Chemical Composition and Light Absorption of PM2.5 Observed at Two Sites near a Busy Road during Summer and Winter



Abstract
:1. Introduction
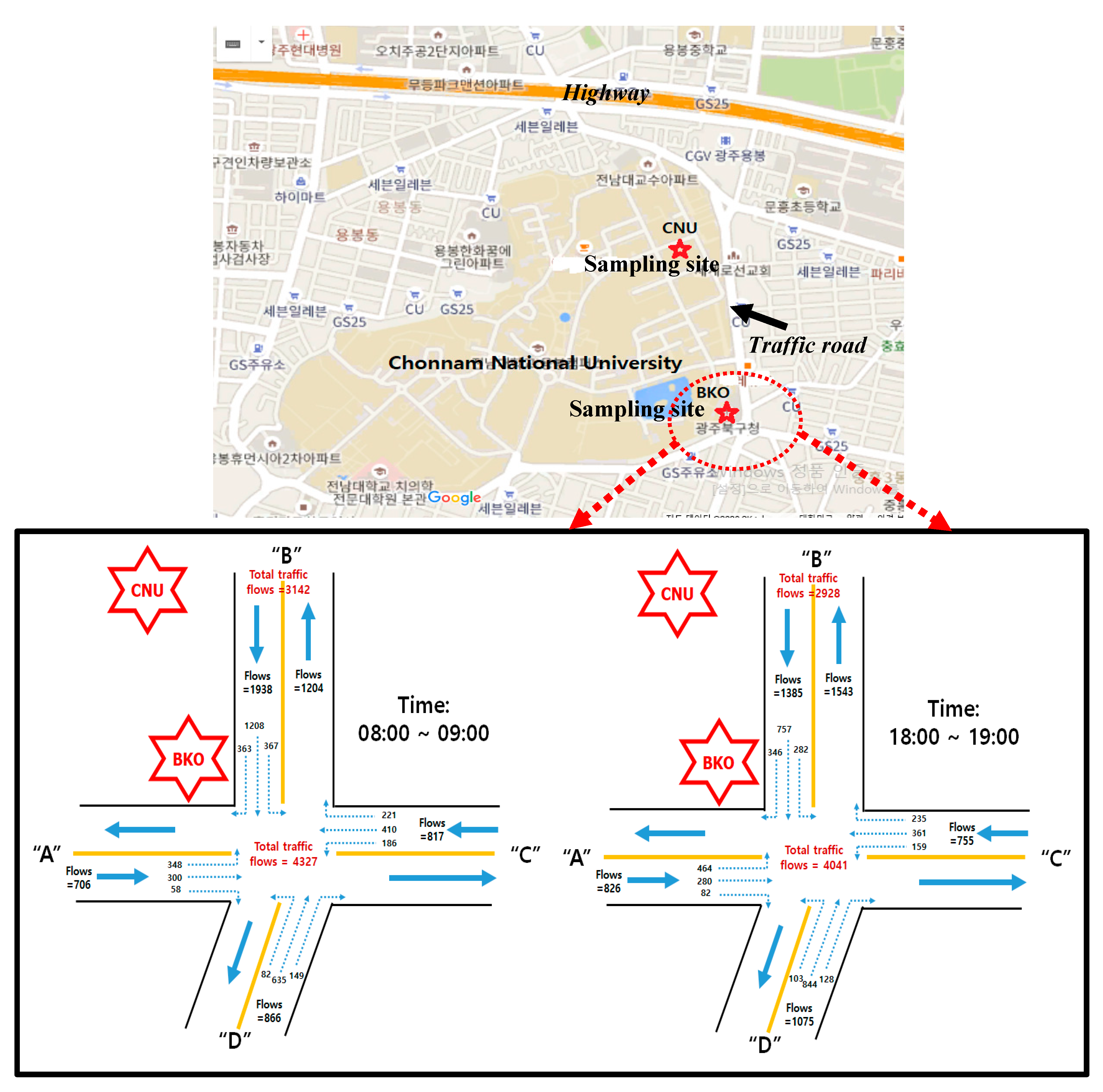
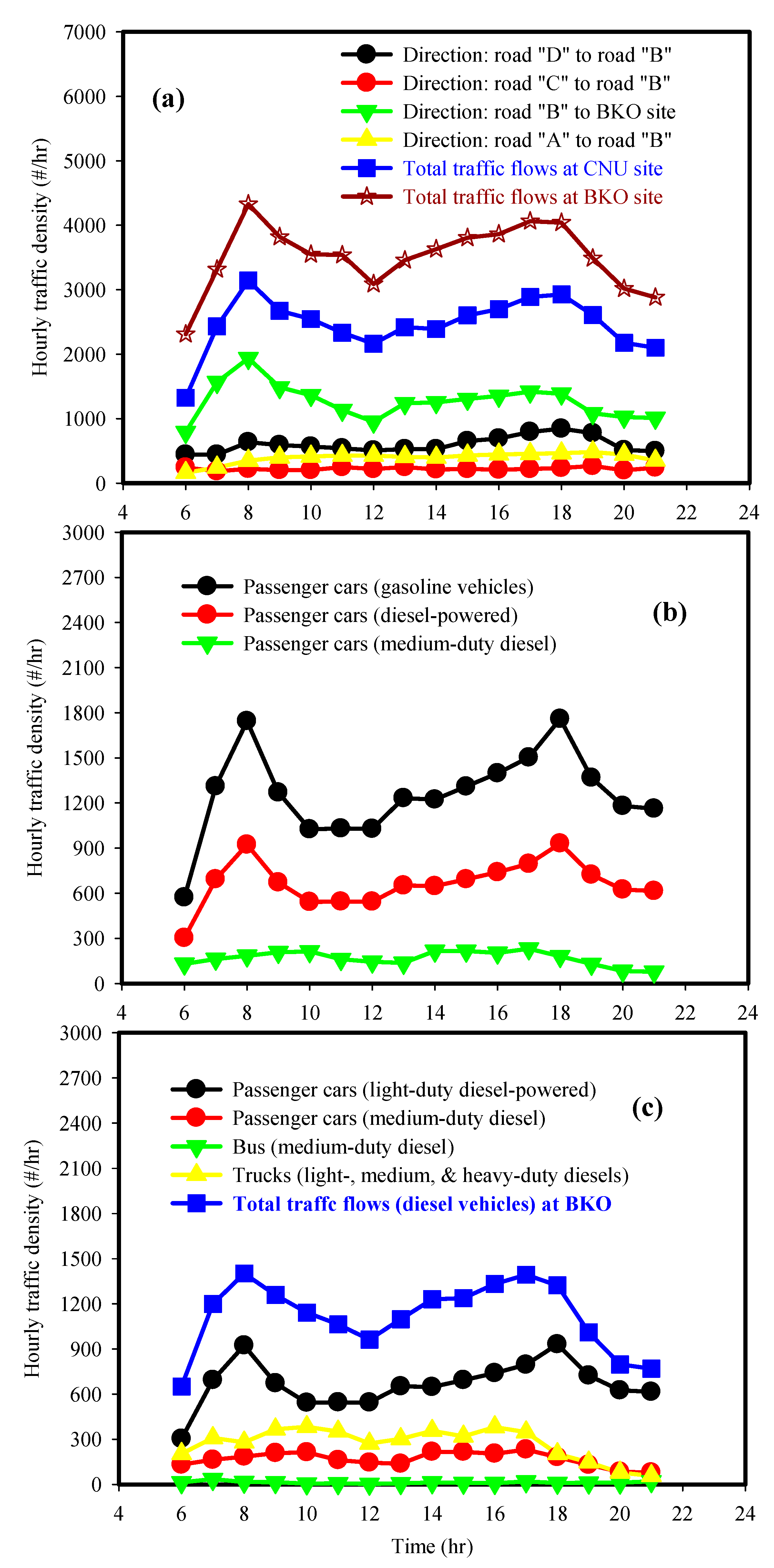
2. Experimental Methods
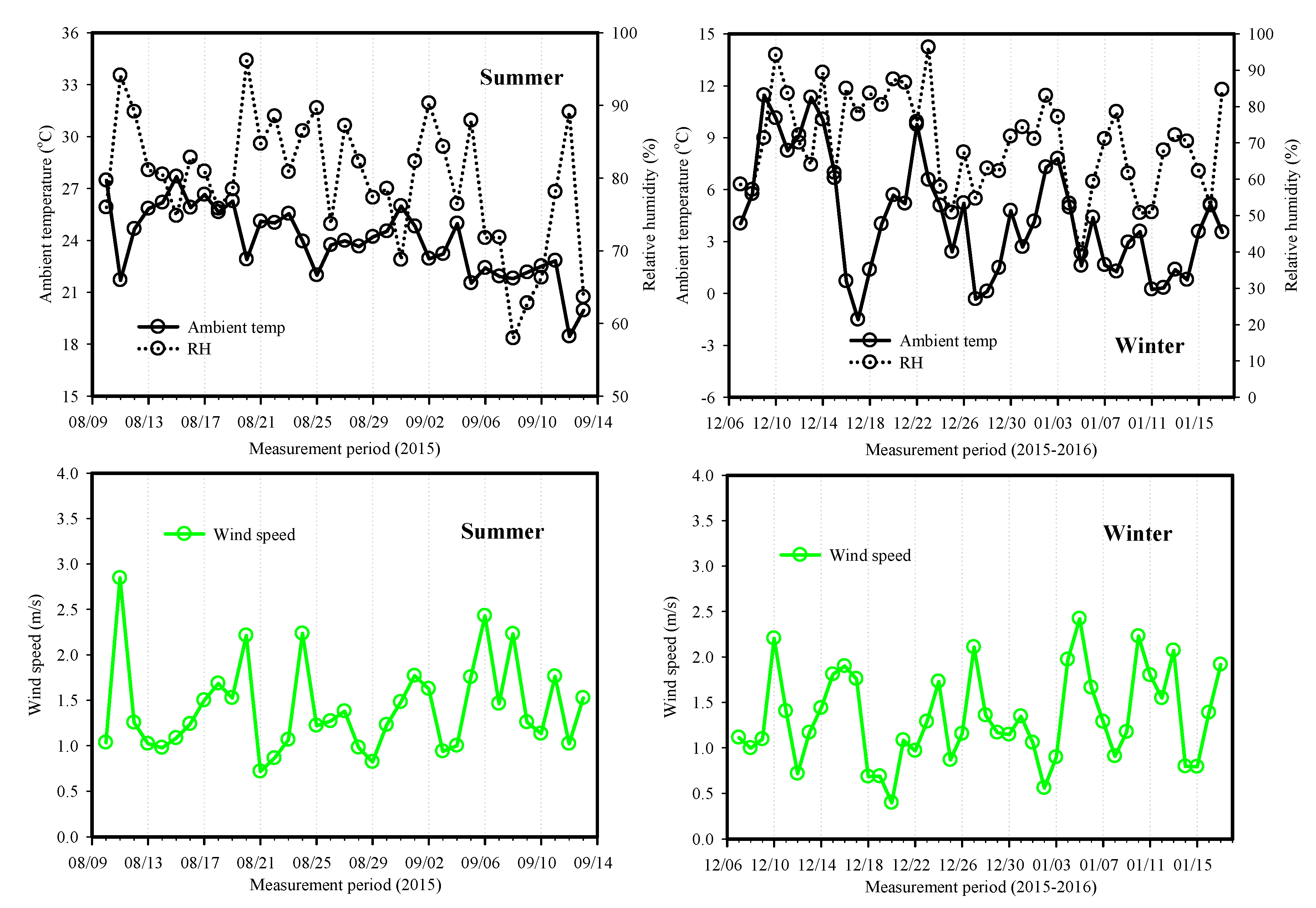
2.1. Measurement of 24 h Integrated PM2.5 and Aerosol Light Absorptions
2.2. Determination of Carbonaceous and Water-Soluble Inorganic Species Concentrations
3. Results and Discussion
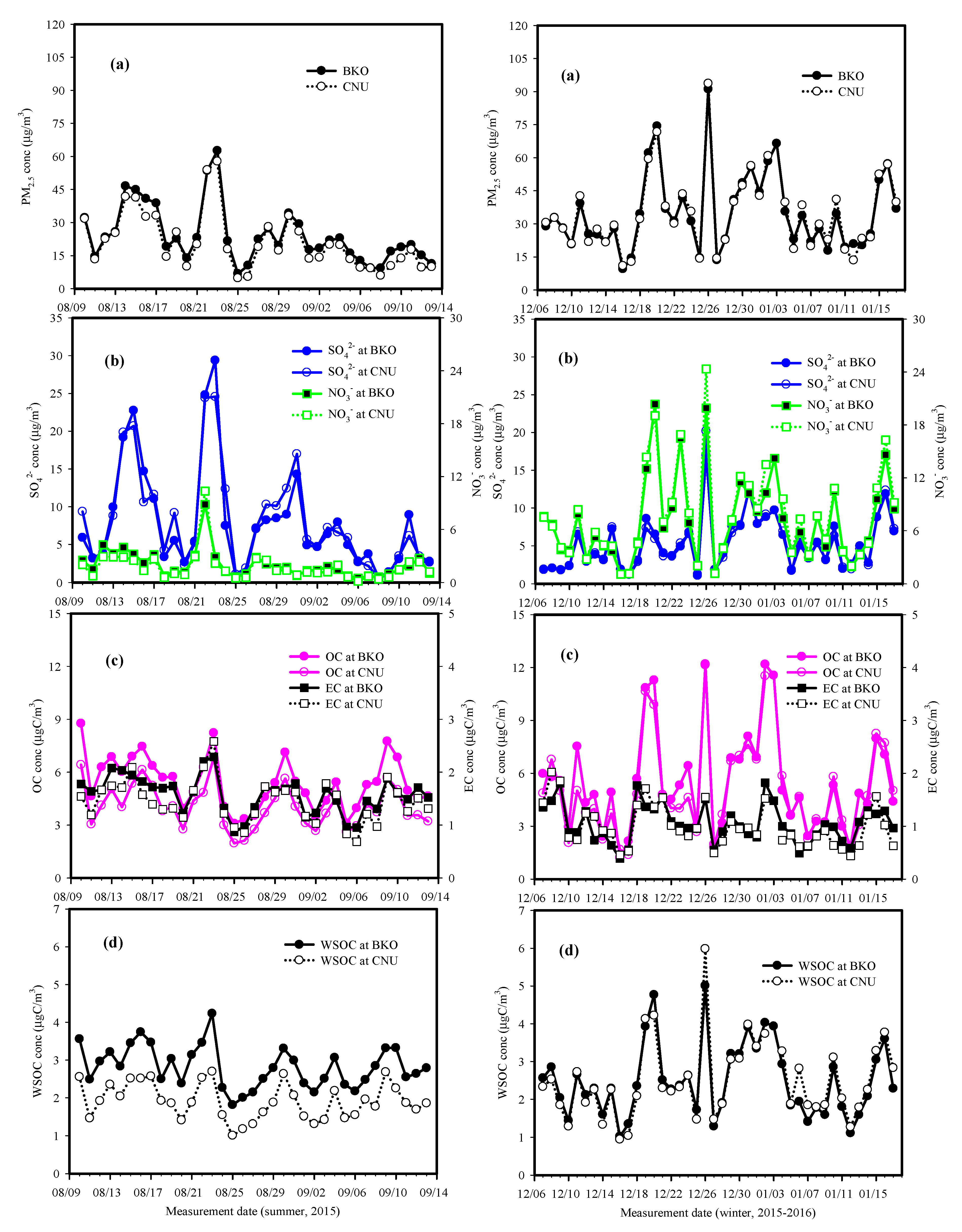
3.1. General Characteristics of PM2.5 and Its Chemical Components
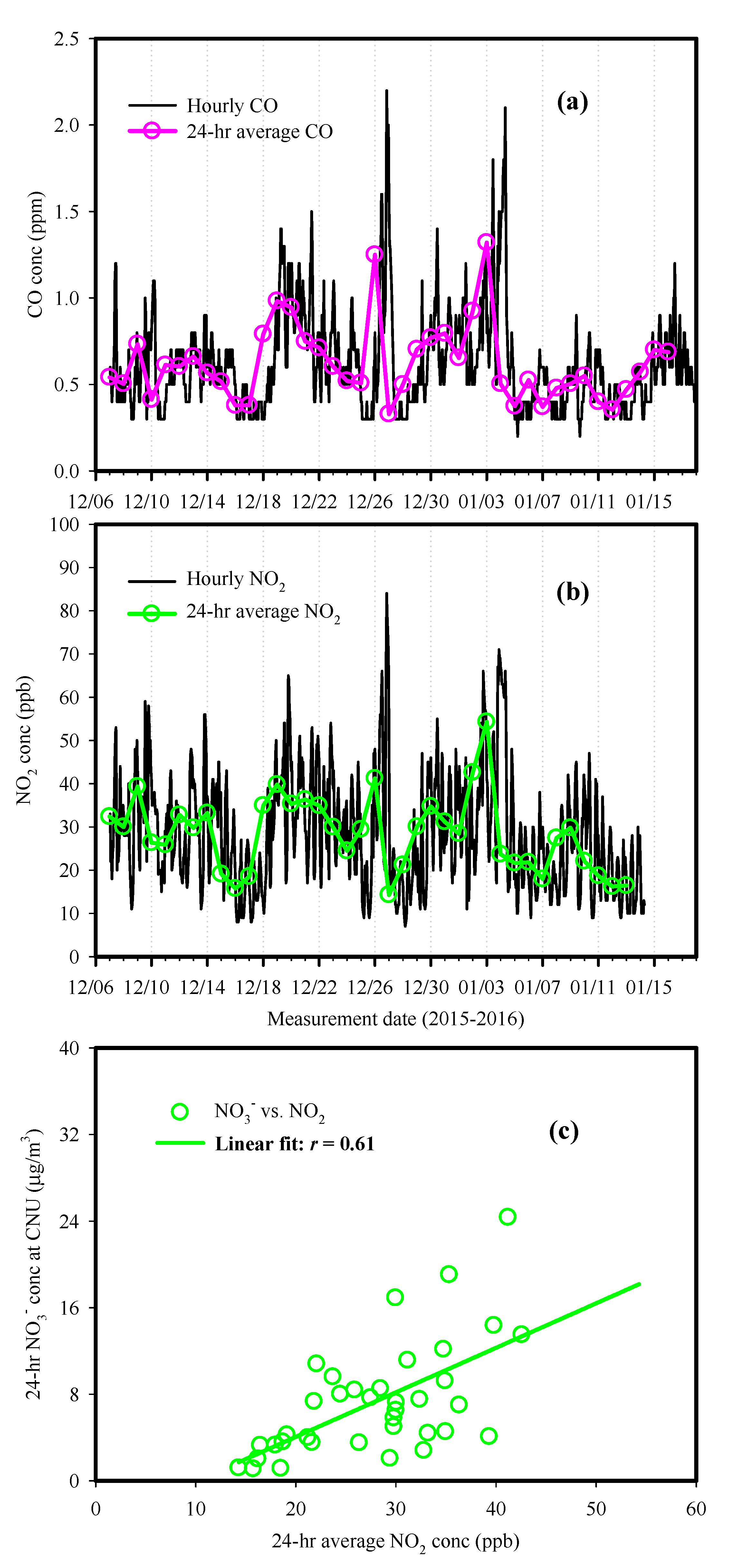
3.2. Possible Sources of Secondary Inorganic Species
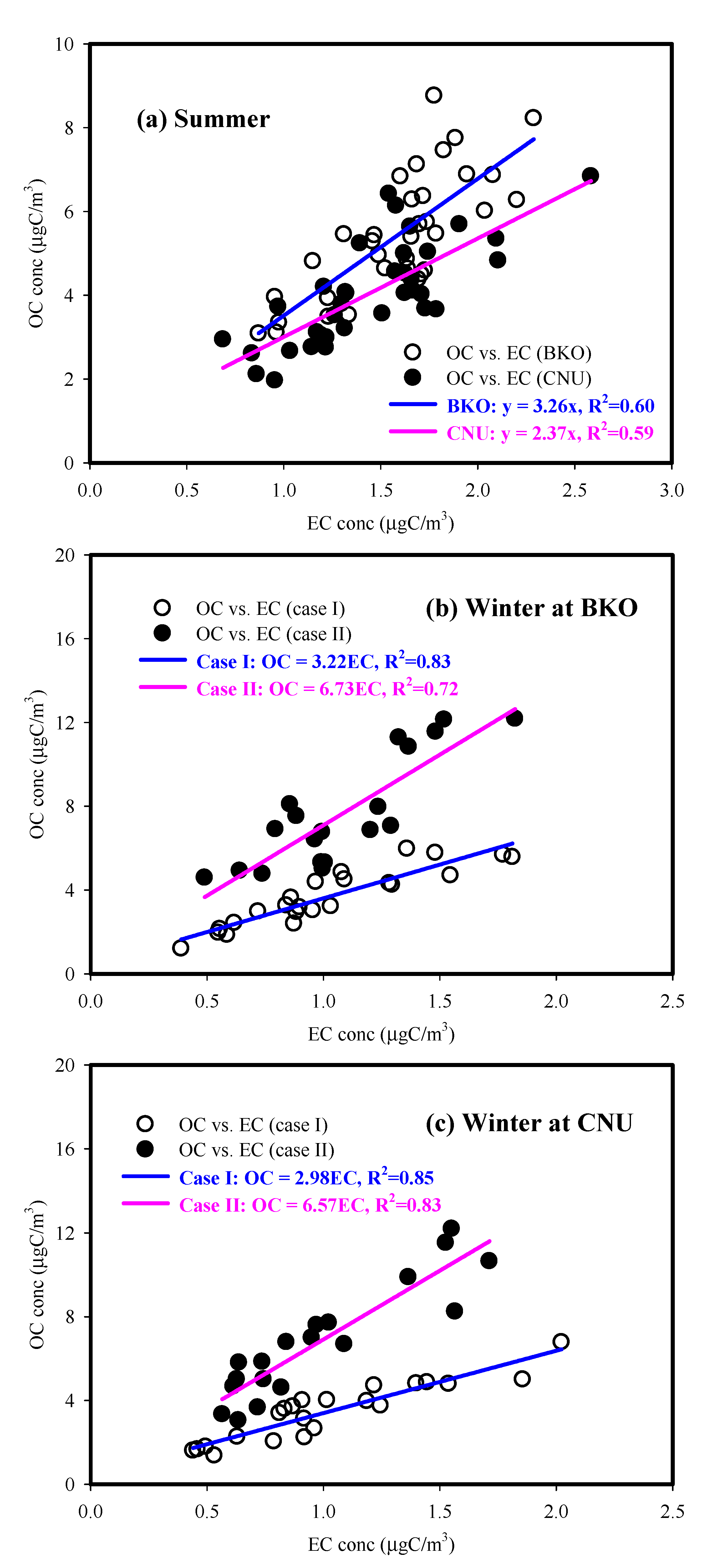
3.3. Characteristics of Carbonaceous Components
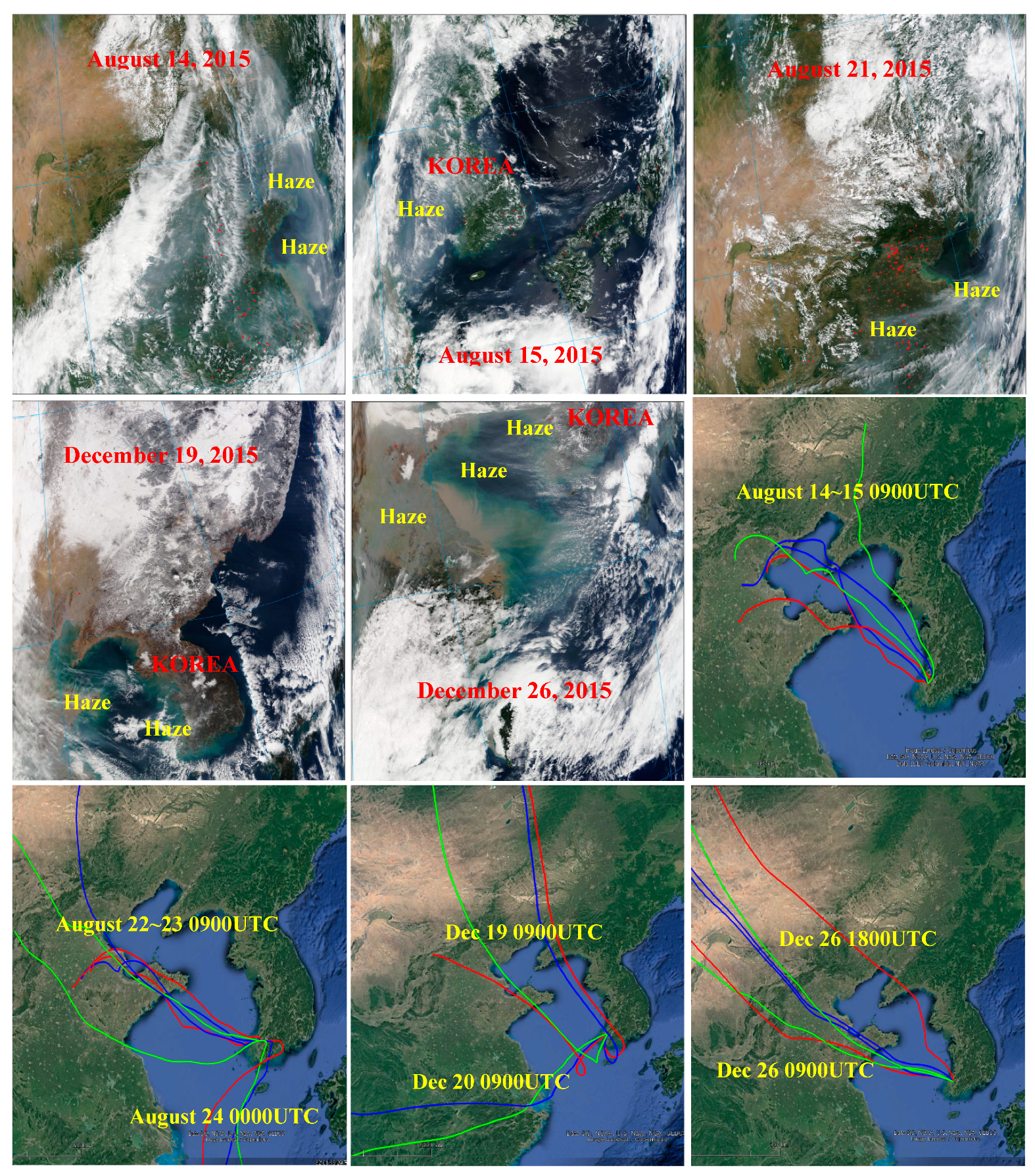
3.4. Characteristics of Pollution Episodes during Summer and Winter
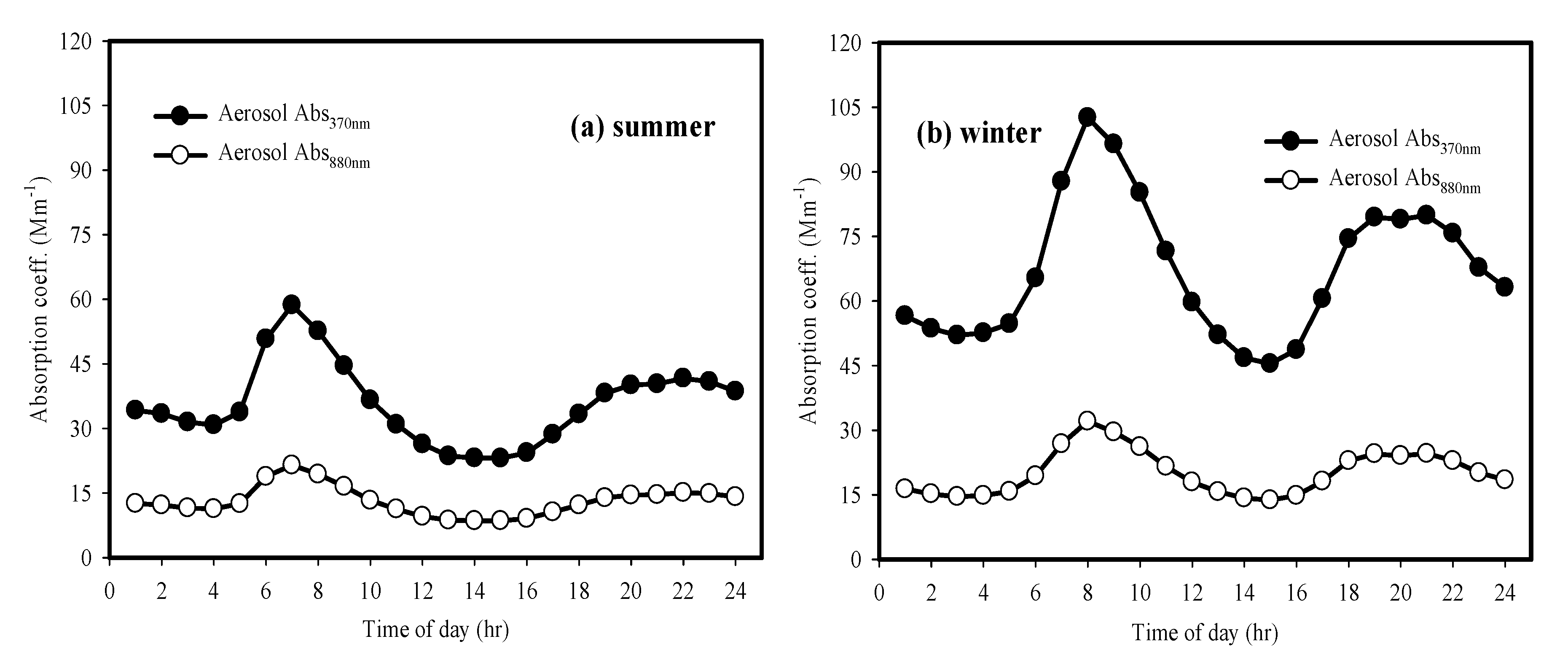
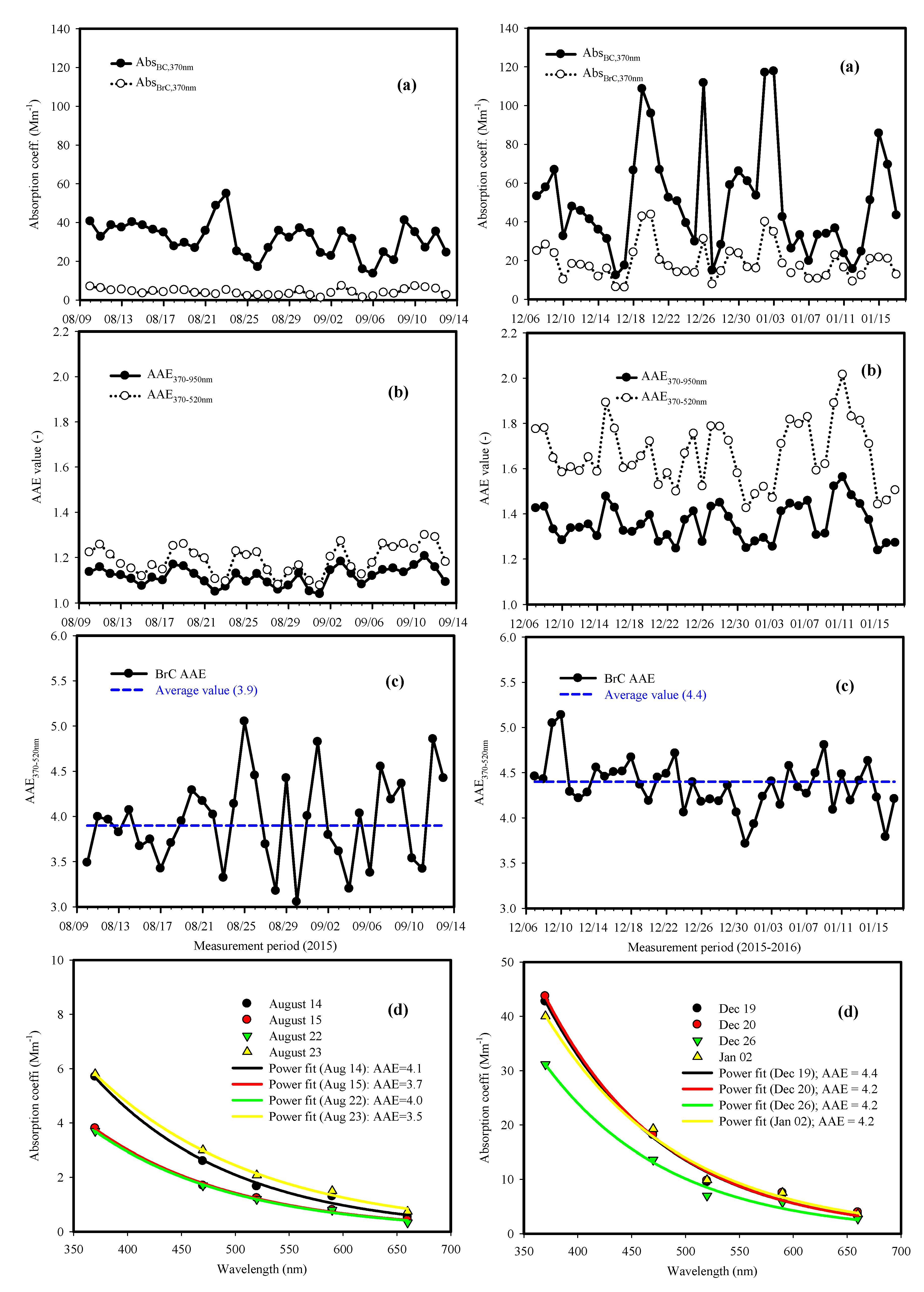
3.5. Optical Properties of Brown Light Absorbing Carbon at the CNU Site
4. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cao, J.J.; Lee, S.C.; Ho, K.F.; Zhang, X.Y.; Zou, S.C.; Fung, K.; Chow, J.C.; Watson, J.G. Characteristics of carbonaceous aerosol in Pearl River Delta Region, China during 2001 winter period. Atmos. Environ. 2003, 37, 1451–1460. [Google Scholar] [CrossRef]
- Park, S.S.; Harrison, D.; Pancras, J.P.; Ondov, J. Time resolved elemental and organic carbon measurements at the Baltimore Supersite in 2002. J. Geophys. Res. Atmos. 2005, 110, D07S06. [Google Scholar] [CrossRef] [Green Version]
- Fuzzi, S.; Andreae, M.O.; Huebert, B.J.; Kulmala, M.; Bond, T.C.; Boy, M.; Doherty, S.J.; Guenther, A.; Kanakidou, M.; Kawamura, K.; et al. Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosols in the atmosphere, climate, and global change. Atmos. Chem. Phys. 2006, 6, 2017–2038. [Google Scholar] [CrossRef] [Green Version]
- Polidori, A.; Turpin, B.J.; Lim, H.J.; Cabada, J.C.; Subramanian, R.; Pandis, S.N.; Robinson, A.L. Local and regional secondary organic aerosol: Insights from a year of semi-continuous carbon measurements at Pittsburgh. Aerosol Sci. Technol. 2006, 40, 861–872. [Google Scholar] [CrossRef]
- Ramanathan, V.; Carmichael, G. Global and regional climate changes due to black carbon. Nat. Geosci. 2008, 1, 221–227. [Google Scholar] [CrossRef]
- Kumar, A.; Ramb, K.; Ojhac, N. Variations in carbonaceous species at a high-altitude site in western India: Role of synoptic scale transport. Atmos. Environ. 2016, 125, 371–382. [Google Scholar] [CrossRef]
- Laskin, A.; Laskin, J.; Nizkorodov, S.A. Chemistry of atmospheric brown carbon. Chem. Rev. 2015, 115, 4335–4382. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Qiao, T.; Huang, Z.; Zhu, M.; Xu, W.; Xiu, G.; Tao, J.; Lee, S. Comparison of ionic and carbonaceous compositions of PM 2.5 in 2009 and 2012 in Shanghai, China. Sci. Total Environ. 2015, 536, 695–703. [Google Scholar] [CrossRef]
