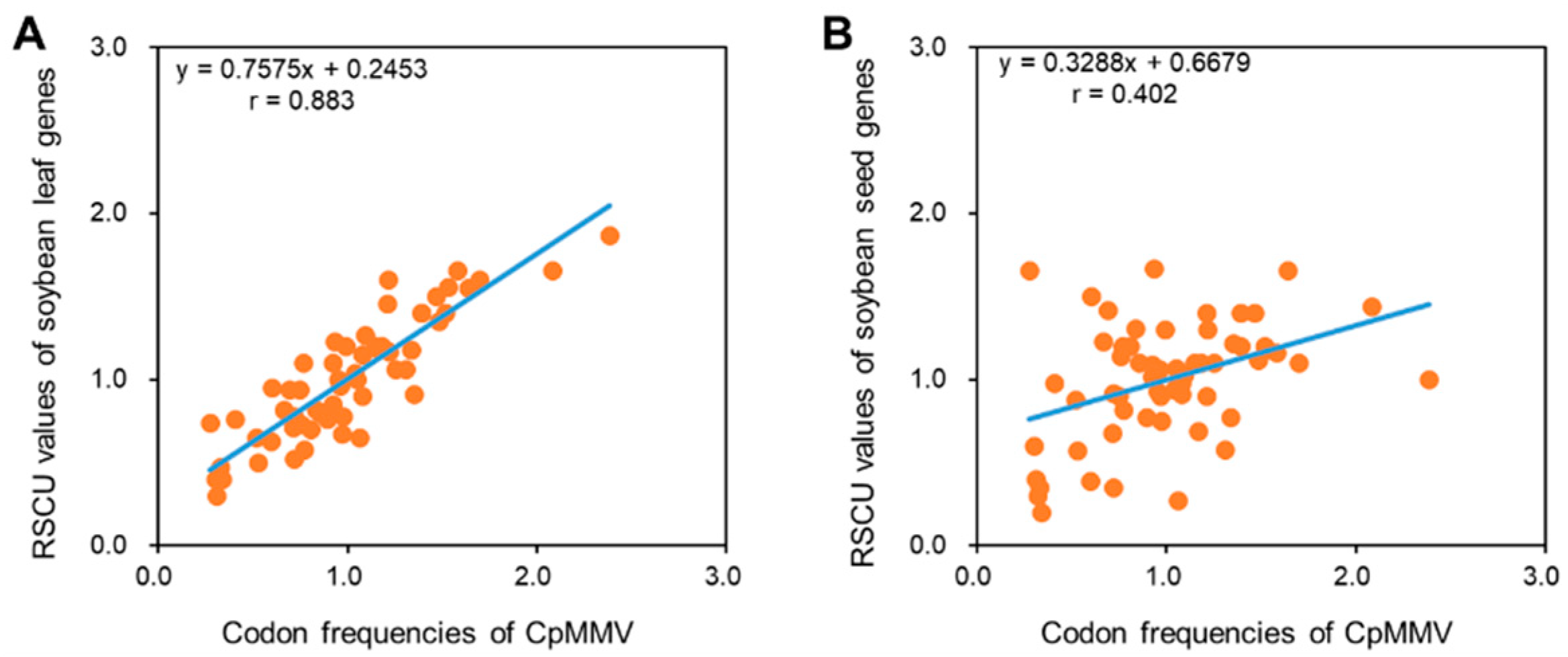
To further gain insight into the codon adaptation, we compared the codon pattern of CpMMV with that of soybean leaf and seed. It has been reported that the codon usage of genes that were highly expressed in leaf tissues dramatically differed from that of genes expressed in seed [26]; therefore, it is expected that the codon usage of CpMMV is closer to that of leaf-specific genes than seed-specific genes since CpMMV is a phloem-limited virus. As expected, we found that the codon usage of CpMMV is highly correlated to that of leaf-specific genes (r = 0.883; p < 0.01), but not seed-specific genes (r = 0.402; p > 0.05) (Figure 5); thus, the codon pattern of CpMMV is highly adapted to the host it infects.
