
Role of Diversity and Recombination in the Emergence of Chilli Leaf Curl Virus


 ,
,  ,
,
Abstract
:1. Introduction
2. Results
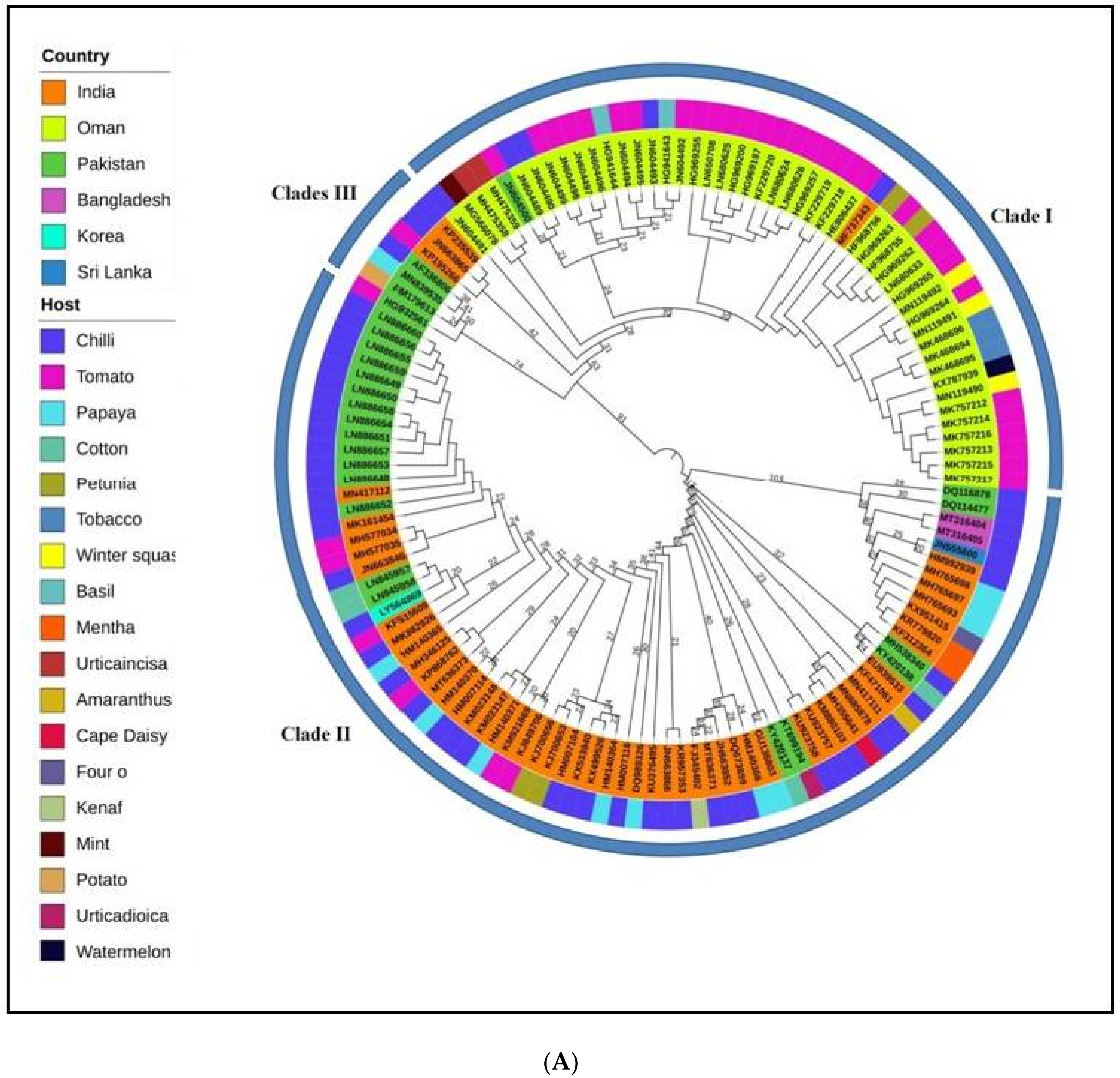
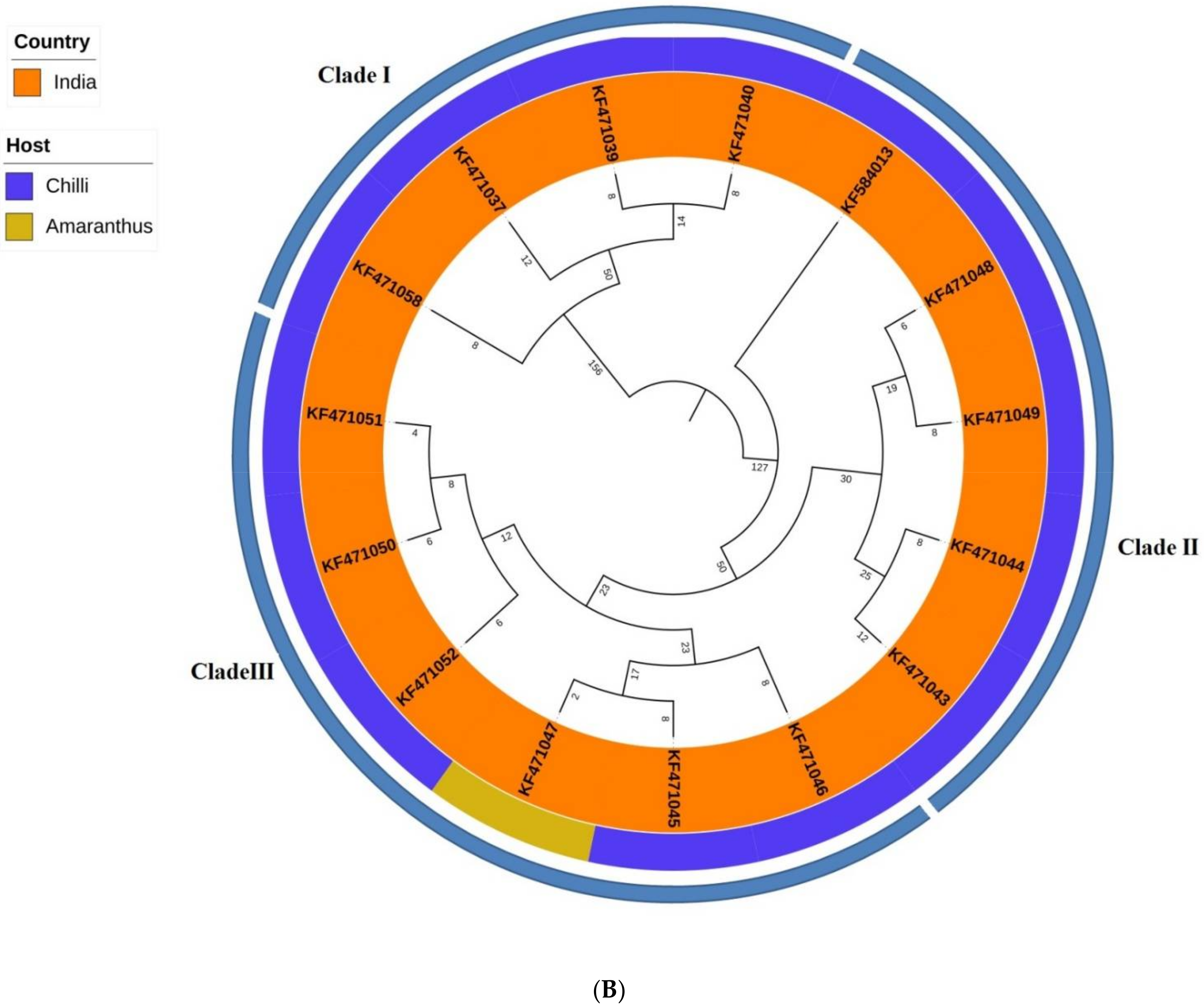
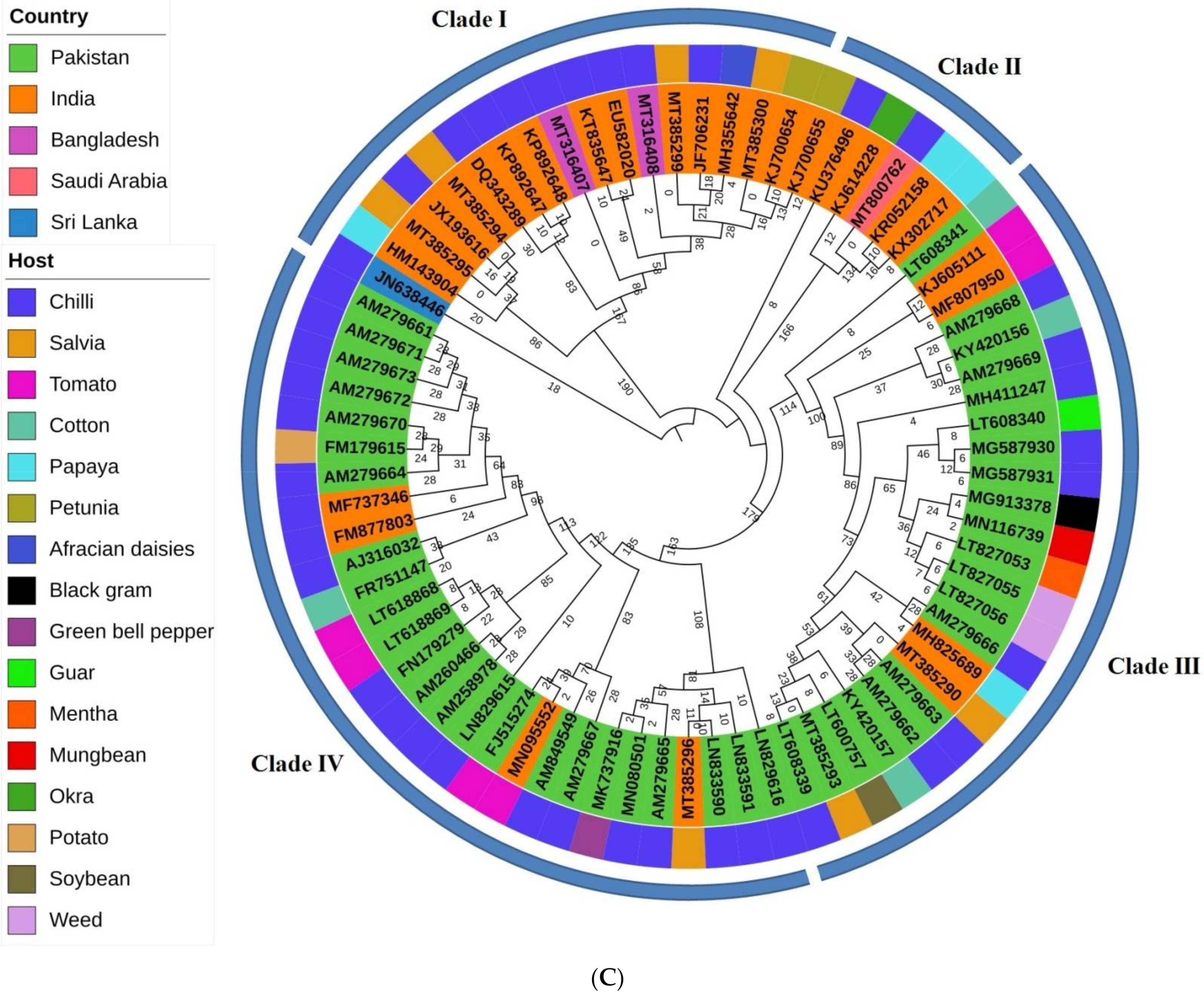
2.1. Phylogenetics and Estimation of Nucleotide Substitution Rates
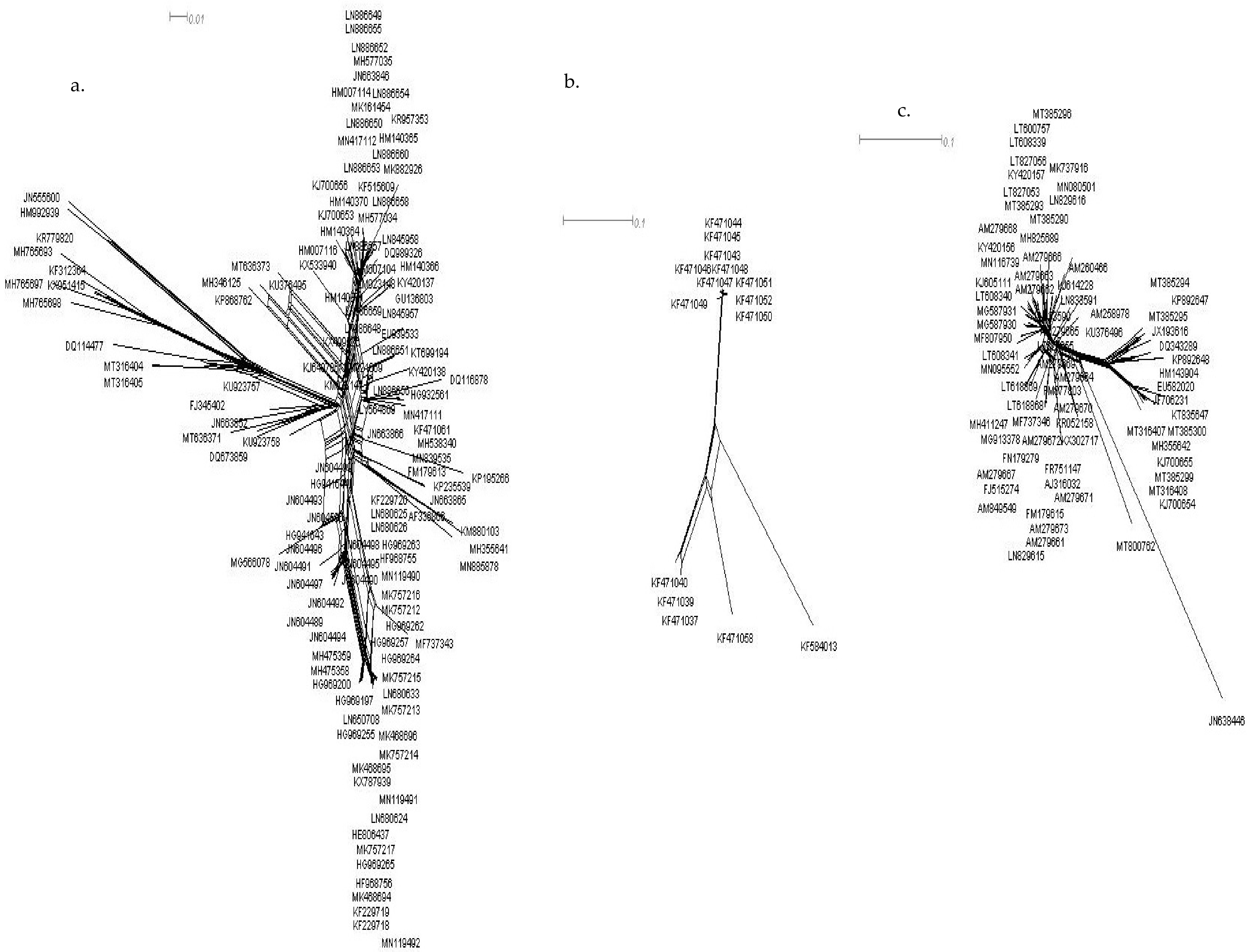
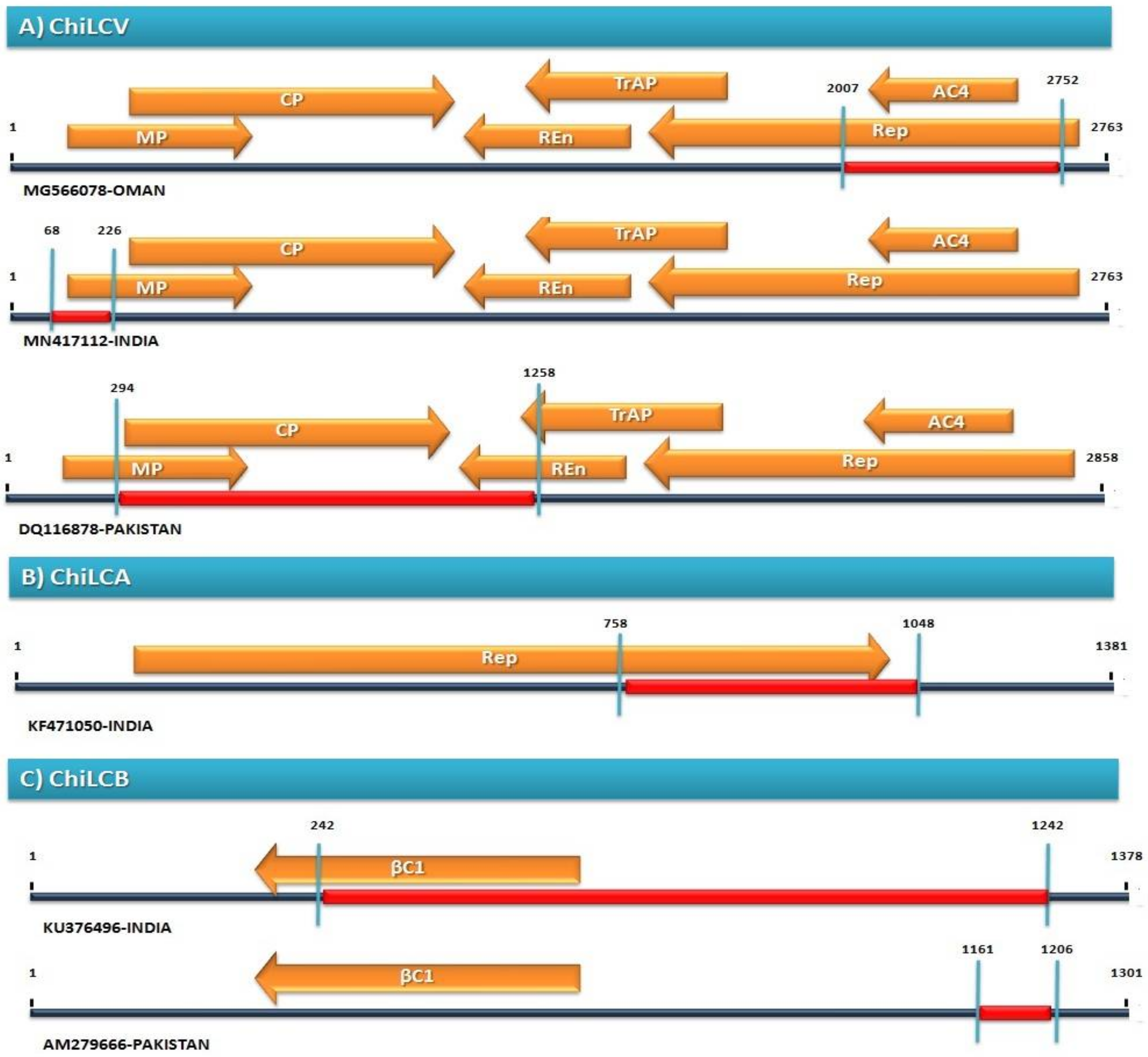
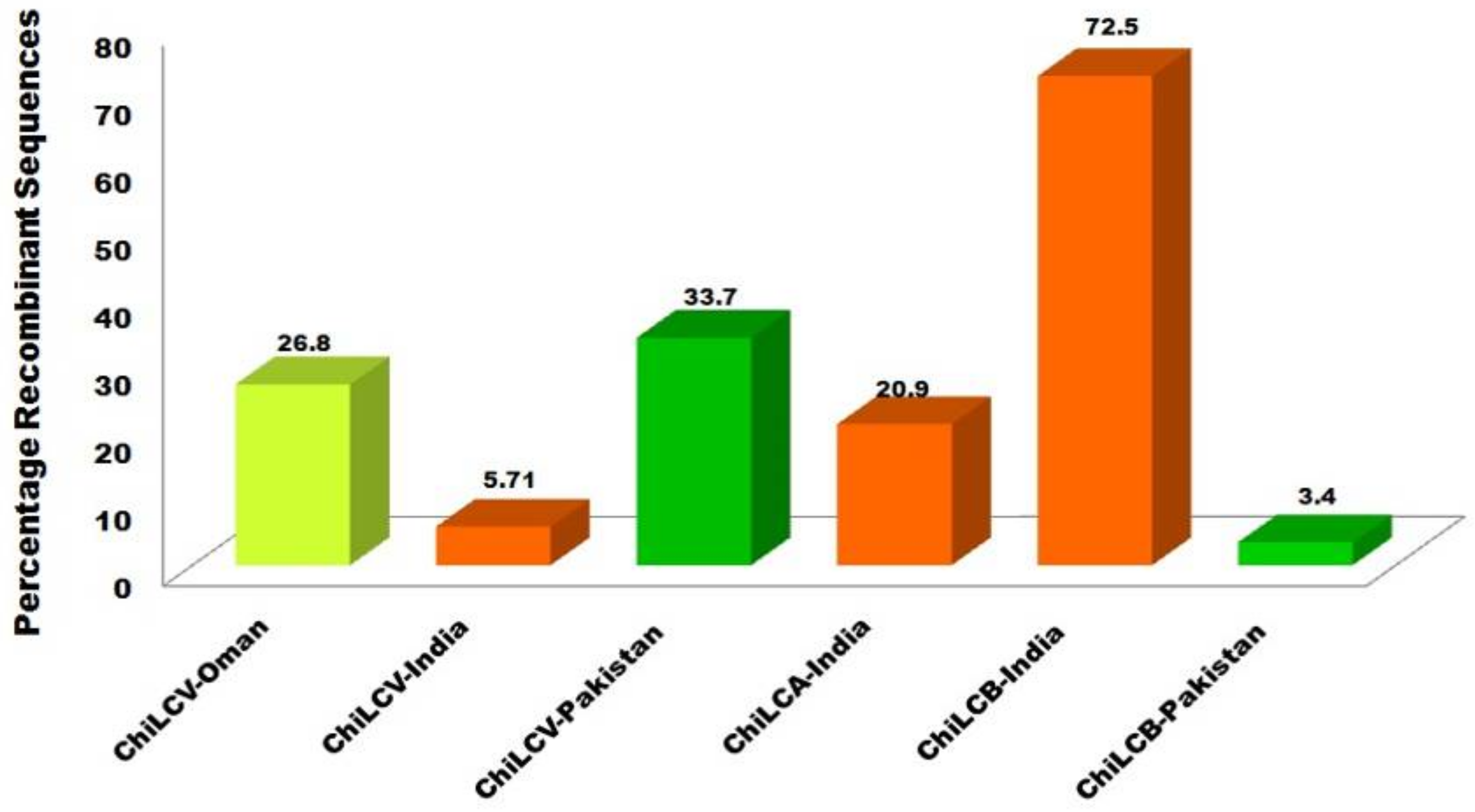
2.2. Recombination Analysis
2.3. Population Demography Analysis
2.4. Amino Acid Sites under Selections
3. Discussion
4. Material and Methods
4.1. Sequence Datasets and Multiple Sequence Alignments
4.2. Phylogenetic and Coalescent Analysis
4.3. Recombination Analysis
4.4. Population Demography Analysis
4.5. Detection of Positive and Negative Selection at Amino Acid Sites
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roossinck, M.J.; Ali, A. Mechanisms of plant virus evolution and identification of genetic bottlenecks: Impact on disease management. In Biotechnology and Plant Disease Management; Punja, Z.K., De Boer, S.H., Sanfaçon, H., Eds.; CABI: Wallingford, UK, 2007; pp. 109–124. ISBN 978-1-84593-288-6. [Google Scholar]
- García-Arenal, F.; Zerbini, F.M. Life on the Edge: Geminiviruses at the Interface Between Crops and Wild Plant Hosts. Annu. Rev. Virol. 2019, 6, 411–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.; Holmes, E.C. Validation of High Rates of Nucleotide Substitution in Geminiviruses: Phylogenetic Evidence from East African Cassava Mosaic Viruses. J. Gen. Virol. 2009, 90, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.T.M.; Silva, J.C.F.; Silva, F.N.; Castillo-Urquiza, G.P.; Silva, F.F.; Seah, Y.M.; Mizubuti, E.S.G.; Duffy, S.; Zerbini, F.M. The Diversification of Begomovirus Populations Is Predominantly Driven by Mutational Dynamics. Virus Evol. 2017, 3, vex005. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.E.; Vanden, B.F.; Jeger, M.J. Factors Influencing Begomovirus Evolution and Their Increasing Global Significance: Implications for Sustainable Control. Crit. Rev. Plant Sci. 2006, 25, 23–46. [Google Scholar] [CrossRef]
- Li, P.; Su, F.; Meng, Q.; Yu, H.; Wu, G.; Li, M.; Qing, L. The C5 protein encoded by Ageratum leaf curl Sichuan virus is a virulence factor and contributes to the virus infection. Mol. Plant Pathol. 2021, 22, 1149–1158. [Google Scholar] [CrossRef]
- Martin, D.P.; Lefeuvre, P.; Varsani, A.; Hoareau, M.; Semegni, J.-Y.; Dijoux, B.; Vincent, C.; Reynaud, B.; Lett, J.-M. Complex Recombination Patterns Arising during Geminivirus Coinfections Preserve and Demarcate Biologically Important Intra-Genome Interaction Networks. PloS Pathog. 2011, 7, e1002203. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.J.C.; Castillo-Urquiza, G.P.; Hora-Júnior, B.T.; Assunção, I.P.; Lima, G.S.A.; Pio-Ribeiro, G.; Mizubuti, E.S.G.; Zerbini, F.M. Species Diversity, Phylogeny and Genetic Variability of Begomovirus Populations Infecting Leguminous Weeds in Northeastern Brazil: Begomovirus Diversity in Leguminous Weeds in Brazil. Plant Pathol. 2012, 61, 457–467. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Roumagnac, P.; Varsani, A. ICTV Report Consortium ICTV Virus Taxonomy Profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef]
