The Host-Microbe Interplay in Human Papillomavirus-Induced Carcinogenesis


Abstract
:1. Introduction
2. Fundamental Aspects of HPV Virology
3. Immune Response to HPV Infection
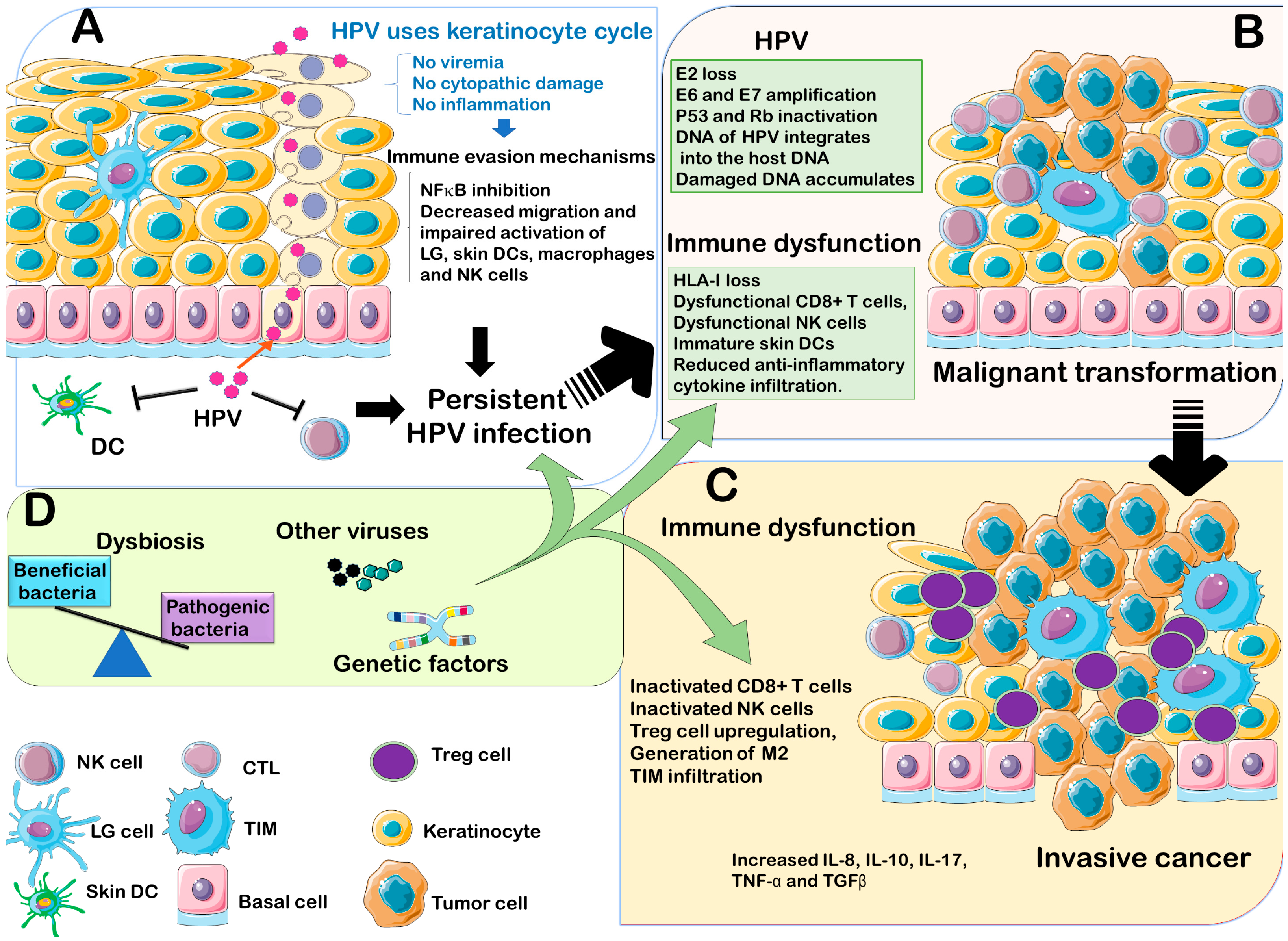
3.1. HPV Interaction with Innate Immune System
3.2. HPV Interaction with Adaptive Immunity
4. HPV Interactions with the Host Genetics
5. HPV Coinfection with Other Microorganisms
6. Concluding Remarks and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gravitt, P.E.; Winer, R.L. Natural history of hpv infection across the lifespan: Role of viral latency. Viruses 2017, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Bonagura, V.R.; Du, Z.; Ashouri, E.; Luo, L.; Hatam, L.J.; DeVoti, J.A.; Rosenthal, D.W.; Steinberg, B.M.; Abramson, A.L.; Gjertson, D.W.; et al. Activating killer cell immunoglobulin-like receptors 3ds1 and 2ds1 protect against developing the severe form of recurrent respiratory papillomatosis. Hum. Immunol. 2010, 71, 212–219. [Google Scholar] [CrossRef] [PubMed]
- DeVoti, J.; Hatam, L.; Lucs, A.; Afzal, A.; Abramson, A.; Steinberg, B.; Bonagura, V. Decreased langerhans cell responses to il-36γ: Altered innate immunity in patients with recurrent respiratory papillomatosis. Mol. Med. 2014, 20, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Miyague, A.H.; Mauad, F.M.; Martins, W.D.P.; Benedetti, A.C.; Ferreira, A.E.; Mauad-Filho, F. Ultrasound scan as a potential source of nosocomial and crossinfection: A literature review. Radiol. Bras. 2015, 48, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, X.; Zhang, Y. Involvement of human papillomaviruses in cervical cancer. Front. Microbiol. 2018, 9, 2896. [Google Scholar] [CrossRef]
- Ong, K.J.; Checchi, M.; Burns, L.; Pavitt, C.; Postma, M.J.; Jit, M. Systematic review and evidence synthesis of non-cervical human papillomavirus-related disease health system costs and quality of life estimates. Sex. Transm. Infect. 2019, 95, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hisamatsu, K.; Suzui, N.; Hara, A.; Tomita, H.; Miyazaki, T. A review of hpv-related head and neck cancer. J. Clin. Med. 2018, 7, 241. [Google Scholar] [CrossRef]
- Yang, L.; Xie, S.; Feng, X.; Chen, Y.; Zheng, T.; Dai, M.; Zhou, C.K.; Hu, Z.; Li, N.; Hang, D. Worldwide prevalence of human papillomavirus and relative risk of prostate cancer: A meta-analysis. Sci. Rep. 2015, 5, 14667. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Ando, M.; Kubo, A.; Isa, S.; Yamamoto, S.; Tsujino, K.; Kurata, T.; Ou, S.H.; Takada, M.; Kawaguchi, T. Human papilloma virus in non-small cell lung cancer in never smokers: A systematic review of the literature. Lung Cancer 2014, 83, 8–13. [Google Scholar] [CrossRef]
- Bae, J.M.; Kim, E.H. Human papillomavirus infection and risk of lung cancer in never-smokers and women: An ‘adaptive’ meta-analysis. Epidemiol. Health 2015, 37, e2015052. [Google Scholar] [CrossRef]
- Liu, S.H.; Cummings, D.A.; Zenilman, J.M.; Gravitt, P.E.; Brotman, R.M. Characterizing the temporal dynamics of human papillomavirus DNA detectability using short-interval sampling. Cancer Epidemiol. Biomark. Prev. 2014, 23, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E. Evidence and impact of human papillomavirus latency. Open Virol. J. 2012, 6, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ambühl, L.M.M.; Villadsen, A.B.; Baandrup, U.; Dybkær, K.; Sørensen, S. Hpv16 e6 and e7 upregulate interferon-induced antiviral response genes isg15 and ifit1 in human trophoblast cells. Pathogens 2017, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, S.; You, J. Targeting persistent human papillomavirus infection. Viruses 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (hpv) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chen, J.; Xu, M.; Yang, H.; Luo, J.; Pan, Y.; Wang, X.; Qiu, L.; Yang, J.; Sun, Q. Genetic variability and functional implication of the long control region in hpv-16 variants in southwest china. PLoS ONE 2017, 12, e0182388. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K. Subcellular trafficking of the papillomavirus genome during initial infection: The remarkable abilities of minor capsid protein l2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef]
- Raj, K.; Berguerand, S.; Southern, S.; Doorbar, J.; Beard, P. E1 empty set e4 protein of human papillomavirus type 16 associates with mitochondria. J. Virol. 2004, 78, 7199–7207. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of hpv transcription. Clinics (Sao Paulo) 2018, 73, e486s. [Google Scholar] [CrossRef]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R.; Venuti, A. Hrhpv e5 oncoprotein: Immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36, 71. [Google Scholar] [CrossRef]
- Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of human papillomavirus genomes in head and neck cancer: Is it time to consider a paradigm shift? Viruses 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; Murall, C.L.; Bravo, I.G. Why human papillomavirus acute infections matter. Viruses 2017, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, V.; Di Domenico, E.G.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimpinelli, F.; Mariani, L.; Ensoli, F. How human papillomavirus replication and immune evasion strategies take advantage of the host DNA damage repair machinery. Viruses 2017, 9, 390. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M. Immune responses to human papillomavirus. Vaccine 2006, 24 (Suppl. 1), S16–S22. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.S.; Stepp, W.H.; Stamos, J.D.; McBride, A.A. Host cell restriction factors that limit transcription and replication of human papillomavirus. Virus Res. 2017, 231, 10–20. [Google Scholar] [CrossRef]
- Richards, K.H.; Wasson, C.W.; Watherston, O.; Doble, R.; Blair, G.E.; Wittmann, M.; Macdonald, A. The human papillomavirus (hpv) e7 protein antagonises an imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci. Rep. 2015, 5, 12922. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, L.; Medeiros, L.J.; Young, K.H. Nf-κb signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev. 2017, 31, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via e6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef]
- Zauner, L.; Melroe, G.T.; Sigrist, J.A.; Rechsteiner, M.P.; Dorner, M.; Arnold, M.; Berger, C.; Bernasconi, M.; Schaefer, B.W.; Speck, R.F.; et al. Tlr9 triggering in burkitt’s lymphoma cell lines suppresses the ebv bzlf1 transcription via histone modification. Oncogene 2010, 29, 4588–4598. [Google Scholar] [CrossRef]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. Tlr9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 e7 oncoprotein induces a transcriptional repressor complex on the toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M. Antigen presentation by mhc-dressed cells. Front. Immunol. 2014, 5, 672. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Whitney, P.G.; Waithman, J.; Eidsmo, L.; Wakim, L.; Caminschi, I.; Allan, R.S.; Wojtasiak, M.; Shortman, K.; Carbone, F.R.; et al. Cross-presentation of viral and self antigens by skin-derived cd103+ dendritic cells. Nat. Immunol. 2009, 10, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Bashaw, A.A.; Leggatt, G.R.; Chandra, J.; Tuong, Z.K.; Frazer, I.H. Modulation of antigen presenting cell functions during chronic hpv infection. Papillomavirus Res. 2017, 4, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Manipulation of the innate immune response by human papillomaviruses. Virus Res. 2017, 231, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummers, B.; Goedemans, R.; Pelascini, L.P.; Jordanova, E.S.; van Esch, E.M.; Meyers, C.; Melief, C.J.; Boer, J.M.; van der Burg, S.H. The interferon-related developmental regulator 1 is used by human papillomavirus to suppress nfκb activation. Nat. Commun. 2015, 6, 6537. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U. Human papillomavirus (hpv) deregulation of toll-like receptor 9. Oncoimmunology 2014, 3, e27257. [Google Scholar] [CrossRef]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.C.; van Rooijen, N.; Caux, C.; Aït-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic cells rapidly recruited into epithelial tissues via ccr6/ccl20 are responsible for cd8+ t cell crosspriming in vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, N.; Watanabe, E.; Norose, Y.; Watari, E.; Kawana, S.; Geijtenbeek, T.B.; Takahashi, H. E-cadherin interactions are required for langerhans cell differentiation. Eur. J. Immunol. 2013, 43, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Sperling, T.; Ołdak, M.; Walch-Rückheim, B.; Wickenhauser, C.; Doorbar, J.; Pfister, H.; Malejczyk, M.; Majewski, S.; Keates, A.C.; Smola, S. Human papillomavirus type 8 interferes with a novel c/ebpβ-mediated mechanism of keratinocyte ccl20 chemokine expression and langerhans cell migration. PLoS Pathog. 2012, 8, e1002833. [Google Scholar] [CrossRef] [PubMed]
- Guess, J.C.; McCance, D.J. Decreased migration of langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 e6/e7 is related to reduced macrophage inflammatory protein-3alpha production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef] [PubMed]