- Novakov, T.; Menon, S.; Kirchstetter, T.W.; Koch, D.; Hansen, J.E. Aerosol organic carbon to black carbon ratios: Analysis of published data and implications for climate forcing. J. Geophys. Res. 2005, 110, D21205. [Google Scholar] [CrossRef]
- Park, S.S.; Cho, S.Y. Tracking sources and behaviors of water-soluble organic carbon in fine particulate matter measured at an urban site in Korea. Atmos. Environ. 2011, 45, 60–72. [Google Scholar] [CrossRef]
- Park, S.S.; Schauer, J.J.; Cho, S.Y. Sources and their contribution to two water-soluble organic carbon fractions at a roadway site. Atmos. Environ. 2013, 77, 348–357. [Google Scholar] [CrossRef]
- Park, S.S.; Cho, S.Y.; Bae, M.S. Source identification of water-soluble organic aerosols at a roadway site using positive matrix factorization analysis. Sci. Total Environ. 2015, 533, 410–421. [Google Scholar] [CrossRef]
- Park, S.S.; Lee, K.H. Characterization and sources of black carbon in PM2.5 at a site close to a roadway in Gwangju, Korea, during winter. Environ. Sci. Process. Impacts 2015, 17, 1794–1805. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2006. [Google Scholar]
- Park, S.S.; Kleissl, J.; Harrison, D.; Kumar, V.; Nair, N.P.; Adam, M.; Ondov, J.; Parlange, M. Characteristics of PM2.5 episodes revealed by semi-continuous measurements at the Baltimore Supersite at Ponca St. Aerosol Sci. Technol. 2006, 40, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.J.; Shen, Z.X.; Chow, J.C.; Watson, J.G.; Lee, S.C.; Tie, X.X.; Ho, K.F.; Wang, G.H.; Han, Y.M. Winter and summer PM2.5 chemical compositions in fourteen Chinese Cities. J. Air Waste Manag. Assoc. 2012, 62, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cao, J.J.; Tie, X.X.; Shen, Z.X.; Liu, S.X.; Ding, H.; Han, Y.M.; Wang, G.H.; Ho, K.F.; Qiang, J.; et al. Water-soluble ions in atmospheric aerosols measured in Xi’an, China: Seasonal variations and sources. Atmos. Res. 2011, 102, 110–119. [Google Scholar] [CrossRef]
- Zhang, R.; Jing, J.; Tao, J.; Hsu, S.C.; Wang, G.; Cao, J.; Lee, C.S.L.; Zhu, L.; Chen, Z.; Zhao, Y.; et al. Chemical characterization and source apportionment of PM2.5 in Beijing: Seasonal perspective. Atmos. Chem. Phys. 2013, 13, 7053–7074. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.K.; Sun, Y.; Liu, Z.R.; Ji, D.S.; Hu, B.; Liu, Q.; Wang, Y.S. Characterization of submicron aerosols during a month of serious pollution in Beijing, 2013. Atmos. Chem. Phys. 2014, 14, 2887–2903. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Wang, Y.S.; Ma, Q.X.; Ma, J.Z.; Chu, B.W.; Ji, D.S.; Tang, G.Q.; Liu, C.; Zhang, H.X.; Hao, J.M. Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days. Sci. Rep. 2014, 4, 4172. [Google Scholar] [CrossRef]