- Sanchez-Chavez, S.; Regla-Marquez, C.F.; Cardenas-Conejo, Z.E.; Garcia-Rodriguez, D.A.; Centeno-Leija, S.; Serrano-Posada, H.; Liñan-Rico, A.; Partida-Palacios, B.L.; Cardenas-Conejo, Y. First Report of Begomoviruses Infecting Cucumis sativus L. in North America and Identification of a Proposed New Begomovirus Species. PeerJ 2020, 8, e9245. [Google Scholar] [CrossRef]
- Gnanasekaran, P.; KishoreKumar, R.; Bhattacharyya, D.; Vinoth Kumar, R.; Chakraborty, S. Multifaceted Role of Geminivirus Associated Betasatellite in Pathogenesis. Mol. Plant Pathol. 2019, 20, 1019–1033. [Google Scholar] [CrossRef]
- Mishra, M.; Verma, R.K.; Marwal, A.; Sharma, P.; Gaur, R.K. Biology and Interaction of the Natural Occurrence of Distinct Monopartite Begomoviruses Associated with Satellites in Capsicum Annum From India. Front. Microbiol. 2020, 11, 2366. [Google Scholar] [CrossRef] [PubMed]
- Roumagnac, P.; Lett, J.M.; Fiallo-Olivé, E.; Navas-Castillo, J.; Zerbini, F.M.; Martin, D.P.; Varsani, A. Establishment of five new genera in the family Geminiviridae: Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus. Arch. Virol. 2021, 167, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.V.; Singh, A.K.; Singh, A.K.; Yadav, T.; Basu, S.; Kushwaha, N.; Chattopadhyay, B.; Chakraborty, S. Complexity of begomovirus and betasatellite populations associated with chilli leaf curl disease in India. J. Gen. Virol. 2015, 96, 3143–3158. [Google Scholar] [CrossRef] [PubMed]
- Briddon, R.W.; Martin, D.P.; Roumagnac, P.; Navas-Castillo, J.; Fiallo-Olivé, E.; Moriones, E.; Lett, J.-M.; Zerbini, F.M.; Varsani, A. Alphasatellitidae: A New Family with Two Subfamilies for the Classification of Geminivirus- and Nanovirus-Associated Alphasatellites. Arch. Virol. 2018, 163, 2587–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mubin, M.; Ijaz, S.; Nahid, N.; Hassan, M.; Younus, A.; Qazi, J.; Nawaz-ul-Rehman, M.S. Journey of BegomovirusBetasatellite Molecules: From Satellites to Indispensable Partners. Virus Genes 2020, 56, 16–26. [Google Scholar] [CrossRef]
- Fiallo-Olivé, E.; Tovar, R.; Navas-Castillo, J. Deciphering the Biology of Deltasatellites from the New World: Maintenance by New World Begomoviruses and Whitefly Transmission. New Phytol. 2016, 212, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X. Advances in Understanding Begomovirus Satellites. Annu. Rev. Phytopathol. 2013, 51, 357–381. [Google Scholar] [CrossRef]
- Patil, B.L.; Fauquet, C.M. Differential Interaction between Cassava Mosaic Geminiviruses and Geminivirus Satellites. J. Gen. Virol. 2010, 91, 1871–1882. [Google Scholar] [CrossRef]
- Malathi, V.G.; Renukadevi, P.; Chakraborty, S.; Biswas, K.K.; Roy, A.; Sivalingam, P.N.; Venkataravanappa, V.; Mandal, B. Begomoviruses and Their Satellites Occurring in India: Distribution, Diversity and Pathogenesis. In A Century of Plant Virology in India; Mandal, B., Rao, G.P., Baranwal, V.K., Jain, R.K., Eds.; Springer: Singapore, 2017; pp. 75–177. ISBN 978-981-10-5671-0. [Google Scholar]