- Routes, J.M.; Morris, K.; Ellison, M.C.; Ryan, S. Macrophages kill human papillomavirus type 16 e6-expressing tumor cells by tumor necrosis factor alpha- and nitric oxide-dependent mechanisms. J. Virol. 2005, 79, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.S.; Mittal, D.; Mattarollo, S.R.; Frazer, I.H. Interleukin-17a promotes arginase-1 production and 2,4-dinitrochlorobenzene-induced acute hyperinflammation in human papillomavirus e7 oncoprotein-expressing skin. J. Innate Immun. 2015, 7, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Hacke, K.; Rincon-Orozco, B.; Buchwalter, G.; Siehler, S.Y.; Wasylyk, B.; Wiesmüller, L.; Rösl, F. Regulation of mcp-1 chemokine transcription by p53. Mol. Cancer 2010, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Lepique, A.P.; Daghastanli, K.R.; Cuccovia, I.M.; Villa, L.L. Hpv16 tumor associated macrophages suppress antitumor t cell responses. Clin. Cancer Res. 2009, 15, 4391–4400. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Perez, M.I.; Jave-Suarez, L.F.; Ortiz-Lazareno, P.C.; Bravo-Cuellar, A.; Gonzalez-Ramella, O.; Aguilar-Lemarroy, A.; Hernandez-Flores, G.; Pereira-Suarez, A.L.; Daneri-Navarro, A.; del Toro-Arreola, S. Cervical cancer cell lines expressing nkg2d-ligands are able to down-modulate the nkg2d receptor on nkl cells with functional implications. BMC Immunol. 2012, 13, 7. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Nguyen, V.H.; Ichimura, H.; Pham, T.T.; Nguyen, C.H.; Pham, T.V.; Elbadry, M.I.; Yoshioka, K.; Tanaka, J.; Trung, L.Q.; et al. A functional polymorphism in the nkg2d gene modulates nk-cell cytotoxicity and is associated with susceptibility to human papilloma virus-related cancers. Sci. Rep. 2016, 6, 39231. [Google Scholar] [CrossRef]
- Cho, H.; Chung, J.Y.; Kim, S.; Braunschweig, T.; Kang, T.H.; Kim, J.; Chung, E.J.; Hewitt, S.M.; Kim, J.H. Mica/b and ulbp1 nkg2d ligands are independent predictors of good prognosis in cervical cancer. BMC Cancer 2014, 14, 957. [Google Scholar] [CrossRef]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (mhc) class i heavy chain, tap1 and lmp2 genes by the human papillomavirus (hpv) type 6b, 16 and 18 e7 oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface hla class i. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Minami, M. Sensing bacterial-induced DNA damaging effects. Front. Immunol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Arreygue-Garcia, N.A.; Daneri-Navarro, A.; del Toro-Arreola, A.; Cid-Arregui, A.; Gonzalez-Ramella, O.; Jave-Suarez, L.F.; Aguilar-Lemarroy, A.; Troyo-Sanroman, R.; Bravo-Cuellar, A.; Delgado-Rizo, V.; et al. Augmented serum level of major histocompatibility complex class i-related chain a (mica) protein and reduced nkg2d expression on nk and t cells in patients with cervical cancer and precursor lesions. BMC Cancer 2008, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer immunotherapy by targeting ido1/tdo and their downstream effectors. Front. Immunol. 2014, 5, 673. [Google Scholar] [CrossRef]
- Mittal, D.; Kassianos, A.J.; Tran, L.S.; Bergot, A.S.; Gosmann, C.; Hofmann, J.; Blumenthal, A.; Leggatt, G.R.; Frazer, I.H. Indoleamine 2,3-dioxygenase activity contributes to local immune suppression in the skin expressing human papillomavirus oncoprotein e7. J. Investig. Dermatol. 2013, 133, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of antitumor immune responses by human papillomavirus through epigenetic downregulation of cxcl14. mBio 2016, 7, e00270-16. [Google Scholar] [CrossRef]
- Lu, J.; Chatterjee, M.; Schmid, H.; Beck, S.; Gawaz, M. Cxcl14 as an emerging immune and inflammatory modulator. J. Inflamm. (Lond.) 2016, 13, 1. [Google Scholar] [CrossRef]
- Meuter, S.; Moser, B. Constitutive expression of cxcl14 in healthy human and murine epithelial tissues. Cytokine 2008, 44, 248–255. [Google Scholar] [CrossRef]
- Schaerli, P.; Willimann, K.; Ebert, L.M.; Walz, A.; Moser, B. Cutaneous cxcl14 targets blood precursors to epidermal niches for langerhans cell differentiation. Immunity 2005, 23, 331–342. [Google Scholar] [CrossRef]
- Kurth, I.; Willimann, K.; Schaerli, P.; Hunziker, T.; Clark-Lewis, I.; Moser, B. Monocyte selectivity and tissue localization suggests a role for breast and kidney-expressed chemokine (brak) in macrophage development. J. Exp. Med. 2001, 194, 855–861. [Google Scholar] [CrossRef]
- Peralta-Zaragoza, O.; Bermúdez-Morales, V.; Gutiérrez-Xicotencatl, L.; Alcocer-González, J.; Recillas-Targa, F.; Madrid-Marina, V. E6 and e7 oncoproteins from human papillomavirus type 16 induce activation of human transforming growth factor beta1 promoter throughout sp1 recognition sequence. Viral Immunol. 2006, 19, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Iancu, I.V.; Botezatu, A.; Goia-Ruşanu, C.D.; Stănescu, A.; Huică, I.; Nistor, E.; Anton, G.; Pleşa, A. Tgf-beta signalling pathway factors in hpv-induced cervical lesions. Roum. Arch. Microbiol. Immunol. 2010, 69, 113–118. [Google Scholar] [PubMed]