4. Discussion
In this study, possible forces other than recombination that drive the evolution of CpMMV were analyzed using phylogeny, nucleotide composition, codon pattern, neutrality and ENC plot, dinucleotide bias, and host adaptation. Phylogenetic analyses showed that isolates from Africa and Southwest Asia were present in Clades I and II; isolates from East Asia and America were distributed only in Clade I (Figure 1), suggesting that CpMMV possibly originated from Africa and/or Southwest Asia and was introduced to East Asia and America through the international trade of agricultural products or along with the spread of its vector: silverleaf whitefly (Bemisia tabaci MEAM1). The availability of more sequences from other regions of the world could confirm or rule out this hypothesis. Codon adaptation analysis supported the fact that cowpea is the original host of CpMMV, while papaya may be an important intermediate host of CpMMV. Nevertheless, further studies are needed to confirm these possibilities.
Nucleotide composition, codon pattern, and ENC plot neutrality analysis showed that CpMMV had a codon pattern that prefers A/U-ending codons. Neutrality analysis showed that the RSCU of CpMMV is subjected to GC constraint and that natural selection instead of mutation pressure plays a dominant role in shaping the codon pattern of CpMMV. Dinucleotide frequency analysis showed that CpG and UpA were significantly underrepresented in both the untranslational regions and coding regions of CpMMV (Figure 4). Interestingly, codons with underrepresented dinucleotides were always significantly unpreferred (Table 2), suggesting that the RSCU of CpMMV is heavily affected by dinucleotide bias, especially CpG and UpA. The dinucleotide pattern is a stable genomic signature, which is related to genome compartmentalization, gene evolution, and gene expression regulation [34]. Studies showed that UpA is underrepresented in almost all organisms tested, whereas CpG is differentially represented in the genomes of eukaryotic organisms [35]. It was proposed that the TpA depletion may be caused by its presence in two out of three stop codons and in transcriptional regulatory motifs (e.g., the TATA box sequence), while CpG repression has been directly linked to cytosine methylation in DNA [36]. The dinucleotides CpG and UpA are also significantly underrepresented in the genome of retroviruses and riboviruses [31,33,37]. Importantly, increasing or decreasing the CpG and UpA dinucleotide frequencies in the genome could significantly attenuate or increase viral proliferation, respectively [38,39,40]. Recently, it was found that the attenuation of viruses with elevated frequencies of CpG and UpA dinucleotides in animals was associated with the ZAP and OAS3/RNAseL-mediated antiviral pathways, respectively [41,42]. How CpG and UpA affect the codon pattern of plant RNA viruses such as CpMMV is an interesting question for investigation in the future since neither ZAP nor OAS3/RNAseL counterparts have been identified in plants.
It has been proposed that the translation of mRNA with rare codons is less efficient than those with optimal codons since the latter have more cognate tRNA species [43]. As a result, a higher translation efficiency certainly will benefit the proliferation of viruses. Indeed, studies showed that the codon pattern of many viruses is highly adapted to their hosts [21,44,45]. Our results also showed that the codon pattern of CpMMV was overall highly adapted to its hosts, e.g., cowpea, papaya, and soybean. Moreover, our results showed that the codon pattern of soybean-isolated CpMMV and papaya-isolated CpMMV was closer to that of soybean and papaya, respectively (Figure 5), which further confirmed the impact of the host on the codon pattern of CpMMV. Moreover, the codon usage of CpMMV resembles the codon usage of leaf-specific genes but not that of seed-specific genes (Figure 6). Thus, mimicking the host codon pattern for maximum translation efficiency is also an important force shaping the codon pattern of CpMMV.
5. Conclusions
The synonymous codon pattern of CpMMV is shaped by the host it infects, possibly through pressures from dinucleotide bias and translational machinery.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/pathogens11040419/s1, Table S1: RSCU values of CpMMV; Table S2: RSCU values of hosts.
