
PLGA-CS-PEG Microparticles for Controlled Drug Delivery in the Treatment of Triple Negative Breast Cancer Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Drug-Loaded PLGA-CS-PEG Microspheres
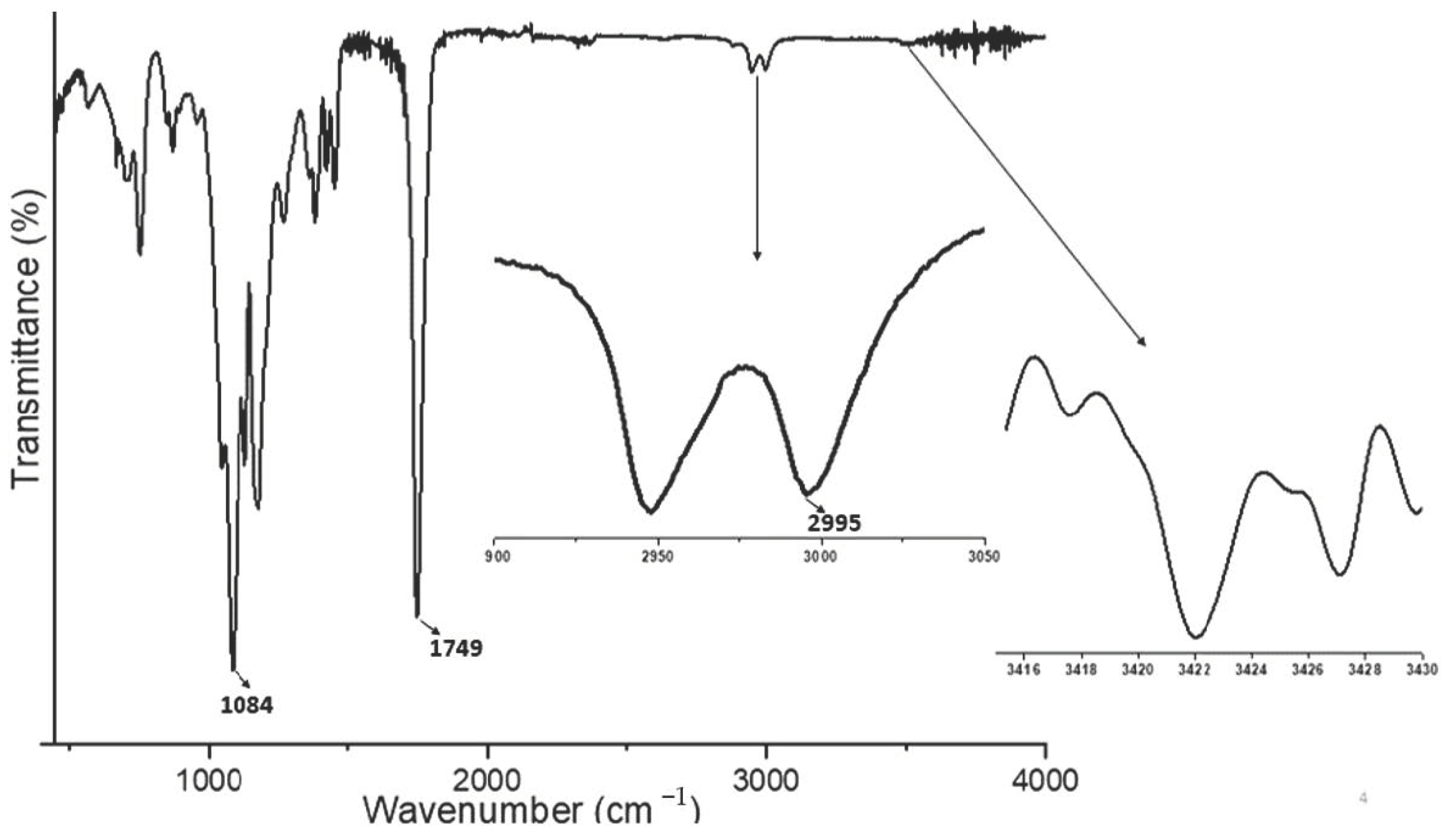