- Piersma, S.J.; Jordanova, E.S.; van Poelgeest, M.I.; Kwappenberg, K.M.; van der Hulst, J.M.; Drijfhout, J.W.; Melief, C.J.; Kenter, G.G.; Fleuren, G.J.; Offringa, R.; et al. High number of intraepithelial cd8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007, 67, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Oguejiofor, K.; Hall, J.; Slater, C.; Betts, G.; Hall, G.; Slevin, N.; Dovedi, S.; Stern, P.L.; West, C.M. Stromal infiltration of cd8 t cells is associated with improved clinical outcome in hpv-positive oropharyngeal squamous carcinoma. Br. J. Cancer 2015, 113, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, B.; Lou, H.; Shen, Z.; Tong, F.; Zhai, A.; Wei, L.; Zhang, F. Immunological network analysis in hpv associated head and neck squamous cancer and implications for disease prognosis. Mol. Immunol. 2018, 96, 28–36. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Henderson, S.; Thirdborough, S.M.; Ottensmeier, C.H.; Su, X.; Lechner, M.; Feber, A.; Thomas, G.J.; Fenton, T.R. Human papillomavirus drives tumor development throughout the head and neck: Improved prognosis is associated with an immune response largely restricted to the oropharynx. J. Clin. Oncol. 2016, 34, 4132–4141. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef]
- Yang, W.; Song, Y.; Lu, Y.L.; Sun, J.Z.; Wang, H.W. Increased expression of programmed death (pd)-1 and its ligand pd-l1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology 2013, 139, 513–522. [Google Scholar] [CrossRef]
- Chang, H.; Hong, J.H.; Lee, J.K.; Cho, H.W.; Ouh, Y.T.; Min, K.J.; So, K.A. Programmed death-1 (pd-1) expression in cervical intraepithelial neoplasia and its relationship with recurrence after conization. J. Gynecol. Oncol. 2018, 29, e27. [Google Scholar] [CrossRef]
- Meng, Y.; Liang, H.; Hu, J.; Liu, S.; Hao, X.; Wong, M.S.K.; Li, X.; Hu, L. Pd-l1 expression correlates with tumor infiltrating lymphocytes and response to neoadjuvant chemotherapy in cervical cancer. J. Cancer 2018, 9, 2938–2945. [Google Scholar] [CrossRef]
- Zhang, J.; Burn, C.; Young, K.; Wilson, M.; Ly, K.; Budhwani, M.; Tschirley, A.; Braithwaite, A.; Baird, M.; Hibma, M. Microparticles produced by human papillomavirus type 16 e7-expressing cells impair antigen presenting cell function and the cytotoxic t cell response. Sci. Rep. 2018, 8, 2373. [Google Scholar] [CrossRef] [PubMed]
- Loenenbach, A.D.; Poethko-Müller, C.; Pawlita, M.; Thamm, M.; Harder, T.; Waterboer, T.; Schröter, J.; Deleré, Y.; Wichmann, O.; Wiese-Posselt, M. Mucosal and cutaneous human papillomavirus seroprevalence among adults in the prevaccine era in germany—results from a nationwide population-based survey. Int. J. Infect. Dis. 2019, 83, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M. Hpv—immune response to infection and vaccination. Infect. Agents Cancer 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; DeMars, L.R. Hpv vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Song, L.Y.; Saah, A.; Brown, M.; Moscicki, A.B.; Meyer, W.A.; Bryan, J.; Levin, M.J.; Team, I.P.P. Humoral, mucosal, and cell-mediated immunity against vaccine and nonvaccine genotypes after administration of quadrivalent human papillomavirus vaccine to hiv-infected children. J. Infect. Dis. 2012, 206, 1309–1318. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Johansson, M.; Waterboer, T.; Kaaks, R.; Chang-Claude, J.; Drogen, D.; Tjønneland, A.; Overvad, K.; Quirós, J.R.; González, C.A.; et al. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J. Clin. Oncol. 2013, 31, 2708–2715. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Brennan, P.; Lang Kuhs, K.A.; Waterboer, T.; Clifford, G.; Franceschi, S.; Michel, A.; Willhauck-Fleckenstein, M.; Riboli, E.; Castellsagué, X.; et al. Human papillomavirus antibodies and future risk of anogenital cancer: A nested case-control study in the european prospective investigation into cancer and nutrition study. J. Clin. Oncol. 2015, 33, 877–884. [Google Scholar] [CrossRef]
- Scott, M.E.; Ma, Y.; Kuzmich, L.; Moscicki, A.B. Diminished ifn-gamma and il-10 and elevated foxp3 mrna expression in the cervix are associated with cin 2 or 3. Int. J. Cancer 2009, 124, 1379–1383. [Google Scholar] [CrossRef]
- Sun, W.; Wei, F.Q.; Li, W.J.; Wei, J.W.; Zhong, H.; Wen, Y.H.; Lei, W.B.; Chen, L.; Li, H.; Lin, H.Q.; et al. A positive-feedback loop between tumour infiltrating activated treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 2017, 117, 1631–1643. [Google Scholar] [CrossRef]
- Loddenkemper, C.; Hoffmann, C.; Stanke, J.; Nagorsen, D.; Baron, U.; Olek, S.; Huehn, J.; Ritz, J.P.; Stein, H.; Kaufmann, A.M.; et al. Regulatory (foxp3+) t cells as target for immune therapy of cervical intraepithelial neoplasia and cervical cancer. Cancer Sci. 2009, 100, 1112–1117. [Google Scholar] [CrossRef]
- Patel, S.; Chiplunkar, S. Host immune responses to cervical cancer. Curr. Opin. Obstet. Gynecol. 2009, 21, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, W.J.; Wu, C.Y.; Zhong, H.; Wen, W.P. Cd45ra-foxp3high but not cd45ra+foxp3low suppressive t regulatory cells increased in the peripheral circulation of patients with head and neck squamous cell carcinoma and correlated with tumor progression. J. Exp. Clin. Cancer Res. 2014, 33, 35. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.; Duurland, C.L.; Jordanova, E.S.; van Ham, J.J.; Ehsan, I.; van Egmond, S.L.; Welters, M.J.P.; van der Burg, S.H. Tbet-positive regulatory t cells accumulate in oropharyngeal cancers with ongoing tumor-specific type 1 t cell responses. J. Immunother. Cancer 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional repression of e-cadherin by human papillomavirus type 16 e6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef] [PubMed]