Author Contributions
Conceptualization, X.W. (Xiaoxia Wu) and X.C.; data acquirement: S.Y.; data analysis S.Y., Y.L. and X.W. (Xiaoyun Wu); supervision, X.W. (Xiaoxia Wu) and X.C.; original draft, S.Y.; review and editing, X.W. (Xiaoxia Wu), X.W. (Xiaoyun Wu) and X.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work is financially supported by the Natural Science Foundation of Heilongjiang Province (Grant No. LH2019C027), the Program for the Scientific Activities of Selected Returned Overseas Professionals in Heilongjiang Province (Grant No. 2018QD0002), and the “Young Talents” Project of Northeast Agricultural University (Grant No. 18XG04).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jeyanandarajah, P.; Brunt, A.A. The natural occurrence, transmission, properties and possible affinities of cowpea mild mottle virus. J. Phytopathol. 1993, 137, 148–156. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Xiang, H.; Abouelnasr, H.; Li, D.; Yu, J.; McBeath, J.H.; Han, C. Discovery and characterization of a novel carlavirus infecting potatoes in China. PLoS ONE 2013, 8, e69255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senshu, H.; Yamaji, Y.; Minato, N.; Shiraishi, T.; Maejima, K.; Hashimoto, M.; Miura, C.; Neriya, Y.; Namba, S. A dual strategy for the suppression of host antiviral silencing: Two distinct suppressors for viral replication and viral movement encoded by Potato virus M. J. Virol. 2011, 85, 10269–10278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Komatsu, K.; Ayukawa, Y.; Matsuo, Y.; Hashimoto, M.; Netsu, O.; Teraoka, T.; Yamaji, Y.; Namba, S.; Arie, T. N-terminal region of cysteine-rich protein (CRP) in carlaviruses is involved in the determination of symptom types. Mol. Plant Pathol. 2018, 19, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, L.G.; Carvalho, C.M. Cowpea mild mottle virus (Carlavirus, Betaflexiviridae): A review. Trop. Plant Pathol. 2017, 42, 417–430. [Google Scholar] [CrossRef]
- Brunt, A.A.; Kenten, R.H. Cowpea mild mottle, a newly recognized virus infecting cowpeas (Vigna unguiculata) in Ghana. Ann. Appl. Biol. 1973, 74, 67–74. [Google Scholar] [CrossRef]
- Iwaki, M.; Thongeearkom, P.; Prommin, M.; Honda, Y.; Hibi, T. Whitefly transmission and some properties of cowpea mild mottle virus on soybean in Thailand. Plant Dis. 1982, 66, 365–368. [Google Scholar] [CrossRef]
- Iizuka, N.; Rajeshwari, R.; Reddy, D.V.R.; Goto, T.; Muniyappa, V.; Bharathan, N.; Ghanekar, A.M. Natural occurrence of a strain of cowpea mild mottle virus on groundnut (Arachis hypogaea) in India. J. Phytopathol. 1984, 109, 245–253. [Google Scholar] [CrossRef]
- Antignus, Y.; Cohen, S. Purification and some properties of a new strain of cowpea mild mottle virus in Israel. Ann. Appl. Biol. 1987, 110, 563–569. [Google Scholar] [CrossRef]
- Mansour, A.; Al-Musa, A.; Vetten, H.J.; Lesemann, D.E. Properties of a cowpea mild mottle virus (CpMMV) isolate from eggplant in Jordan and evidence for biological and serological differences between CPMMV isolates from Leguminons and Solanaceous hosts. J. Phytopathol. 1998, 146, 539–547. [Google Scholar] [CrossRef]
- Pardina, P.E.R.; Arneodo, J.D.; Truol, G.A.; Herrera, P.S.; Laguna, I.G. First report of cowpea mild mottle virus in bean crops in Argentina. Australas. Plant Pathol. 2004, 33, 129–130. [Google Scholar]