- Guo, S.; Hu, M.; Zamora, M.L.; Peng, J.F.; Shang, D.J.; Zheng, J.; Fu, Z.F.; Wu, Z.J.; Shao, M.; Zeng, L.M.; et al. Elucidating severe urban haze formation in China. Proc. Natl. Acad. Sci. USA 2014, 111, 17373–17378. [Google Scholar] [CrossRef] [Green Version]
- Ji, D.S.; Li, L.; Wang, Y.S.; Zhang, J.K.; Cheng, M.T.; Sun, Y.; Liu, Z.R.; Wang, L.L.; Tang, G.Q.; Hu, B.; et al. The heaviest particulate air-pollution episodes occurred in northern China in January, 2013: Insights gained from observation. Atmos. Environ. 2014, 92, 546–556. [Google Scholar] [CrossRef]
- Sun, Y.L.; Jiang, Q.; Wang, Z.F.; Yin, Y. Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013. J. Geophys. Res. 2014, 119, 420–425. [Google Scholar] [CrossRef]
- Yu, G.H.; Cho, S.Y.; Bae, M.S.; Lee, K.H.; Park, S.S. Investigation of PM2.5 pollution episodes in Gwangju. J. Korean Soc. Atmos. Environ. 2015, 31, 269–286, (In Korean with English abstract). [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Wu, Z.; Wang, Y.; Xiao, Y.; Gu, F.; Zheng, J.; Tan, T.; Shang, D.; Wu, Y.; Zeng, L.; et al. Submicrometer particles are in the liquid state during heavy haze episodes in the urban atmosphere of Beijing, China. Environ. Sci. Technol. Lett. 2017, 4, 427–432. [Google Scholar] [CrossRef]
- Yu, G.-H.; Park, S.S.; Park, J.S.; Park, S.M.; Song, I.H.; Oh, J.; Shin, H.J.; Lee, M.D.; Lim, H.B.; Kim, H.W.; et al. Pollution characteristics of PM2.5 observed during winter and summer in Baengryeongdo and Seoul. J. Korean Soc. Atmos. Environ. 2018, 34, 38–55, (In Korean with English abstract). [Google Scholar] [CrossRef]
- Park, S.S.; Jung, S.A.; Gong, B.J.; Cho, S.Y.; Lee, S.J. Characteristics of PM2.5 haze episodes revealed by highly time-resolved measurements at an air pollution monitoring Supersite in Korea. Aerosol Air Qual. Res. 2013, 13, 957–976. [Google Scholar] [CrossRef]
- Park, S.S.; Kim, S.J.; Gong, B.J.; Cho, S.Y.; Kim, J.C.; Lee, S.J. Investigation on a haze episode of fine particulate matter using semi-continuous chemical composition data. J. Korean Soc. Atmos. Environ. 2013, 29, 642–655, (In Korean with English abstract). [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, Z.; Fu, P.; Jiang, Q.; Yang, T.; Li, J.; Ge, X. The impact of relative humidity on aerosol composition and evolution processes during wintertime in Beijing, China. Atmos. Environ. 2013, 77, 927–934. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Zhang, M.; Yang, Y.; Shen, X.; Wang, Y.; Chen, D.; Guo, J. A study of the meteorological causes of a prolonged and severe haze episode in January 2013 over central eastern China. Atmos. Environ. 2014, 98, 146–157. [Google Scholar] [CrossRef]
- Wang, J.D.; Wang, S.X.; Jiang, J.K.; Ding, A.J.; Zheng, M.; Zhao, B.; Wong, D.C.; Zhou, W.; Zheng, G.; Wang, L. Impact of aerosol-meteorology interactions on fine particle pollution during China’s severe haze episode in January 2013. Environ. Res. Lett. 2014, 9, 094002. [Google Scholar] [CrossRef]