- Iijima, N.; Goodwin, E.C.; Dimaio, D.; Iwasaki, A. High-risk human papillomavirus e6 inhibits monocyte differentiation to langerhans cells. Virology 2013, 444, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Morales, V.H.; Peralta-Zaragoza, O.; Alcocer-González, J.M.; Moreno, J.; Madrid-Marina, V. Il-10 expression is regulated by hpv e2 protein in cervical cancer cells. Mol. Med. Rep. 2011, 4, 369–375. [Google Scholar] [PubMed]
- Berti, F.C.B.; Pereira, A.P.L.; Cebinelli, G.C.M.; Trugilo, K.P.; Brajão de Oliveira, K. The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor Rev. 2017, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Monnier-Benoit, S.; Mauny, F.; Riethmuller, D.; Guerrini, J.S.; Căpîlna, M.; Félix, S.; Seillès, E.; Mougin, C.; Prétet, J.L. Immunohistochemical analysis of cd4+ and cd8+ t-cell subsets in high risk human papillomavirus-associated pre-malignant and malignant lesions of the uterine cervix. Gynecol. Oncol. 2006, 102, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The e5 protein of human papillomavirus type 16 perturbs mhc class ii antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef]
- Suprynowicz, F.A.; Disbrow, G.L.; Krawczyk, E.; Simic, V.; Lantzky, K.; Schlegel, R. Hpv-16 e5 oncoprotein upregulates lipid raft components caveolin-1 and ganglioside gm1 at the plasma membrane of cervical cells. Oncogene 2008, 27, 1071–1078. [Google Scholar] [CrossRef]
- Cuzick, J.; Terry, G.; Ho, L.; Monaghan, J.; Lopes, A.; Clarkson, P.; Duncan, I. Association between high-risk hpv types, hla drb1* and dqb1* alleles and cervical cancer in british women. Br. J. Cancer 2000, 82, 1348–1352. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Cartier, I.; Jourdan-Da Silva, N.; De Crémoux, P.; Lepage, V.; Charron, D. Regression of low-grade cervical intraepithelial neoplasia in patients with hla-drb1*13 genotype. Obstet. Gynecol. 2004, 104, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Maeda, H.; Oki, A.; Takatsuka, N.; Yasugi, T.; Furuta, R.; Hirata, R.; Mitsuhashi, A.; Kawana, K.; Fujii, T.; et al. Human leukocyte antigen class ii drb1*1302 allele protects against cervical cancer: At which step of multistage carcinogenesis? Cancer Sci. 2015, 106, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Wang, H.; Liu, J.; Cheng, Q.; Chen, X.; Chen, H. Association of smug1 snps in intron region and linkage disequilibrium with occurrence of cervical carcinoma and hpv infection in chinese population. J. Cancer 2019, 10, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of nkp30 and nkg2d receptors: Consequences for the nk-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Shen, Z.; Hu, X.; Sun, L.; Zhu, X. Transforming growth factor-β1 in carcinogenesis, progression, and therapy in cervical cancer. Tumour Biol. 2016, 37, 7075–7083. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Sturgis, E.M.; Lei, D.; Liu, Z.; Dahlstrom, K.R.; Wei, Q.; Li, G. Association of tgf-beta1 genetic variants with hpv16-positive oropharyngeal cancer. Clin. Cancer Res. 2010, 16, 1416–1422. [Google Scholar] [CrossRef]
- Tao, Y.; Sturgis, E.M.; Huang, Z.; Wang, Y.; Wei, P.; Wang, J.R.; Wei, Q.; Li, G. Genetic variants predict clinical outcomes of hpv-positive oropharyngeal cancer patients after definitive radiotherapy. Clin. Cancer Res. 2018, 24, 2225–2233. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Onizuka, M.; Sao, H.; Akiyama, H.; Miyamura, K.; Okamoto, S.; Inoue, M.; Kanda, Y.; Ohtake, S.; et al. Nkg2d gene polymorphism has a significant impact on transplant outcomes after hla-fully-matched unrelated bone marrow transplantation for standard risk hematologic malignancies. Haematologica 2009, 94, 1427–1434. [Google Scholar] [CrossRef]
- Hayashi, T.; Imai, K.; Morishita, Y.; Hayashi, I.; Kusunoki, Y.; Nakachi, K. Identification of the nkg2d haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. 2006, 66, 563–570. [Google Scholar] [CrossRef]