- Brito, M.; Fernández-Rodríguez, T.; Garrido, M.J.; Mejías, A.; Romano, M.; Marys, E. First report of cowpea mild mottle carlavirus on yardlong bean (Vigna unguiculata subsp. sesquipedalis) in Venezuela. Viruses 2012, 4, 3804–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.A.; Chien, L.Y.; Tsai, C.F.; Lin, Y.Y.; Cheng, Y.H. First report of cowpea mild mottle virus in cowpea and french bean in Taiwan. Plant Dis. 2013, 97, 1001. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, L.G.; Silva, F.N.; Lima, A.T.M.; Milanesi, D.F.; Castilho-Urquiza, G.P.; Almeida, A.M.R.; Zerbini, F.M.; Carvalho, C.M. Molecular variability of cowpea mild mottle virus infecting soybean in Brazil. Arch. Virol. 2014, 159, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Lamas, N.S.; Matos, V.O.R.L.; Alves-Freitas, D.M.T.; Melo, F.L.; Costa, A.F.; Faria, J.C.; Ribeiro, S.G. Occurrence of cowpea mild mottle virus in common bean and associated weeds in Northeastern Brazil. Plant Dis. 2017, 101, 1828. [Google Scholar] [CrossRef]
- Wei, Z.Y.; Wu, G.W.; Ye, Z.X.; Jiang, C.; Mao, C.Y.; Zhang, H.H.; Miao, R.P.; Yan, F.; Li, J.M.; Chen, J.P.; et al. First report of cowpea mild mottle virus infecting soybean in China. Plant Dis. 2020, 104, 2534. [Google Scholar] [CrossRef]
- Wei, Z.; Mao, C.; Jiang, C.; Zhang, H.; Chen, J.; Sun, Z. Identification of a new genetic clade of cowpea mild mottle virus and characterization of its interaction with soybean mosaic virus in co-infected soybean. Front. Microbiol. 2021, 12, 650773. [Google Scholar] [CrossRef]
- Zanardo, L.G.; Silva, F.N.; Bicalho, A.A.C.; Urquiza, G.P.C.; Lima, A.T.M.; Almeida, A.M.R.; Zerbini, F.M.; Carvalho, C.M. Molecular and biological characterization of cowpea mild mottle virus isolates infecting soybean in Brazil and evidence of recombination. Plant Pathol. 2014, 63, 456–465. [Google Scholar] [CrossRef]
- Zanardo, L.; Trindade, T.; Mar, T.; Barbosa, T.; Milanesi, D.; Alves, M.; Lima, R.; Zerbini, F.; Janssen, A.; Mizubuti, E.; et al. Experimental evolution of cowpea mild mottle virus reveals recombination-driven reduction in virulence accompanied by increases in diversity and viral fitness. Virus Res. 2021, 303, 198389. [Google Scholar] [CrossRef]
- McInerney, J.O. GCUA: General codon usage analysis. Bioinformatics 1998, 14, 372–373. [Google Scholar] [CrossRef]
- Cheng, X.F.; Wu, X.Y.; Wang, H.; Sun, Y.; Qian, Y.; Luo, L. High codon adaptation in citrus tristeza virus to its citrus host. Virol. J. 2012, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chen, X.; Ushijima, H.; Frey, T.K. Analysis of base and codon usage by rubella virus. Arch. Virol. 2012, 157, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.M.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L.M. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, Y.; Luo, L.; Wang, Y.P.; Dong, Z.M.; Sun, S.H.; Qiu, L.J. Analysis of nuclear gene codon bias on soybean genome and transcriptome. Zuo Wu Xue Bao 2011, 37, 965–974. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G + C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Cheng, X.F.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG usage in RNA viruses: Data and hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Cheng, S.; Li, S.; Gong, Y.; Zhang, Z.; Lu, Q.; Hu, R.; Liu, Y.; Zhang, D. Genomic sequences measure and molecular characteristics of cowpea mild mottle virus of Hainan isolate. China Veg. 2019, 6, 35–38. [Google Scholar]
- Rima, B.K.; McFerran, N.V. Dinucleotide and stop codon frequencies in single-stranded RNA viruses. J. Gen. Virol. 1997, 78, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Grover, A.; Kahl, G. Mining microsatellites in eukaryotic genomes. Trends Biotechnol. 2007, 25, 490–498. [Google Scholar] [CrossRef]