- Senanayake, D.M.J.B.; Varma, A.; Mandal, B. Virus-Vector Relationships, Host Range, Detection and Sequence Comparison of Chilli Leaf Curl Virus Associated with an Epidemic of Leaf Curl Disease of Chilli in Jodhpur, India: Chilli Leaf Curl Virus in Jodhpur. J. Phytopathol. 2012, 160, 146–155. [Google Scholar] [CrossRef]
- Zaidi, S.S.-A.; Tashkandi, M.; Mansoor, S.; Mahfouz, M.M. Engineering Plant Immunity: Using CRISPR/Cas9 to Generate Virus Resistance. Front. Plant Sci. 2016, 7, 1673. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Abulfaraj, A.; Idris, A.; Ali, S.; Tashkandi, M.; Mahfouz, M.M. CRISPR/Cas9-Mediated Viral Interference in Plants. Genome Biol. 2015, 16, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, T.; Unckless, R.L. Baculovirus Molecular Evolution via Gene Turnover and Recurrent Positive Selection of Key Genes. J. Virol. 2017, 91, e01319-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, A.K.; Verma, R.K.; Gaur, R.K.; Sanan-Mishra, N. Complexity and Recombination Analysis of Novel Begomovirus Associated with Spinach Yellow Vein Disease in India. Plant Gene 2018, 13, 42–49. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Harkins, G.W.; Lett, J.-M.; Briddon, R.W.; Chase, M.W.; Moury, B.; Martin, D.P. Evolutionary Time-Scale of the Begomoviruses: Evidence from Integrated Sequences in the Nicotiana Genome. PLoS ONE 2011, 6, e19193. [Google Scholar] [CrossRef]
- Kumar, R.V.; Prasanna, H.C.; Singh, A.K.; Ragunathan, D.; Garg, G.K.; Chakraborty, S. Molecular Genetic Analysis and Evolution of Begomoviruses and Betasatellites Causing Yellow Mosaic Disease of Bhendi. Virus Genes 2017, 53, 275–285. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, P.; Liu, P.; Gong, H.; Zhou, X. Characterization of Alphasatellites Associated with Monopartite Begomovirus/Betasatellite Complexes in Yunnan, China. Virol. J. 2010, 7, 178. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, D.; Gnanasekaran, P.; Kumar, R.K.; Kushwaha, N.K.; Sharma, V.K.; Yusuf, M.A.; Chakraborty, S. A GeminivirusBetasatellite Damages the Structural and Functional Integrity of Chloroplasts Leading to Symptom Formation and Inhibition of Photosynthesis. J. Exp. Bot. 2015, 66, 5881–5895. [Google Scholar] [CrossRef] [Green Version]
- Jridi, C.; Martin, J.F.; Marie-Jeanne, V.; Labonne, G.; Blanc, S. Distinct viral populations differentiate and evolve independently in a single perennial host plant. J. Virol. 2006, 80, 2349–2357. [Google Scholar] [CrossRef] [Green Version]
- Conflon, D.; Granier, M.; Tiendrébéogo, F.; Gentit, P.; Peterschmitt, M.; Urbino, C. Accumulation and transmission of alphasatellite, betasatellite and tomato yellow leaf curl virus in susceptible and Ty-1-resistant tomato plants. Virus Res. 2018, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouattara, A.; Tiendrébéogo, F.; Becker, N.; Urbino, C.; Thébaud, G.; Hoareau, M.; Allibert, A.; Chiroleu, F.; Vernerey, M.-S.; Traoré, E.V.; et al. Synergy between an emerging monopartite begomovirus and a DNA-B component. Sci. Rep. 2022, 12, 695. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Pita, J.S.; Fondong, V.N.; Sangaré, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, Pseudorecombination and Synergism of Geminiviruses Are Determinant Keys to the Epidemic of Severe Cassava Mosaic Disease in Uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, H.; Rai, M. Detection and Frequency of Recombination in Tomato-Infecting Begomoviruses of South and Southeast Asia. Virol. J. 2007, 4, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez, M.; Rabadán, M.P.; Martínez, L.D.; Tayahi, M.; Grande-Pérez, A.; Gómez, P. Natural Hosts and Genetic Diversity of the Emerging Tomato Leaf Curl New Delhi Virus in Spain. Front. Microbiol. 2019, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Venkataravanappa, V.; Lakshminarayana Reddy, C.; Swaranalatha, P.; Jalali, S.; Briddon, R.W.; Reddy, M.K. Diversity and Phylogeography of Begomovirus-Associated Beta Satellites of Okra in India. Virol. J. 2011, 8, 555. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Verma, R.K.; Gaur, R.K. Identification of Chilli Leaf Curl Virus and Associated Betasatellite Infecting OsteospermumFruticosum in Rajasthan, India. 3 Biotech 2020, 10, 169. [Google Scholar] [CrossRef]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and Genetic Structure of Plant Virus Populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool forphylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Component | DNA-A | CP | Pre-CP | Rep | TrAP | REn | C4 | Alphasatellite | Betasatellite | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mean substitution rate (at 95% HPD interval) | Relaxed clock | 3.34 × 10−3 (1.72 × 10−3, 6.62 × 10−3) | 1.00 × 10−2 (1.64 × 10−3, 0.027) | 0.012 (1.56 × 10−3, 0.030) | 2.72 × 10−3 (1.11 × 10−3, 5.76 × 10−3) | 5.75 × 10−3 (2.25 × 10−3, 0.011) | 3.58 × 10−3 (1.06 × 10−3, 6.51 × 10−3) | 4.08 × 10−3 (5.52 × 10−5, 0.03) | 6.19 × 10−3 (4.11 × 10−6, 0.016) | 9.17 × 10−4 (4.92 × 10−4, 1.35 × 10−3) |
| Strict clock | 6.17 × 10−4 (4.67 × 10−4, 8.29 × 10−4) | 6.57 × 10−9 (2.34 × 10−17, 4.31 × 10−8) | 1.11 × 10−4 (2.15 × 10−9, 2.55 × 10−4) | 3.95 × 10−4 (2.73 × 10−4, 5.49 × 10−4) | 3.64 × 10−4 (1.79 × 10−4, 5.28 × 10−4) | 3.41 × 10−4 (1.43 × 10−4, 5.50 × 10−4) | 1.12 × 10−4 (3.037 × 10−11, 3.69 × 10−4) | 4.23 × 10−5 (1.10 × 10−18, 2.56 × 10−4) | 1.06 × 10−3 (8.47 × 10−4, 1.279 × 10−3) | |
| Mutation at 3 different codon positions (CP1, CP2, CP3 respectively) | Relaxed clock | 0.92, 1.13, 0.86 | 0.58, 0.32, 2.10 | 0.41, 1.44, 1.16 | 1.32, 0.85, 0.83 | 0.97, 0.87, 1.17 | 1.02, 0.69, 1.11 | 0.84, 1.17, 0.99 | 1.05, 1.09, 0.85 | 0.99, 1.09, 0.91 |
| Strict clock | 0.97, 1.20, 0.88 | 0.61, 0.36, 2.05 | 0.47, 1.39, 1.14 | 1.31, 0.87, 0.824 | 0.93, 0.84, 1.23 | 1.16, 1.03, 0.81 | 0.85, 1.67, 0.98 | 1.06, 1.11, 0.83 | 0.992, 1.10, 0.91 | |
| a | |||||||
| Events | Breakpoints | Recombinant | Parents | Methods $ | p-Value # | ||
| Begin | End | Major | Minor | ||||
| ChiLCV DNA-A | |||||||
| 1 | 294 | 1258 | DQ116878_PepLC | MH538340__ChiL | DQ114477_ChiLC | R, G, B, M, C, S, 3S | 4.58 × 10−66 |
| 2 | 522 | 1032 | JN604496_ChiLC | KF229719_ChiLC | MK882926_ChiLC | R, G, B, M, C, S, 3S | 3.68 × 10−47 |
| 3 | 2704 | 537 | KM880103_ChiLC | MF737343_ChiLC | Unknown (MT636373_ChiLC) | R, G, M, C, S, 3S | 4.54 × 10−40 |
| 4 | 2748 | 632 | HM140366_ChiLC | HM007104_ChiLC | Unknown (KP195266__ChiL) | R, G, B, M, C, S, 3S | 3.22 × 10−36 |
| 5 | 2741 | 958 | KP195266__ChiL | HG969255__ChiL | KU376495_ChiLC | R, G, B, M, C, S, 3S | 4.69 × 10−33 |