- Furue, H.; Kumimoto, H.; Matsuo, K.; Suzuki, T.; Hasegawa, Y.; Shinoda, M.; Sugimura, T.; Mitsudo, K.; Tohnai, I.; Ueda, M.; et al. Opposite impact of nkg2d genotype by lifestyle exposure to risk of aerodigestive tract cancer among japanese. Int. J. Cancer 2008, 123, 181–186. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human microrna-1245 downregulates the nkg2d receptor in nk cells and impairs nkg2d-mediated functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Legarda, L.Y.; Khakoo, S.I. Conserved and variable natural killer cell receptors: Diverse approaches to viral infections. Immunology 2019, 156, 319–328. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, S.; Mattei, J.; Bunn, P.A.; Zhou, C.; Chan, D. The combination of anti-kir monoclonal antibodies with anti-pd-1/pd-l1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in nsclc. Drug Des. Dev. Ther. 2018, 12, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Guha, U.; Mitra, S.; Ghosh, S.; Bhattacharjee, S.; Sengupta, M. Meta-analysis of polymorphic variants conferring genetic risk to cervical cancer in indian women supports cyp1a1 as an important associated locus. Asian Pac. J. Cancer Prev. 2018, 19, 2071–2081. [Google Scholar]
- Palefsky, J. Human papillomavirus-related tumors in hiv. Curr. Opin. Oncol. 2006, 18, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.J.; Burk, R.D.; Xie, X.; Anastos, K.; Massad, L.S.; Minkoff, H.; Xue, X.; D’Souza, G.; Watts, D.H.; Levine, A.M.; et al. Risk of cervical precancer and cancer among hiv-infected women with normal cervical cytology and no evidence of oncogenic hpv infection. JAMA 2012, 308, 362–369. [Google Scholar] [CrossRef]
- Keller, M.J.; Burk, R.D.; Massad, L.S.; Eltoum, I.E.; Hessol, N.A.; Castle, P.E.; Anastos, K.; Xie, X.; Minkoff, H.; Xue, X.; et al. Cervical precancer risk in hiv-infected women who test positive for oncogenic human papillomavirus despite a normal pap test. Clin. Infect. Dis. 2015, 61, 1573–1581. [Google Scholar] [CrossRef]
- Lee, J.E.; Lee, S.; Lee, H.; Song, Y.M.; Lee, K.; Han, M.J.; Sung, J.; Ko, G. Association of the vaginal microbiota with human papillomavirus infection in a korean twin cohort. PLoS ONE 2013, 8, e63514. [Google Scholar] [CrossRef]
- Klein, C.; Gonzalez, D.; Samwel, K.; Kahesa, C.; Mwaiselage, J.; Aluthge, N.; Fernando, S.; West, J.T.; Wood, C.; Angeletti, P.C. Relationship between the cervical microbiome, hiv status, and precancerous lesions. mBio 2019, 10, e02785-18. [Google Scholar] [CrossRef]
- Brusselaers, N.; Shrestha, S.; van de Wijgert, J.; Verstraelen, H. Vaginal dysbiosis and the risk of human papillomavirus and cervical cancer: Systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2018, 221, 9–18.e8. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, S.Y.; Throop, A.L.; Herbst-Kralovetz, M.M. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 2014, 209, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical microbiome and cytokine profile at various stages of cervical cancer: A pilot study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [PubMed]
- Kroon, S.J.; Ravel, J.; Huston, W.M. Cervicovaginal microbiota, women’s health, and reproductive outcomes. Fertil. Steril. 2018, 110, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Borgdorff, H.; Tsivtsivadze, E.; Verhelst, R.; Marzorati, M.; Jurriaans, S.; Ndayisaba, G.F.; Schuren, F.H.; van de Wijgert, J.H. Lactobacillus-dominated cervicovaginal microbiota associated with reduced hiv/sti prevalence and genital hiv viral load in african women. ISME J. 2014, 8, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Tyssen, D.; Wang, Y.Y.; Hayward, J.A.; Agius, P.A.; DeLong, K.; Aldunate, M.; Ravel, J.; Moench, T.R.; Cone, R.A.; Tachedjian, G. Anti-hiv-1 activity of lactic acid in human cervicovaginal fluid. mSphere 2018, 3, e00055-18. [Google Scholar] [CrossRef] [PubMed]
- Torcia, M.G. Interplay among vaginal microbiome, immune response and sexually transmitted viral infections. Int. J. Mol. Sci. 2019, 20, 266. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Tian, T.; Wei, Z.; Peck, K.N.; Shih, N.; Chalian, A.A.; O’Malley, B.W.; Weinstein, G.S.; Feldman, M.D.; Alwine, J.; et al. Microbial signatures associated with oropharyngeal and oral squamous cell carcinomas. Sci. Rep. 2017, 7, 4036. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Matsumoto, A.; Tanaka, H.; Matsumura, I. Gastric microbiota: An emerging player in helicobacter pylori-induced gastric malignancies. Cancer Lett. 2018, 414, 147–152. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, X.; Wang, Y.; Zhao, J.; Shi, H.; Zhang, H.; Wei, Y.; Xue, W.; Zhang, J. Proteomic screening for serum biomarkers for cervical cancer and their clinical significance. Med. Sci. Monit. 2019, 25, 288–297. [Google Scholar] [CrossRef]
- Inan, H.; Wang, S.; Inci, F.; Baday, M.; Zangar, R.; Kesiraju, S.; Anderson, K.S.; Cunningham, B.T.; Demirci, U. Isolation, detection, and quantification of cancer biomarkers in hpv-associated malignancies. Sci. Rep. 2017, 7, 3322. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L. Machine learning for tackling microbiota data and infection complications in immunocompromised patients with cancer. J. Intern. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Owais, M.; Arsalan, M.; Choi, J.; Park, K.R. Effective diagnosis and treatment through content-based medical image retrieval (cbmir) by using artificial intelligence. J. Clin. Med. 2019, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Xu, L.; Simoens, C.; Martin-Hirsch, P.P. Prophylactic vaccination against human papillomaviruses to prevent cervical cancer and its precursors. Cochrane Database Syst. Rev. 2018, 5, CD009069. [Google Scholar] [CrossRef] [PubMed]