- Karlin, S.; Burge, C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995, 11, 283–290. [Google Scholar] [PubMed]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- González de Prádena, A.; Sánchez Jimenez, A.; San León, D.; Simmonds, P.; García, J.A.; Valli, A.A. Plant virus genome is shaped by specific dinucleotide restrictions that influence viral infection. mBio 2020, 11, e02818–e02819. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Fros, J.; Bertran, A.; Sechan, F.; Odon, V.; Torrance, L.; Kormelink, R.; Simmonds, P. A functional investigation of the suppression of CpG and UpA dinucleotide frequencies in plant RNA virus genomes. Sci. Rep. 2019, 9, 18359. [Google Scholar] [CrossRef]
- Tulloch, F.; Atkinson, N.J.; Evans, D.J.; Ryan, M.D.; Simmonds, P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 2014, 3, e04531. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Fros, J.J.; Goonawardane, N.; Dietrich, I.; Ibrahim, A.; Alshaikhahmed, K.; Nguyen, D.; Simmonds, P. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res. 2019, 47, 8061–8083. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gan, H.; Liang, X. Analysis of synonymous codon usage bias in potato virus M and its adaption to hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Phylogeny of CpMMV. (A) Neighbor-joining phylogenetic tree based on full-genome sequences of CpMMV. (B) Neighbor-joining phylogenetic tree of CpMMV based on sequences of CP gene. Numbers on branches indicate the percentage support values of 1000 bootstrap replicates. The GenBank accession number, origin, and host of each sequence are indicated. Isolates from soybean, other legumes, and non-legumes are in blue, purple, and black, respectively. Clades I and II are also highlighted in light blue and light green backgrounds, respectively.
Figure 1.
Phylogeny of CpMMV. (A) Neighbor-joining phylogenetic tree based on full-genome sequences of CpMMV. (B) Neighbor-joining phylogenetic tree of CpMMV based on sequences of CP gene. Numbers on branches indicate the percentage support values of 1000 bootstrap replicates. The GenBank accession number, origin, and host of each sequence are indicated. Isolates from soybean, other legumes, and non-legumes are in blue, purple, and black, respectively. Clades I and II are also highlighted in light blue and light green backgrounds, respectively.

Figure 2.
ENC plot and neutrality analysis. (A) ENC plot of the thirty-three CpMMV isolates. Blue curve indicates the expected curve when all codons are used randomly (no selection). (B,C) An enlarged image of the ENC plot to show the distribution of CpMMV isolates according to their phylogenetic relationships (B) or hosts (C). (D–F) Correlation between nucleotide composition GC12 and GC3 of all thirty-three isolates (D) or according to their phylogenetic relationships (E) or hosts (F). S, P, M, F, D, C, and B represent soybean, papaya, mungbean, French bean, Florida beggarweed (D. tortuosum), cowpea, and whitefly, respectively.
Figure 2.
ENC plot and neutrality analysis. (A) ENC plot of the thirty-three CpMMV isolates. Blue curve indicates the expected curve when all codons are used randomly (no selection). (B,C) An enlarged image of the ENC plot to show the distribution of CpMMV isolates according to their phylogenetic relationships (B) or hosts (C). (D–F) Correlation between nucleotide composition GC12 and GC3 of all thirty-three isolates (D) or according to their phylogenetic relationships (E) or hosts (F). S, P, M, F, D, C, and B represent soybean, papaya, mungbean, French bean, Florida beggarweed (D. tortuosum), cowpea, and whitefly, respectively.