| 6 | 2348 | 2752 | KP868762_ChiLC | Unknown (MN417111_ChiLC) | HM007116_ChiLC | R, G, B, M, C, S, 3S | 3.82 × 10−31 |
| 7 | 2206 | 2720 | EU939533_ChiLC | HM140366_ChiLC | Unknown (DQ673859_ChiLC) | R, G, B, M, C, S, 3S | 1.16 × 10−44 |
| 8 | 2111 | 2590 | DQ114477_ChiLC | KP235539_ChiLC | HM992939_ChiLC | R, G, M, C, S, 3S | 1.21 × 10−27 |
| 9 | 521 | 1001 | KP235539_ChiLC | KF229719_ChiLC | MK882926_ChiLC | R, G, M, C, S, 3S | 1.07 × 10−27 |
| 10 | 56 | 614 | KP868762_ChiLC | MT636373_ChiLC | MK882926_ChiLC | R, G, B, M, C, S, 3S | 1.81 × 10−26 |
| 11 | 538 | 962 | KF229719_ChiLC | MH355641_ChiLC | Unknown (HM992939_ChiLC) | R, G, M, C, S, 3S | 3.21 × 10−26 |
| 12 | 2145 | 2652 | HM992939_ChiLC | Unknown (KU923758__ChiL) | MH765698__ChiL | R, G, M, C, S, 3S | 2.86 × 10−23 |
| 13 | 1378 | 1542 | KJ700656_ChiLC | MH577034_ChiLC | Unknown (FJ345402_ChiLC) | R, G, B, M, C, S, 3S | 4.80 × 10−22 |
| 14 | 479 | 1029 | KU923757__ChiL | KU923758__ChiL | Unknown (MK882926_ChiLC) | R, G, M, C, S, 3S | 2.50 × 10−24 |
| 15 | 1030 | 1178 | MH765697_ChiLC | Unknown (KM880103_ChiLC) | JN604491_ChiLC | R, G, M, C, S, 3S | 1.14 × 10−20 |
| 16 | 2007 | 2752 | MG566078_ChiLC | KP235539_ChiLC | Unknown (KP195266__ChiL) | R, G, B, M, C, S, 3S | 5.50 × 10−29 |
| 17 | 2174 | 2516 | KU923758__ChiL | HM140366_ChiLC | Unknown (DQ673859_ChiLC) | R, G, M, C, S, 3S | 2.85 × 10−23 |
| 18 | 484 | 920 | MT636371_ChiLC | KT699194__ChiL | MH765693__ChiL | R, G, M, C, S, 3S | 3.80 × 10−32 |
| 19 | 2754 | 481 | MT636373_ChiLC | Unknown (JN663852__ChiL) | MK882926_ChiLC | R, G, M, C, S, 3S | 1.26 × 10−17 |
| 20 | 2732 | 1076 | DQ673859_ChiLC | MK882926_ChiLC | Unknown (FM179613__ChiL) | R, G, M, C, S, 3S | 8.76 × 10−16 |
| 21 | 111 | 513 | MF737343_ChiLC | JN604491_ChiLC | MK882926_ChiLC | R, G, M, C, S, 3S | 2.56 × 10−14 |
| 22 | 1943 | 2740 | KP195266__ChiL | Unknown (HM007116_ChiLC) | KP235539_ChiLC | R, G, B, M, C, S, 3S | 4.05 × 10−21 |
| 23 | 1069 | 1302 | JN555600__ChiL | Unknown (DQ114477_ChiLC) | MT636371_ChiLC | R, G, M, C, S, 3S | 9.52 × 10−14 |
| 24 | 1751 | 2036 | MH355641_ChiLC | LY564869__ChiL | Unknown (MH765698__ChiL) | R, G, B, M, C, S, 3S | 1.52 × 10−12 |
| 25 | 466 | 958 | FJ345402_ChiLC | KT699194__ChiL | MH765693__ChiL | R, M, C, S, 3S | 1.78 × 10−12 |
| 26 | 509 | 1393 | KU376495_ChiLC | MK882926_ChiLC | Unknown (GU136803_ChiLC) | R, G, B, M, C, S, 3S | 9.47 × 10−12 |
| 27 | 2614 | 151 | KR779820__ChiL | MH765697_ChiLC | Unknown (KX951415__ChiL) | R, G, M, C, S, 3S | 3.55 × 10−11 |
| 28 | 1211 | 1726 | MK882926_ChiLC | HM140371_ChiLC | JN604491_ChiLC | R, G, B, M, C, S, 3S | 8.45 × 10−12 |
| 29 | 2747 | 446 | MT636371_ChiLC | KR957353_ChiLC | FJ345402_ChiLC | R, G, M, C, S, 3S | 6.33 × 10−8 |
| 30 | 1807 | 2205 | MH538340__ChiL | MK882926_ChiLC | FM179613__ChiL | R, G, B, M, C, S, 3S | 4.46 × 10−8 |
| 31 | 1091 | 1540 | FM179613__ChiL | HM007104_ChiLC | KT699194__ChiL | R, G, M, C, S, 3S | 2.94 × 10−7 |
| 32 | 1411 | 2071 | DQ114477_ChiLC | KF312364__ChiL | KT699194__ChiL | R, G, B, M, C, S, 3S | 4.46 × 10−14 |
| 33 | 1209 | 1533 | MF737343_ChiLC | KF229719_ChiLC | HM007104_ChiLC | R, G, B, M, C, S, 3S | 8.96 × 10−9 |