- Orbegoso, C.; Murali, K.; Banerjee, S. The current status of immunotherapy for cervical cancer. Rep. Pract. Oncol. Radiother. 2018, 23, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Le Tourneau, C. Pembrolizumab in cervical cancer: Latest evidence and clinical usefulness. Ther. Adv. Med. Oncol. 2017, 9, 431–439. [Google Scholar] [CrossRef]
- van Poelgeest, M.I.; Welters, M.J.; van Esch, E.M.; Stynenbosch, L.F.; Kerpershoek, G.; van Persijn van Meerten, E.L.; van den Hende, M.; Löwik, M.J.; Berends-van der Meer, D.M.; Fathers, L.M.; et al. Hpv16 synthetic long peptide (hpv16-slp) vaccination therapy of patients with advanced or recurrent hpv16-induced gynecological carcinoma, a phase ii trial. J. Transl. Med. 2013, 11, 88. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef]
- Colmenares, V.; Noyola, D.E.; Monsiváis-Urenda, A.; Salgado-Bustamante, M.; Estrada-Capetillo, L.; González-Amaro, R.; Baranda, L. Human papillomavirus immunization is associated with increased expression of different innate immune regulatory receptors. Clin. Vaccine Immunol. 2012, 19, 1005–1011. [Google Scholar] [CrossRef]
- Chouhy, D.; Bolatti, E.M.; Pérez, G.R.; Giri, A.A. Analysis of the genetic diversity and phylogenetic relationships of putative human papillomavirus types. J. Gen. Virol. 2013, 94, 2480–2488. [Google Scholar] [CrossRef]
- Sias, C.; Salichos, L.; Lapa, D.; Del Nonno, F.; Baiocchini, A.; Capobianchi, M.R.; Garbuglia, A.R. Alpha, beta, gamma human papillomaviruses (hpv) detection with a different sets of primers in oropharyngeal swabs, anal and cervical samples. Virol. J. 2019, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Farzan, S.F.; Waterboer, T.; Gui, J.; Nelson, H.H.; Li, Z.; Michael, K.M.; Perry, A.E.; Spencer, S.K.; Demidenko, E.; Green, A.C.; et al. Cutaneous alpha, beta and gamma human papillomaviruses in relation to squamous cell carcinoma of the skin: A population-based study. Int. J. Cancer 2013, 133, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The biology of beta human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Müller-Decker, K.; Accardi, R.; Robitaille, A.; Dürst, M.; Beer, K.; Jansen, L.; Flechtenmacher, C.; Bozza, M.; Harbottle, R.; et al. Beta hpv38 oncoproteins act with a hit-and-run mechanism in ultraviolet radiation-induced skin carcinogenesis in mice. PLoS Pathog. 2018, 14, e1006783. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
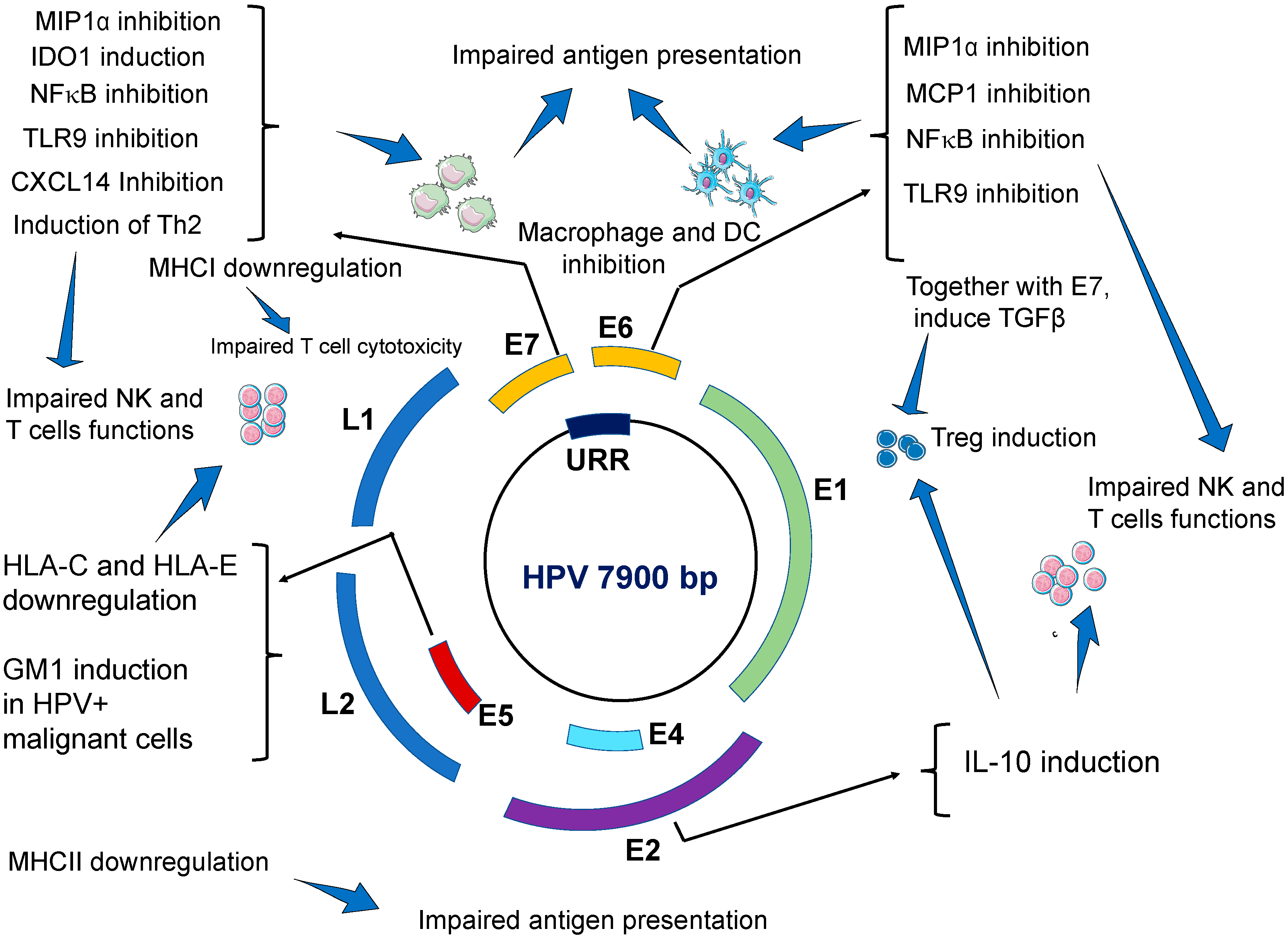
| Effects on Innate Immune System | Viral Factor | Main Findings/Mechanisms | Ref. |
| Impairing antiviral activities of keratinocytes. | E6 and E7 | E6 and E7 oncogenes repress IFN-κ transcription independently from binding to PDZ proteins. As a result, interferon stimulated genes (ISG), such as IFIT1 and MX1, which have antiviral activities, are inhibited. Similarly, the expression of the pathogen recognition receptors TLR3, RIG-I, and MDA5 in keratinocytes is inhibited. Taken together, our results suggest that HR-HPVs target IFN-κ, which is a master regulator of ISG expression and inducible IFNs in keratinocytes. The expression of proapoptotic genes (TRAIL and XAF1) are also inhibited. | [29] |
| E6 | Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. Impairs migration and inhibits the maturation of Langerhans cells in the epidermis. | [84] | |
| E7 | HPV decreases CCL20 secretion by keratinocytes by inhibiting CCL20 promoter. E7 oncoviral protein prevents the binding of C/EBP to the CCL20 promoter. | [41] | |
| E6 and E7 | CCL20 production is also affected via downregulation of NFκB signal by E6 and E7 | [42] | |
| Impaired antigen presenting cells (APCs) | E6 and E7 | HPVs impair the migration, recruitment and localization of dendritic cells to HPV-infected epidermis. HPV impairs the maturation and the function of skin DCs. HR-HPV positive cancer cells, and E6- and E7-expressing cells can inhibit differentiation of monocytes into Langerhans cells. | [35,36] |
| Induction of dendritic cells with immunosuppressive activities. Increased expression of programmed death (PD)-1 and its ligand PD-L1 on the surface of dendritic cells. | [85] | ||
| Impairing macrophage immune responses | E6 | E6 of HPV inhibits the release of monocyte chemotactic protein MCP-1 from infected keratinocytes Downregulate the secretion of MIP1α from infected keratinocytes. As a result, HPV-inhibits macrophage infiltration during acute infection. | [45] |