Figure 3.
Graphic illustration the variations of CpMMV synonymous codon usage. (A) The average RSCU values of CpMMV isolates of Clades I and II. (B) The average RSCU values of CpMMV isolates of seven hosts. (C) The range of RSCU values of CpMMV isolates of seven hosts.
Figure 3.
Graphic illustration the variations of CpMMV synonymous codon usage. (A) The average RSCU values of CpMMV isolates of Clades I and II. (B) The average RSCU values of CpMMV isolates of seven hosts. (C) The range of RSCU values of CpMMV isolates of seven hosts.

Figure 4.
Histogram showing the dinucleotide pattern of CpMMV. (A) Overall dinucleotide pattern of CpMMV. (B) Dinucleotide pattern of untranslated regions of CpMMV. (C) Dinucleotide pattern of coding region of CpMMV.
Figure 4.
Histogram showing the dinucleotide pattern of CpMMV. (A) Overall dinucleotide pattern of CpMMV. (B) Dinucleotide pattern of untranslated regions of CpMMV. (C) Dinucleotide pattern of coding region of CpMMV.

Figure 5.
Correlations of the codon abundances of CpMMV and soybean, cowpea, and papaya. (A–C) RSCU values of cowpea-infecting CpMMV versus overall RSCU values of cowpea (A), soybean (B), and papaya (C). (D–F) RSCU values of papaya-infecting CpMMV versus RSCU values of papaya (D), soybean (E), and cowpea (F). (G–I) RSCU values of soybean-infecting CpMMV versus RSCU values of soybean (G), papaya (H), and cowpea (I).
Figure 5.
Correlations of the codon abundances of CpMMV and soybean, cowpea, and papaya. (A–C) RSCU values of cowpea-infecting CpMMV versus overall RSCU values of cowpea (A), soybean (B), and papaya (C). (D–F) RSCU values of papaya-infecting CpMMV versus RSCU values of papaya (D), soybean (E), and cowpea (F). (G–I) RSCU values of soybean-infecting CpMMV versus RSCU values of soybean (G), papaya (H), and cowpea (I).

Figure 6.
The codon pattern of CpMMV is adapted to leaf-specific genes of soybean. (A,B) RSCU values of CpMMV versus RSCU values of leaf-specific genes of soybean (A) and seed-specific genes of soybean (B).
Figure 6.
The codon pattern of CpMMV is adapted to leaf-specific genes of soybean. (A,B) RSCU values of CpMMV versus RSCU values of leaf-specific genes of soybean (A) and seed-specific genes of soybean (B).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The information of CpMMV isolates used in this study.