- Ma, J.; Chu, B.; Liu, J.; Liu, Y.; Zhang, H.; He, H. NOx promotion of SO2 conversion to sulfate: An important mechanism for the occurrence of heavy haze during winter in Beijing. Environ. Pollut. 2018, 233, 662–669. [Google Scholar] [CrossRef]
- Yu, G.H.; Park, S.S.; Jung, S.A.; Jo, M.R.; Lim, Y.J.; Shin, H.J.; Lee, S.B.; Ghim, Y.S. Investigation on Characteristics of high PM2.5 pollution occurred during October 2015 in Gwangju. J. Korean Soc. Atmos. Environ. 2018, 34, 567–587, (In Korean with English abstract). [Google Scholar] [CrossRef]
- Yu, G.H.; Lee, B.J.; Park, S.S.; Jung, S.A.; Jo, M.R.; Lim, Y.J.; Kim, S. A case study of severe PM2.5 event in the Gwangju urban area during February 2014. J. Korean Soc. Atmos. Environ. 2019, 35, 195–213, (In Korean with English abstract). [Google Scholar] [CrossRef]
- Saxena, M.; Sharma, A.; Sen, A.; Saxena, P.; Saraswati, P.; Mandal, T.K.; Sharma, S.K.; Sharma, C. Water soluble inorganic species of PM10 and PM2.5 at an urban site of Delhi, India: Seasonal variability and sources. Atmos. Res. 2017, 184, 112–125. [Google Scholar] [CrossRef]
- Zhao, P.S.; Dong, F.; He, D.; Zhao, X.J.; Zhang, X.L.; Zhang, W.Z.; Yao, Q.; Liu, H.Y. Characteristics of concentrations and chemical compositions for PM2.5 in the region of Beijing, Tianjin, and Hebei, China. Atmos. Chem. Phys. 2013, 13, 4631–4644. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.L.; Pan, Y.P.; Wang, Y.S. Size-resolved source apportionment of particulate matter in urban Beijing during haze and non-haze episodes. Atmos. Chem. Phys. 2016, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhang, R.; Gomez, M.E.; Yang, L.; Levy Zamora, M.; Hu, M.; Lin, Y.; Peng, J.; Guo, S.; Meng, J.; et al. Persistent sulfate formation from London Fog to Chinese haze. Proc. Natl. Acad. Sci. USA 2016, 113, 13630–13635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Sun, X.; Shi, A.; Huang, Y.; Yan, J.; Nie, T.; Yan, X.; Li, X. Secondary inorganic aerosols formation during haze episodes at an urban site in Beijing, China. Atmos. Environ. 2018, 177, 275–282. [Google Scholar] [CrossRef]
- Park, S.S.; Son, S.C. Relationship between carbonaceous components and aerosol light absorption during winter at an urban site of Gwangju, Korea. Atmos. Res. 2017, 185, 73–83. [Google Scholar] [CrossRef]
- Zhu, Y.; Hinds, W.C. Concentration and size distribution of ultrafine particles near a major highway. J. Air Waste Manag. Assoc. 2002, 52, 1032–1042. [Google Scholar] [CrossRef]
- Zhu, Y.; Hinds, W.C.; Kim, S.; Shen, S.; Sioutas, C. Study of ultrafine particles near a major highway with heavy-duty diesel traffic. Atmos. Environ. 2002, 36, 4323–4335. [Google Scholar] [CrossRef]
- Morawska, L.; Tomas, S.; Gilbert, D.; Greenaway, C.; Rijnders, E. A study of the horizontal and vertical profile of submicrometer particles in relation to a busy road. Atmos. Environ. 1999, 33, 1261–1274. [Google Scholar] [CrossRef]