| E6 and E7 | During advanced carcinogenesis, HPV tumor infiltrating macrophages (TIM) promote cancer growth and metastasis. | [42] | |
| Impairing NK cells cytotoxicity | E5 | E5 induces downregulation of HLA-C and HLA-E on cell surface by sequestering these proteins in intracellular compartments. | [20,51] |
| E7 | E7 induces the secretion of the immunosuppressive molecule IDO1 from dendritic cells. High levels of IDO1 also found in the serum of patients with HPV-induced CIN 2/3. | [15,55] | |
| HPV+ tumor cells shed NKG2D-Ls leading to elevated levels of these ligands in the blood of patients with HPV-induced cancers, which ultimately leads to downregulation of NKG2D receptor expression on NK cells thus impairing NKG2D-mediated cytotoxicity. | [53] | ||
| Inhibition of NFκB signal pathway | E6 and E7 | E7 oncogene, and to a lesser extend E6, strongly reduce NFκB activation and this strongly impairs immune response. As a result, the production of proinflammatory cytokines (IL-1, IL-6 TNF-α, IFN-α and IFN-β) is severely compromised. | [26] |
| Downregulation of TLR9 expression | E6 and E7 | E6 and E7 oncoproteins directly bind to TLR9 promoter and down-regulate TLR9 transcriptional. | [32] |
| Inhibition of CXCL14 | E7 | E7 induces hypermethylation of CXCL14 promoter which downregulates CXCL14 expression. CXCL14 deficiency impairs immune cells infiltration to the site of HPV-infection. NK cells, and T cells are especially affected by CXCL14 deficiency. | [56] |
| CXCL14 deficiency impairs the differentiation of CD14+ monocytes into Langerhans cells | [59] | ||
| Low levels of CXCL14 may impair macrophage maturation | [60] | ||
| Induction of TGF-β1 secretion | E6 and E7 | E6 and E7 indirectly interact with the TGF-β1 regulatory element site (GGGGCGG) and activate TGF-β1 promoter. High levels of TGF-β1 in the blood impairs immune cells responses. For example, the expression of activator receptors NKp30 NKp46 and NKG2D on NK cells are severely downregulated resulting in impaired cytotoxicity. High levels of TGF-β1 downregulates MHC class II expression which impairs antigen presenting function by APC. | [61,62] |
| Induction of IL-10 secretion | E2 | E6 and E7 bind to IL-10 promoter thus increasing IL-10 transcription. E2 protein binds to the regulatory region of the human IL-10 gene (-2054 nt) and induces high promoter activity in epithelial cells. High levels of IL-10 impair the cytotoxicity of NK and T cells. IL-10 also promotes TIM infiltration. | [61,86] |
| IL-10 stimulates HPV E6 and E7 expression. | [87] | ||
| Effects on Adaptive Immune System | Viral Factor | Main Findings | Ref. |
| Impairment of humoral immune response | E7 | Induction of a shift from a Th1 response to a Th2 immune response. Upregulation of Th2 cytokines (IL-6, IL-8, and IL-10). | [88] |
| E7 | CD4+ T cells from HPV-associated lesions have impaired production of IL-1β, IL-18, IL2, IFN-γ, and TNF-α | [23] | |
| Promoting immunosuppressive Treg cells | E2, E6, E7 | HPV-induced IL-10 and TGF-β1 promote the proliferation of Treg cells. Treg cells accumulate in HPV-transformed tissues. Treg cells suppress Th1 immune responses. Treg cells (CD25/FOXP3 and CD4/TGF-β) secrete immunosuppressive cytokines IL-10 and TGF-β harnessing antiviral and antitumor response of CD8+ CTL and CD4+T cells | [83] |
| Activated Treg cells promote the differentiation of monocytes into an immunosuppressive M2-like phenotype | [79] | ||
| Impaired cytotoxic T cells activity | E7 | E7 directly interacts and block MHC-I heavy chain promoter leading to reduced MHC-I expression on infected keratinocytes and reduces target cell recognition by CD8+ T cells. | [50] |
| E5 | Downregulation or MHC class II in human keratinocytes leading to impaired APC and poor antigen recognition | [89] | |
| E5 | Upregulation of ganglioside GM1, on the cell surface of HPV-transformed cells leading to cytotoxic T lymphocytes inhibition Induction of PD1 in CD8+ T cells and PDL-1 in DCs and HPV-transformed cells leading to impaired immune surveillance and immune escape. | [68,70,90] | |
| E7 | Microparticles shed from HPVs infected keratinocytes may suppress the cytotoxicity of CD8+ T cells | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakabayashi, R.; Nakahama, Y.; Nguyen, V.; Espinoza, J.L. The Host-Microbe Interplay in Human Papillomavirus-Induced Carcinogenesis. Microorganisms 2019, 7, 199. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7070199
Wakabayashi R, Nakahama Y, Nguyen V, Espinoza JL. The Host-Microbe Interplay in Human Papillomavirus-Induced Carcinogenesis. Microorganisms. 2019; 7(7):199. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7070199
Chicago/Turabian StyleWakabayashi, Rei, Yusuke Nakahama, Viet Nguyen, and J. Luis Espinoza. 2019. "The Host-Microbe Interplay in Human Papillomavirus-Induced Carcinogenesis" Microorganisms 7, no. 7: 199. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7070199