| No. | GenBank Accession No | Country | Isolate Name | Host |
|---|---|---|---|---|
| 1 | AF024628 | India | CPMMV-H | Arachis hypogaea |
| 2 | AF024629 | India | CPMMV-M | A. hypogaea |
| 3 | DQ444266 | Brazil | Arachis | A. repens |
| 4 | DQ885940 | Brazil | Barreiras | Glycine max |
| 5 | GU191840 | Puerto Rico | CpMMVPR | Vigna unguiculata |
| 6 | HQ184471 | Ghana | Ghana | V. unguiculata |
| 7 | JX524198 | India | D1 | G. max |
| 8 | KC774019 | USA | Whiteflies_2007 | Bemisia tabaci |
| 9 | KC774020 | USA | Florida_Bean_2011 | Desmodium tortuosum |
| 10 | KC884244 | Brazil | CPMMV:BR:MG:09:2 | G. max |
| 11 | KC884245 | Brazil | CPMMV:BR:MG:09:3 | G. max |
| 12 | KC884246 | Brazil | CPMMV:BR:MT:02:1 | G. max |
| 13 | KC884247 | Brazil | CPMMV:BR:BA:02 | G. max |
| 14 | KC884248 | Brazil | CPMMV:BR:GO:01:1 | G. max |
| 15 | KC884249 | Brazil | CPMMV:BR:GO:10:5 | G. max |
| 16 | KF554101 | Brazil | BR:GO:10:4 | G. max |
| 17 | KF554102 | Brazil | BR:MA:02 | G. max |
| 18 | KF554103 | Brazil | CPMMV:BR:MG:09:1 | G. max |
| 19 | KF554104 | Brazil | CPMMV:BR:MG:09:4 | G. max |
| 20 | KF554105 | Brazil | CPMMV:BR:MG:09:5 | G. max |
| 21 | KF554106 | Brazil | CPMMV:BR:MG:09:6 | G. max |
| 22 | KF554107 | Brazil | BR:MG:09:7 | G. max |
| 23 | KF554108 | Brazil | BR:MG:09:11 | G. max |
| 24 | KF554109 | Brazil | BR:MG:09:12 | G. max |
| 25 | KF554110 | Brazil | BR:MG:09:15 | G. max |
| 26 | KF554111 | Brazil | BR:MG:09:16 | G. max |
| 27 | KF554112 | Brazil | BR:PA:02 | G. max |
| 28 | KJ534276 | India | MUNGBEAN1 | V. radiata |
| 29 | KJ534277 | India | URDBEAN1 | V. mungo |
| 30 | KP402890 | Argentina | MA01 | Salvia hispanica |
| 31 | KY420906 | China | Hainan1 | V. unguiculata |
| 32 | MH345698 | India | CpMMV-Urd-Kanpur | V. mungo |
| 33 | MK069989 | Mexico | CN2 | Phaseolus vulgaris |
| 34 | MK202583 | Brazil | CPMMV:BR:GO:14 | P. vulgaris |
| 35 | MK984605 | Kenya | KE-Kit_01 | Carica papaya |
| 36 | MN908944 | China | Anhui_SZ_DN1383 | G. max |
| 37 | MT232837 | China | cpmmv-anhui-sz | G. max |
| 38 | MT366555 | China | CPMMV-JS | G. max |
| 39 | MT473963 | Brazil | Casa Branca_BR | G. max |
| 40 | MW354936 | China | CPMMV_HN_LH | G. max |
| 41 | MW354937 | China | CPMMV_SD_JX | G. max |
| 42 | MW354938 | China | CPMMV_HN_XC | G. max |
| 43 | MW354939 | China | CPMMV_HB_JZ | G. max |
| 44 | MW354940 | China | CPMMV_HN_SQ | G. max |
| 45 | MW354941 | China | CPMMV_SD_JN | G. max |
| 46 | MW354942 | China | CPMMV_JL_GZL | G. max |
| 47 | MW354943 | China | CPMMV_JL_CC | G. max |
| 48 | MW354944 | China | CPMMV_JS_NJ | G. max |
| 49 | MW354945 | China | CPMMV_AH_FY | G. max |
| 50 | MW371117 | Pakistan | PK1 | G. max |
| 51 | MW371118 | Pakistan | PK2 | G. max |
| 52 | MW371119 | Pakistan | PK3 | G. max |
| 53 | MW961149 | Ghana | DSMZ PV-0090 | V. unguiculate |
| 54 | MZ202343 | Sudan | DSMZ PV-0907 | P. vulgaris |
| 55 | OL631603 | China | AH-FY | G. max |
Table 2.