- Hitchins, J.; Morawska, L.; Wolff, R.; Gilbert, D. Concentrations of submicrometre particles from vehicle emissions near a major road. Atmos. Environ. 2000, 34, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Ghim, Y.S.; Won, S.R.; Choi, Y.; Chang, Y.S.; Jin, H.C.; Kim, Y.P.; Kang, C.H. Concentration variations in primary and secondary particulate matter near a major road in Korea. Asian J. Atmos. Environ. 2016, 10, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Gwangju Metropolitan City. A Report of Basic Survey on the Volume Traffic in Gwangju in 2015; Gwangju City: Gwangju, Korea, 2016. [Google Scholar]
- Drinovec, L.; Močnik, G.; Zotter, P.; Prévôt, A.S.H.; Ruckstuhl, C.; Coz, E.; Rupakheti, M.; Sciare, J.; Müller, T.; Wiedensohler, A.; et al. The “dualspot” Aethalometer: An improved measurement of aerosol black carbon with real-time loading compensation. Atmos. Meas. Tech. 2015, 8, 1965–1979. [Google Scholar] [CrossRef] [Green Version]
- Martinsson, J.; Eriksson, A.C.; Elbæk Nielsen, I.; Berg Malmborg, V.; Ahlberg, E.; Andersen, C.; Lindgren, R.; Nyström, R.; Nordin, E.Z.; Brune, W.H.; et al. Impacts of combustion conditions and photochemical processing on the light absorption of biomass combustion aerosol. Environ. Sci. Technol. 2015, 49, 14663–14671. [Google Scholar] [CrossRef]
- Park, S.; Yu, G.H.; Bae, M.S. Effects of combustion condition and biomass type on the light absorption of fine organic aerosols from fresh biomass burning emissions over Korea. Environ. Pollut. 2020, 265, 114841. [Google Scholar] [CrossRef]
- NIOSH. Method 5040 issue 1: Elemental carbon (diesel exhaust). In NIOSH Manual of Analytical Methods, 4th ed.; National Institute of Occupational Safety and Health: Cincinnati, OH, USA, 1996. [Google Scholar]
- Park, S.; Yu, G.H.; Lee, S. Optical absorption characteristics of brown carbon aerosols during the KORUS-AQ campaign at an urban site. Atmos. Res. 2018, 203, 16–27. [Google Scholar] [CrossRef]
- Park, S.; Son, S.C.; Lee, S. Characterization, sources, and light absorption of fine organic aerosols during summer and winter at an urban site. Atmos. Res. 2018, 213, 370–380. [Google Scholar] [CrossRef]
- Kim, B.M.; Teffera, S.; Zeldin, M.D. Characterization of PM25 and PM10 in the South Coast Air Basin of Southern California: Part 1—Spatial variations. J. Air Waste Manag. Assoc. 2000, 50, 2034–2044. [Google Scholar] [CrossRef]
- Yao, X.; Chan, C.K.; Fang, M.; Cadle, S.; Chan, T.; Mulawa, P.; He, K.; Ye, B. The water-soluble ionic composition of PM 2.5 in Shanghai and Beijing, China. Atmos. Environ. 2002, 36, 4223–4234. [Google Scholar] [CrossRef]
- Cho, S.Y.; Park, S.S. Resolving sources of water-soluble organic carbon in fine particulate matter measured during winter at an urban site. Environ. Sci. Process. Impacts 2013, 15, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.J.; Lim, H.J. Species Contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass. Aerosol Sci. Technol. 2001, 35, 602–610. [Google Scholar] [CrossRef]