| 34 | 1761 | 2131 | MH765698__ChiL | MH765697_ChiLC | MK882926_ChiLC | R, B, M, C, 3S | 8.71 × 10−07 |
| 35 | 178 | 974 | MT316405_ChiLC | KF471061_ChiLC | MH765697_ChiLC | R, M, C, S, 3S | 1.28 × 10−24 |
| 36 | 2693 | 429 | MH765693__ChiL | Unknown (JN555600__ChiL) | GU136803_ChiLC | R, G, B, M, C, S, 3S | 3.85 × 10−11 |
| 37 | 76 | 535 | GU136803_ChiLC | KR957353_ChiLC | KT699194__ChiL | R, G, B, M, C, S, 3S | 5.25 × 10−6 |
| 38 | 2693 | 34 | LY564869__ChiL | HM140365_ChiLC | KT699194__ChiL | R, G, 3S | 1.43 × 10−5 |
| 39 | 2088 | 2205 | MT636373_ChiLC | MK882926_ChiLC | KF471061_ChiLC | R, G, B, M, C, S, 3S | 2.39 × 10−8 |
| 40 | 1528 | 1750 | KM880103_ChiLC | MN885878_ChiLC | KF229719_ChiLC | R, G, B, M, C, S, 3S | 1.23 × 10−6 |
| 41 | 1128 | 1527 | KM880103_ChiLC | MT636373_ChiLC | KT699194__ChiL | R, G, B, M, C, S, 3S | 5.94 × 10−8 |
| 42 | 1638 | 2105 | KU923757__ChiL | Unknown (HM140370_ChiLC) | HM140364_ChiLC | R, G, B, M, C, S, 3S | 1.65 × 10−5 |
| 43 | 1064 | 1153 | KT699194__ChiL | MT636371_ChiLC | Unknown (FJ345402_ChiLC) | R, G, M, S, 3S | 1.72 × 10−5 |
| 44 | 2086 | 2203 | MK882926_ChiLC | KY420138__ChiL | Unknown (JN604491_ChiLC) | R, G, 3S | 5.29 × 10−5 |
| 45 | 1234 | 1602 | MT636371_ChiLC | HM140370_ChiLC | Unknown (JN604491_ChiLC) | R, B, M, C, S, 3S | 8.58 × 10−5 |
| 46 | 68 | 226 | MN417112_ChiLC | JN663846_ChiLC | Unknown (KT699194__ChiL) | R, G, B, 3S | 6.15 × 10−5 |
| 47 | 1077 | 1181 | KP195266__ChiL | Unknown (HM007104_ChiLC) | JN604491_ChiLC | R, M, C, 3S | 2.03 × 10−5 |
| 48 | 2709 | 157 | KT699194__ChiL | Unknown (KM023147_ChiLC) | HM140370_ChiLC | R, G, B, M | 4.21 × 10−4 |
| 49 | 2651 | 2738 | KP235539_ChiLC | KM023148_ChiLC | HM992939_ChiLC | R, G, 3S | 7.16 × 10−5 |
| 50 | 2190 | 2731 | DQ673859_ChiLC | Unknown (KM023148_ChiLC) | HM140366_ChiLC | R, S, 3S | 1.37 × 10−3 |
| 51 | 1853 | 2200 | KR957353_ChiLC | Unknown (HM007114_ChiLC) | KJ700653_ChiLC | R, C, 3S | 4.09 × 10−3 |
| b | |||||||
| Events | Breakpoints | Recombinant | Parents | Method $ | p-Value # | ||
| Begin | End | Major | Minor | ||||
| Rep protein AC1 | |||||||
| 1 | 185 | 511 | MH765698 | Unknown (MH355641) | AF336806 | R, G, M, C, S, 3S | 8.45 × 10−35 |
| 2 | 820 | 1071 | KP868762 | Unknown (MN417111) | DQ673859 | R, G, M, C, S, 3S | 1.12 × 10−13 |
| 3 | 35 | 418 | DQ114477 | Unknown (KP195266) | MN417111 | R, G, M, C, S, 3S | 5.34 × 10−16 |
| 4 | 218 | 303 | JN555600 | MH355641 | Unknown (MH475358) | R, G, M, C, S, 3S | 2.98 × 10−11 |
| 5 | 202 | 562 | MH355641 | MH538340 | Unknown (MK882926) | R, G, M, C, S, 3S | 3.84 × 10−18 |
| 6 | 679 | 77 | KY420138 | HM140366 | Unknown (DQ673859) | R, G, M, C, S, 3S | 8.61 × 10−19 |
| 7 | 631 | 942 | MF737343 | MK757217 | Unknown (KP195266) | R, G, M, C, S, 3S | 5.10 × 10−18 |
| 8 | 1068 | 741 | MG566078 | AF336806 | MK757217 | R, G, M, C, S, 3S | 4.93 × 10−18 |
| 9 | 255 | 573 | MK757217 | AF336806 | Unknown (MK882926) | R, G, M, C, S, 3S | 9.62 × 10−11 |
| 10 | 640 | 1068 | AF336806 | HM140366 | KP195266 | R, G, M, C, S, 3S | 5.82 × 10−12 |
| 11 | 735 | 939 | MT636373 | MT636371 | Unknown (MT316404) | R, M, C, S, 3S | 1.03 × 10−4 |
| 12 | 425 | 1068 | KP195266 | Unknown (HM140366) | MN417111 | R, M, C, S, 3S | 9.20 × 10−11 |