The nucleotide composition and properties of CDS of the CpMMV genomes.
| Catalogs | Overall | Clade-I | Clade-II | Soybean | Cowpea | French Bean | Mung Bean | Papaya |
|---|---|---|---|---|---|---|---|---|
| A | 0.29 | 0.29 | 0.30 | 0.29 | 0.30 | 0.29 | 0.30 | 0.30 |
| U | 0.30 | 0.30 | 0.29 | 0.30 | 0.29 | 0.30 | 0.30 | 0.31 |
| C | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.17 |
| G | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.22 |
| GC | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.40 | 0.39 |
| GC1 | 0.41 | 0.41 | 0.42 | 0.42 | 0.41 | 0.42 | 0.39 | 0.39 |
| GC2 | 0.40 | 0.40 | 0.40 | 0.40 | 0.41 | 0.39 | 0.41 | 0.39 |
| GC12 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.40 | 0.39 |
| GC3 | 0.40 | 0.40 | 0.40 | 0.40 | 0.41 | 0.41 | 0.41 | 0.39 |
| A3 | 0.35 | 0.34 | 0.36 | 0.35 | 0.35 | 0.34 | 0.34 | 0.36 |
| U3 | 0.41 | 0.41 | 0.40 | 0.41 | 0.39 | 0.41 | 0.41 | 0.41 |
| C3 | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 | 0.25 | 0.24 | 0.22 |
| G3 | 0.28 | 0.28 | 0.27 | 0.28 | 0.28 | 0.28 | 0.29 | 0.28 |
| ENC | 52.23 | 52.28 | 51.91 | 52.22 | 52.33 | 52.49 | 51.54 | 52.16 |
Table 3.
Relative synonymous codon usage (RSCU) values of CpMMV.
| AA | Codon | Overall | AA | Codon | Overall | AA | Codon | Overall |
|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 1.27 | Pro | CCU | 1.68 | Glu | GAA | 1.00 |
| UUC | 0.73 | CCC | 0.75 | GAG | 1.00 | |||
| Leu | UUA | 1.11 | CCA | 1.31 | Arg | CGU | 0.52 | |
| UUG | 1.81 | CCG | 0.26 | CGC | 0.29 | |||
| CUU | 1.16 | Thr | ACU | 1.71 | CGA | 0.65 | ||
| CUC | 0.53 | ACC | 0.94 | CGG | 0.17 | |||
| CUA | 0.55 | ACA | 1.16 | AGA | 2.12 | |||
| CUG | 0.83 | ACG | 0.20 | AGG | 2.25 | |||
| Val | GUU | 1.72 | Gly | GGU | 1.36 | Cys | UGU | 1.24 |
| GUC | 0.72 | GGC | 0.79 | UGC | 0.76 | |||
| GUA | 0.57 | GGA | 0.85 | Tyr | UAU | 1.04 | ||
| GUG | 0.99 | GGG | 1.00 | UAC | 0.96 | |||
| Ser | UCU | 1.39 | Gln | CAA | 1.37 | Ala | GCU | 2.08 |
| UCC | 0.83 | CAG | 0.63 | GCC | 0.83 | |||
| UCA | 1.17 | His | CAU | 1.02 | GCA | 0.86 | ||
| UCG | 0.13 | CAC | 0.98 | GCG | 0.24 | |||
| AGU | 1.58 | Asn | AAU | 1.46 | Ile | AUU | 1.52 | |
| AGC | 0.91 | AAC | 0.54 | AUC | 0.67 | |||
| Lys | AAA | 1.11 | Asp | GAU | 1.45 | AUA | 0.81 | |
| AAG | 0.89 | GAC | 0.55 |
Note: Significantly preferred codons (RSCU value > 1.5) and unpreferred codons (RSCU value < 0.6) are in bold and underlined, respectively.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, S.; Liu, Y.; Wu, X.; Cheng, X.; Wu, X. Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution. Pathogens 2022, 11, 419. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11040419
AMA Style
Yang S, Liu Y, Wu X, Cheng X, Wu X. Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution. Pathogens. 2022; 11(4):419. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11040419
Chicago/Turabian StyleYang, Siqi, Ye Liu, Xiaoyun Wu, Xiaofei Cheng, and Xiaoxia Wu. 2022. "Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution" Pathogens 11, no. 4: 419. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11040419
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.