- Cao, J.; Lee, S.C.; Ho, K.F.; Zou, S.C.; Fung, K.; Li, Y.; Watson, J.G.; Chow, J.C. Spatial and seasonal variations of atmospheric organic carbon and elemental carbon in Pearl River Delta Region, China. Atmos. Environ. 2004, 38, 4447–4456. [Google Scholar] [CrossRef]
- Ram, K.; Sarin, M.M.; Tripathi, S.N. One-year record of carbonaceous aerosols from an urban location (Kanpur) in the Indo-Gangetic Plain: Characterization, sources and temporal variability. J. Geophys. Res. 2010, 115, D24313. [Google Scholar] [CrossRef]
- Wang, J.; Ho, S.S.H.; Cao, J.; Huang, R.; Zhou, J.; Zhao, Y.; Xu, H.; Liu, S.; Wang, G.; Shen, Z.; et al. Characteristics and major sources of carbonaceous aerosols in PM2.5 from Sanya, China. Sci. Total Environ. 2015, 530–531, 110–119. [Google Scholar] [CrossRef]
- Lim, H.J.; Turpin, B.J. Origins of primary and secondary organic aerosols in Atlanta: Results of time-resolved measurements during the Atlanta supersite experiment. Environ. Sci. Technol. 2002, 36, 4489–4496. [Google Scholar] [CrossRef]
- Jaffrezo, J.L.; Aymoz, G.; Delaval, C.; Cozic, J. Seasonal variation of the water soluble organic carbon mass fraction of aerosol in two valleys of the French Alps. Atmos. Chem. Phys. 2005, 5, 2809–2821. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; Kondo, Y.; Takegawa, N.; Komazaki, Y.; Fukuda, M.; Kawamura, K.; Mochida, M.; Okuzawa, K.; Weber, R.J. Time-resolved measurements of water-soluble organic carbon in Tokyo. J. Geophys. Res. 2006, 111, D23206. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, A.P.; Weber, R.J. Chemical characterization of the ambient organic aerosol soluble in water: 1. Isolation of hydrophilic and hydrophobic fractions with a XAD-8 resin. J. Geophys. Res. 2006, 111, D05314. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.F.; Lee, S.C.; Cao, J.J.; Li, Y.S.; Chow, J.C.; Watson, J.G.; Fung, K. Variability of organic and elemental carbon, water soluble organic carbon, and isotopes in Hong Kong. Atmos. Chem. Phys. 2006, 6, 4569–4576. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.J.; Sullivan, A.P.; Peltier, R.E.; Russell, A.; Yan, B.; Zheng, M.; de Gouw, J.; Warneke, C.; Brock, C.; Holloway, J.S.; et al. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States. J. Geophys. Res. 2007, 112, D13302. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Kondo, Y.; Han, S.; Koike, M.; Kodama, D.; Komazaki, Y.; Tanimoto, H.; Matsueda, H. Chemical characteristics of water-soluble organic carbon in the Asian outflow. J. Geophys. Res. 2007, 112, D22S30. [Google Scholar] [CrossRef]
- Park, S.S.; Sim, S.Y.; Bae, M.S.; Schauer, J.J. Size distribution of water-soluble components in particulate matter emitted from biomass burning. Atmos. Environ. 2013, 73, 62–72. [Google Scholar] [CrossRef]