| 13 | 234 | 619 | KJ700656 | HM007114 | KU923757 | R, G, M, C, S, 3S | 4.62 × 10−7 |
| 14 | 37 | 258 | MK882926 | HM140370 | Unknown (KU923758) | R, G, M, C, 3S | 1.91 × 10−5 |
| 15 | 1035 | 184 | MH765698 | JN555600 | KJ700656 | R, M, C, 3S | 1.30 × 10−5 |
| TrAP protein AC2 | |||||||
| 1 | 404 | 88 | HM992939 | DQ114477 | KY420138 | R, G, M, C, 3S | 2.40 × 10−7 |
| 2 | 205 | 315 | DQ114477 | Unknown (KJ700656) | LN886657 | R, G, M, C, S, 3S | 1.97 × 10−8 |
| 3 | 247 | 320 | KJ700656 | DQ116878 | KP868762 | R, G, M, C, S, 3S | 3.63 × 10−8 |
| REn protein AC3 | |||||||
| 1 | 212 | 406 | DQ116878 | Unknown (JN555600) | MH538340 | R, G, M, C, S, 3S | 7.81 × 10−15 |
| 2 | 106 | 234 | MH765698 | MF737343 | HM992939 | R, M, C, S, 3S | 1.85 × 10−13 |
| 3 | 263 | 61 | MH765693 | HM992939 | Unknown (JN604491) | R, M, C, S, 3S | 3.03 × 10−9 |
| C4 protein | |||||||
| 1 | 279 | 175 | KP868762 | MK882926 | NC028046 | B, M, S, 3S | 3.52 × 10−7 |
| CP-protein AV1 | |||||||
| 1 | 740 | 228 | MF737343 | Unknown (MH355641) | KM921669 | R, G, M, C, S, 3S | 5.68 × 10−23 |
| 2 | 762 | 314 | MT316405 | Unknown (MH355641) | KP868762 | R, G, M, C, S, 3S | 2.04 × 10−26 |
| 3 | 759 | 225 | MH355641 | KU923758 | Unknown (KP868762) | R, G, M, C, S, 3S | 4.01 × 10−11 |
| 4 | 403 | 702 | KU376495 | HM140370 | Unknown (HM140366) | R, M, S, 3S | 1.06 × 10−8 |
| 5 | 38 | 240 | MT636373 | Unknown (MH538340) | DQ989326 | R, S, 3S | 2.32 × 10−2 |
| Pre-CP protein AV2 | |||||||
| 1 | 348 | 128 | KM880103 | MH355641 | Unknown (MG566078) | R, G, M, C, S, 3S | 2.11 × 10−9 |
| Alphasatellite | |||||||
| 1 | 1058 | 1161 | KF471058_ChiLC | KF471037_CHiLC | Unknown (KF471050_ChiLC) | R, G, B, M, C, S, 3S | 5.90 × 10−11 |
| 2 | 758 | 1048 | KF471050_ChiLC | Unknown (KF471058_ChiLC) | KF471037_CHiLC | R, B, M, C, S, 3S | 9.46 × 10−38 |
| 3 | 308 | 624 | KF584013_ChiLC | KF471058_ChiLC | Unknown (KF471049_ChiLC) | R, M, C, S | 1.46 × 10−14 |
| 4 | 929 | 1010 | KF471058_ChiLC | KF471037_CHiLC | Unknown (KF584013_ChiLC) | R, G, 3S | 6.78 × 10−3 |
| Betasatellite | |||||||
| 1 | 1260 | 1310 | MT385290_ChiLC | AM279663_ChiLC | Unknown (JN638446_ChiLC) | R, G, B, M, C, 3S | 2.30 × 10−24 |
| 2 | 141 | 493 | MF737346_ChiLC | KJ614228_ChiLC | MT385300_ChiLC | R, G, B, M, C, S, 3S | 2.03 × 10−23 |
| 3 | 1033 | 1282 | KJ614228_ChiLC | AM279666_ChiLC | MT316408_ChiLC | R, G, B, M, C, S, 3S | 3.77 × 10−13 |
| 4 | 1161 | 1206 | AM279666_ChiLC | LT608340_ChiLC | Unknown (EU582020_ChiLC) | R, G, B, M, C, S, 3S | 7.57 × 10−11 |
| 5 | 1079 | 1142 | AM258978_ChiLC | LT608339_ChiLC | Unknown (KT835647_ChiLC) | R, G, M, C, S, 3S | 3.73 × 10−10 |
| 6 | 723 | 1242 | KU376496_ChiLC | Unknown (KX302717_ChiLC) | MN080501_ChiLC | R, G, 3S | 1.64 × 10−9 |
| 7 | 1342 | 1377 | MN080501_ChiLC | MF737346_ChiLC | Unknown (JN638446_ChiLC) | R, G, 3S | 8.01 × 10−8 |
| 8 | 1293 | 242 | KU376496_ChiLC | Unknown (MH411247_ChiLC) | AM258978_ChiLC | R, G, B, M, C, S, 3S | 8.15 × 10−7 |
| 9 | 243 | 367 | MN080501_ChiLC | MK737916_ChiLC | AJ316032_CHiLC | R, G, M, 3S | 6.82 × 10−6 |
| 10 | 1003 | 1059 | MT800762_ChiLC | Unknown (AM849549_ChiLC) | MK737916_ChiLC | R, G, S, 3S | 1.22 × 10−28 |