- Fan, X.; Wei, S.; Zhu, M.; Song, J.; Peng, P. Comprehensive characterization of humic-like substances in smoke PM2.5 emitted from the combustion of biomass materials and fossil fuels. Atmos. Chem. Phys. 2016, 16, 13321–13340. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Yu, J. Chemical and light absorption properties of humic-like substances from biomass burning emissions under controlled combustion experiments. Atmos. Environ. 2016, 136, 114–122. [Google Scholar] [CrossRef]
- Yu, J.; Yu, G.H.; Park, S.; Bae, M.S. Chemical and absorption characteristics of water-soluble organic carbon and humic-like substances in size-segregated particles from biomass burning emissions. Asian J. Atmos. Environ. 2017, 11, 1–11. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.H.; Surratt, J.D.; Weber, R.J. Sources, composition and absorption Angstrom exponent of light-absorbing organic components in aerosol extracts from the Los Angeles Basin. Environ. Sci. Technol. 2013, 47, 3685–3693. [Google Scholar] [CrossRef]
- Yu, G.H.; Park, S.; Shin, S.K.; Lee, K.H.; Nam, H.G. Enhanced light absorption due to aerosol particles in ship plumes observed at a seashore site. Atmos. Pollut. Res. 2018, 9, 1177–1183. [Google Scholar] [CrossRef]
- Yu, G.H.; Park, S.; Lee, K.H. Light Absorption characteristics of fine particles using a real-time optical absorption measurement instrument at a seashore wharf. J. Korean Soc. Atmos. Environ. 2019, 35, 564–576. [Google Scholar] [CrossRef]
- Jung, J.; Kim, Y. Tracking sources of severe haze episodes and their physicochemical and hygroscopic properties under Asian continental outflow: Long range transport pollution, post-harvest biomass burning, and Asian dust. J. Geophys. Res. 2011, 116, D02206. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time environmental applications and display system: Ready. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Kirchstetter, T.W.; Novakov, T.; Hobbs, P.V. Evidence that spectral dependence of light absorption by aerosols is affected by organic carbon. J. Geophys. Res. 2004, 109, D21208. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Hay, M.D.; Holder, A.L. Light-absorbing organic carbon from prescribed and laboratory biomass burning and gasoline vehicle emissions. Sci. Rep. 2017, 7, 7318. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Bond, T.C. Light absorption by organic carbon from wood combustion. Atmos. Chem. Phys. 2010, 10, 1773–1787. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Shen, Z.; Zhang, T.; Zhang, Q.; Wang, Q.; Sun, J.; Gong, X.; Cao, J.; Xu, H.; Liu, S.; et al. Optical source profiles of brown carbon in size-resolved particulate matter from typical domestic biofuel burning over Guanzhong Plain, China. Sci. Total Environ. 2018, 622–623, 244–251. [Google Scholar] [CrossRef]
- Xie, M.; Chen, X.; Holder, A.L.; Hays, M.D.; Lewandowski, M.; Offenberg, J.H.; Kleindienst, T.E.; Jaoui, M.; Hannigan, M.P. Light absorption of organic carbon and its sources at a southeastern U.S. location in summer. Environ. Pollut. 2019, 244, 38–46. [Google Scholar] [CrossRef]
- Ye, Z.; Qu, Z.; Ma, S.; Luo, S.; Chen, Y.; Chen, H.; Chen, Y.; Zhao, Z.; Chen, M.; Ge, X. A comprehensive investigation of aqueous-phase photochemical oxidation of 4-ethylphenol. Sci. Total Environ. 2019, 685, 976–985. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | CNU Site | BKO Site | ||||||
|---|---|---|---|---|---|---|---|---|
| Summer | Winter | Summer | Winter | |||||
| Parameters | Range (µg/m3) | Mean (µg/m3) | Range (µg/m3) | Mean (µg/m3) | Range (µg/m3) | Mean (µg/m3) | Range (µg/m3) | Mean (µg/m3) |
| PM2.5 | 8.8–61.9 | 24.2 | 11.0–93.7 | 34.6 | 9.8–62.6 | 26.3 | 9.5–91.1 | 34.9 |
| OC | 2.0–6.8 | 4.1 | 1.4–12.2 | 5.0 | 3.1–8.8 | 5.4 | 1.2–12.2 | 5.5 |
| EC | 0.7–2.6 | 1.5 | 0.4–2.0 | 1.0 | 0.9–2.3 | 1.6 | 0.4–1.8 | 1.1 |
| WSOC | 1.0–2.7 | 1.9 | 1.0–6.0 | 2.5 | 1.8–4.2 | 2.8 | 0.8–6.2 | 2.6 |
| OC/EC | 2.1–4.3 | 2.8 | 2.4–9.1 | 5.1 | 2.6–4.9 | 3.4 | 2.7–9.5 | 5.2 |
| WSOC/OC | 0.22–0.46 | 0.38 | 0.32–0.82 | 0.54 | 0.24–0.59 | 0.42 | 0.31–0.70 | 0.49 |
| WSOC/EC | 0.80–2.25 | 1.36 | 1.0–4.88 | 2.67 | 1.39–2.44 | 1.82 | 1.13–4.57 | 2.46 |
| NH4+ | 0.1–11.1 | 2.4 | 1.0–13.4 | 4.2 | 0.2–9.3 | 2.4 | 0.8–12.9 | 4.0 |
| K+ | 0.03–0.61 | 0.15 | 0.07–0.68 | 0.24 | 0.03–0.60 | 0.14 | 0.08–0.71 | 0.25 |
| Cl− | 0.01–0.30 | 0.05 | 0.28–2.05 | 0.60 | 0.01–0.25 | 0.05 | 0.22–1.82 | 0.55 |
| NO3− | 0.2–10.3 | 1.8 | 1.2–27.7 | 8.8 | 0.5–8.8 | 2.0 | 1.2–26.2 | 8.4 |
| SO42− | 0.6–24.6 | 8.1 | 1.4–22.9 | 6.0 | 0.7–29.4 | 7.8 | 1.2–22.3 | 6.2 |
| Oxalate | N.D.–0.26 | 0.14 | N.D.–0.40 | 0.13 | N.D.–0.42 | 0.19 | N.D.–0.37 | 0.14 |
| NSS K+ | 0.01–0.58 | 0.12 | 0.07–0.66 | 0.23 | 0.03–0.55 | 0.13 | 0.07–0.70 | 0.24 |
| ∑SIS (1) | 1.2–45.8 | 12.2 | 3.6–64.0 | 19.0 | 1.3–42.5 | 12.3 | 3.5–61.4 | 18.5 |
| NO3−/SO42− | 0.05–0.79 | 0.29 | 0.57–4.06 | 1.55 | 0.06–1.09 | 0.38 | 0.51–4.0 | 1.47 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Thi Hong, H.D.; Cho, S.Y.; Bae, M.-S. Chemical Composition and Light Absorption of PM2.5 Observed at Two Sites near a Busy Road during Summer and Winter. Appl. Sci. 2020, 10, 4858. https://0-doi-org.brum.beds.ac.uk/10.3390/app10144858
Park S, Thi Hong HD, Cho SY, Bae M-S. Chemical Composition and Light Absorption of PM2.5 Observed at Two Sites near a Busy Road during Summer and Winter. Applied Sciences. 2020; 10(14):4858. https://0-doi-org.brum.beds.ac.uk/10.3390/app10144858
Chicago/Turabian StylePark, Seungshik, Hue Dinh Thi Hong, Sung Yong Cho, and Min-Suk Bae. 2020. "Chemical Composition and Light Absorption of PM2.5 Observed at Two Sites near a Busy Road during Summer and Winter" Applied Sciences 10, no. 14: 4858. https://0-doi-org.brum.beds.ac.uk/10.3390/app10144858