| 11 | 975 | 1073 | MK737916_ChiLC | AM279665_ChiLC | Unknown (AJ316032_CHiLC) | R, G, M, C | 2.33 × 10−5 |
| 12 | 1077 | 1248 | MT385293_ChiLC | FM877803_ChiLC | KT835647_ChiLC | R, G, B, M, 3S | 3.97 × 10−7 |
| 13 | 1019 | 1100 | MT385290_ChiLC | AM279666_ChiLC | Unknown (EU582020_ChiLC) | R, G, S, 3S | 4.00 × 10−26 |
| 14 | 1087 | 1338 | KP892648_ChiLC | DQ343289_ChiLC | Unknown (MT385294_ChiLC) | R, M, C, 3S | 2.93 × 10−5 |
| 15 | 323 | 453 | MT385293_ChiLC | FM179615_ChiLC | Unknown (MT385300_ChiLC) | R, B, M, 3S | 5.34 × 10−4 |
| 16 | 960 | 1138 | KX302717_ChiLC | FM877803_ChiLC | MT385300_ChiLC | R, M, 3S | 4.45 × 10−5 |
| 17 | 148 | 232 | MT385294_ChiLC | MT385295_ChiLC | LT827053_ChiLC | R, S, 3S | 6.43 × 10−4 |
| Virus Component | η | π | k | θ - η | θ - W | H | Hd |
|---|---|---|---|---|---|---|---|
| DNA-A | 2104 | 0.10769 | 257.81691 | 0.16108 | 0.10817 | 113 | 0.997 |
| CP | 563 | 0.12144 | 90.11150 | 0.13906 | 0.09806 | 81 | 0.977 |
| Pre-CP | 305 | 0.13907 | 47.14315 | 0.22340 | 0.22340 | 55 | 0.998 |
| Rep | 399 | 0.17521 | 48.18240 | 0.26592 | 0.16195 | 68 | 0.927 |
| TrAP | 426 | 0.14246 | 35.18726 | 0.31744 | 0.17660 | 72 | 0.965 |
| REn | 282 | 0.15639 | 32.37376 | 0.25038 | 0.15627 | 70 | 0.972 |
| C4 | 322 | 0.18375 | 39.32350 | 0.27577 | 0.16187 | 62 | 0.922 |
| Alphasatellite | 727 | 0.13886 | 178.29524 | 0.17413 | 0.13772 | 14 | 0.990 |
| Betasatellite | 1070 | 0.10878 | 109.43568 | 0.21760 | 0.14317 | 66 | 0.996 |
| Virus Component | dN | dS | dN/dS | Total Number of Amino Acid Sites under Positive Selection | Neutrality Tests | ||
|---|---|---|---|---|---|---|---|
| Tajima’s D | Fu and Li’s D | Fu and Li’s F | |||||
| DNA-A | 0.066 ± 0.003 | 0.064 ± 0.003 | 1.03125 | 101 | −1.105 | −1.970 | −1.869 |
| CP | 0.073 ± 0.006 | 0.065 ± 0.006 | 1.12307 | 2 | −0.420 | −1.192 | −0.997 |
| Pre-CP | 0.043 ± 0.008 | 0.038 ± 0.011 | 1.13157 | 1 | −1.453 | −1.918 | −2.086 |
| Rep | 0.134 ± 0.008 | 0.299 ± 0.015 | 0.44816 | 17 | −1.127 | −0.128 | −0.706 |
| TrAP | 0.094 ± 0.009 | 0.133 ± 0.018 | 0.70676 | 13 | −1.826 | −1.794 | −2.165 |
| REn | 0.128 ± 0.019 | 0.167 ± 0.023 | 0.76646 | 5 | −1.237 | 0.012 | −0.670 |
| C4 | 0.127 ± 0.015 | 0.200 ± 0.028 | 0.635 | 6 | −1.100 | 1.158 | 0.141 |
| Alphasatellite | 0.093 ± 0.006 | 0.086 ± 0.005 | 1.08139 | 3 | −0.902 | −0.964 | −1.092 |
| Betasatellite | 0.072 ± 0.004 | 0.055 ± 0.003 | 1.30909 | 34 | −1.750 | −2.557 | −2.664 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, M.; Verma, R.K.; Pandey, V.; Srivastava, A.; Sharma, P.; Gaur, R.; Ali, A. Role of Diversity and Recombination in the Emergence of Chilli Leaf Curl Virus. Pathogens 2022, 11, 529. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050529
Mishra M, Verma RK, Pandey V, Srivastava A, Sharma P, Gaur R, Ali A. Role of Diversity and Recombination in the Emergence of Chilli Leaf Curl Virus. Pathogens. 2022; 11(5):529. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050529
Chicago/Turabian StyleMishra, Megha, Rakesh Kumar Verma, Vineeta Pandey, Aarshi Srivastava, Pradeep Sharma, Rajarshi Gaur, and Akhtar Ali. 2022. "Role of Diversity and Recombination in the Emergence of Chilli Leaf Curl Virus" Pathogens 11, no. 5: 529. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050529