Material Characterization of Drug-Loaded Microparticles
2.3. In Vitro Drug Release
2.4. Modeling of Drug Release
2.4.1. Kinetics of Drug Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Kinetic Model | Equation |
|---|---|---|
| 1 | Zeroth order | |
| 2 | First order | |
| 3 | Higuchi | |
| 4 | Korsmeyer–Peppas (K–P) |
2.4.2. In Vitro Drug Release Thermodynamics
2.5. In Vitro Cell Viability and Cytotoxicity
2.6. Statistical Analysis
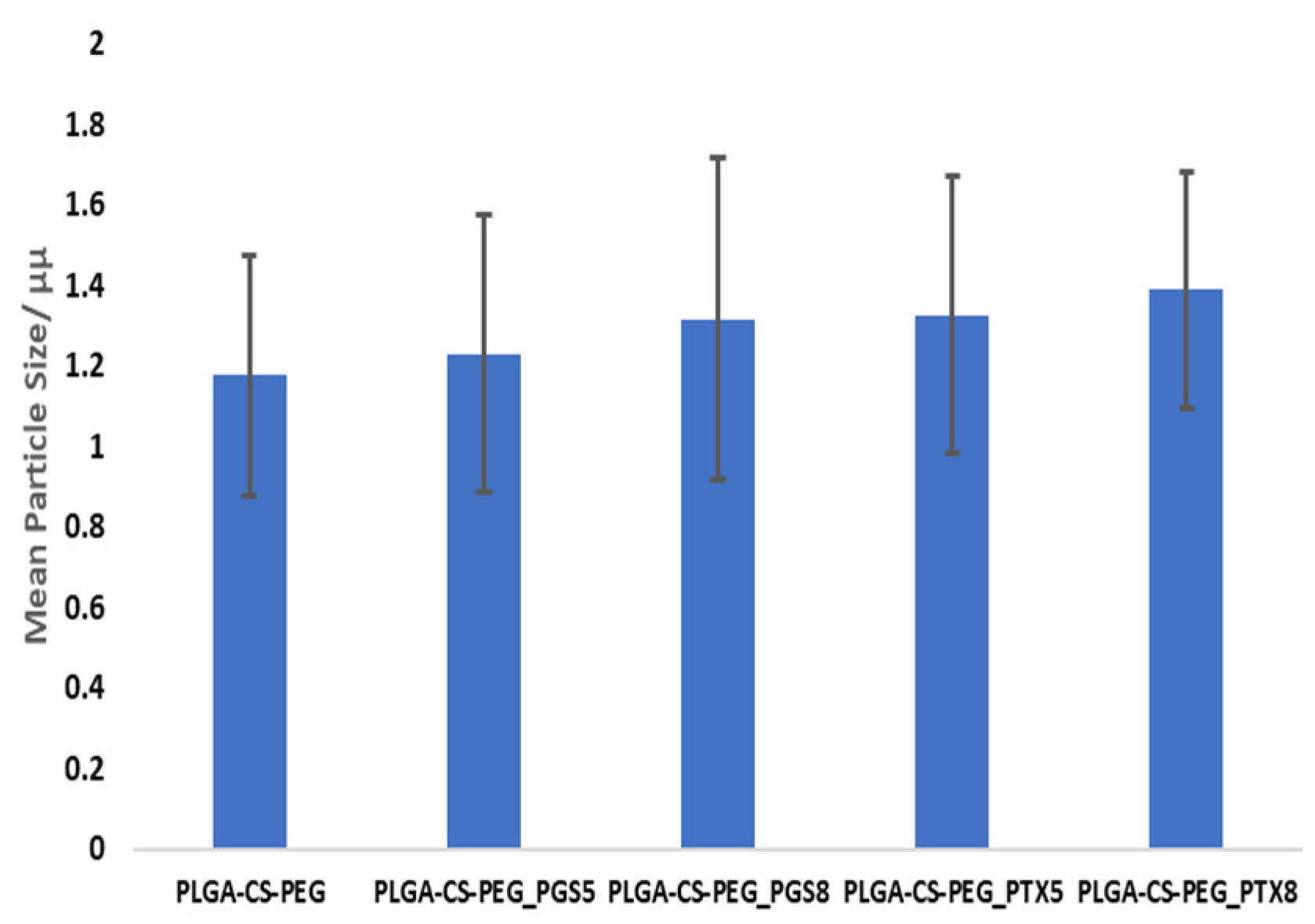
3. Results
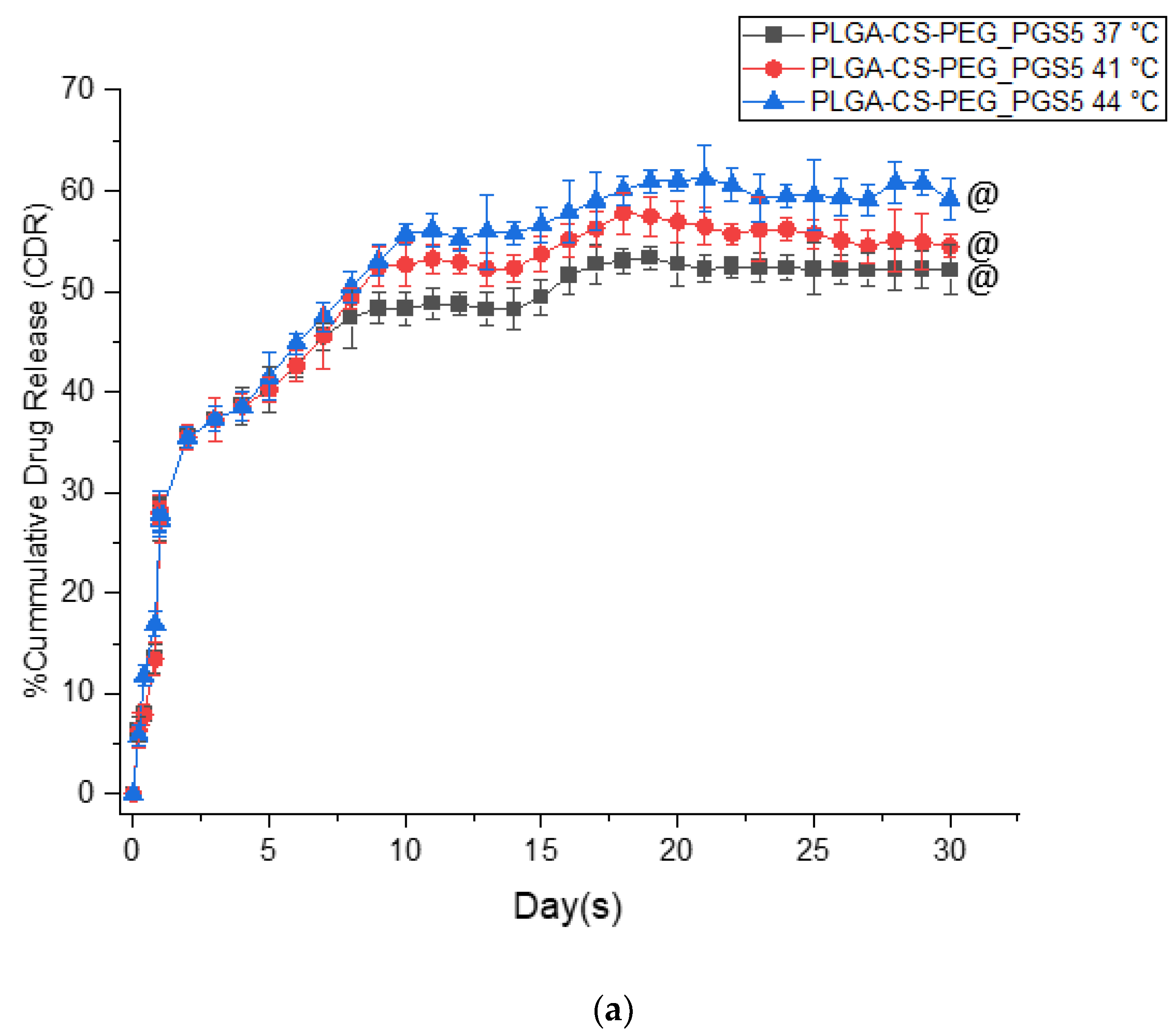
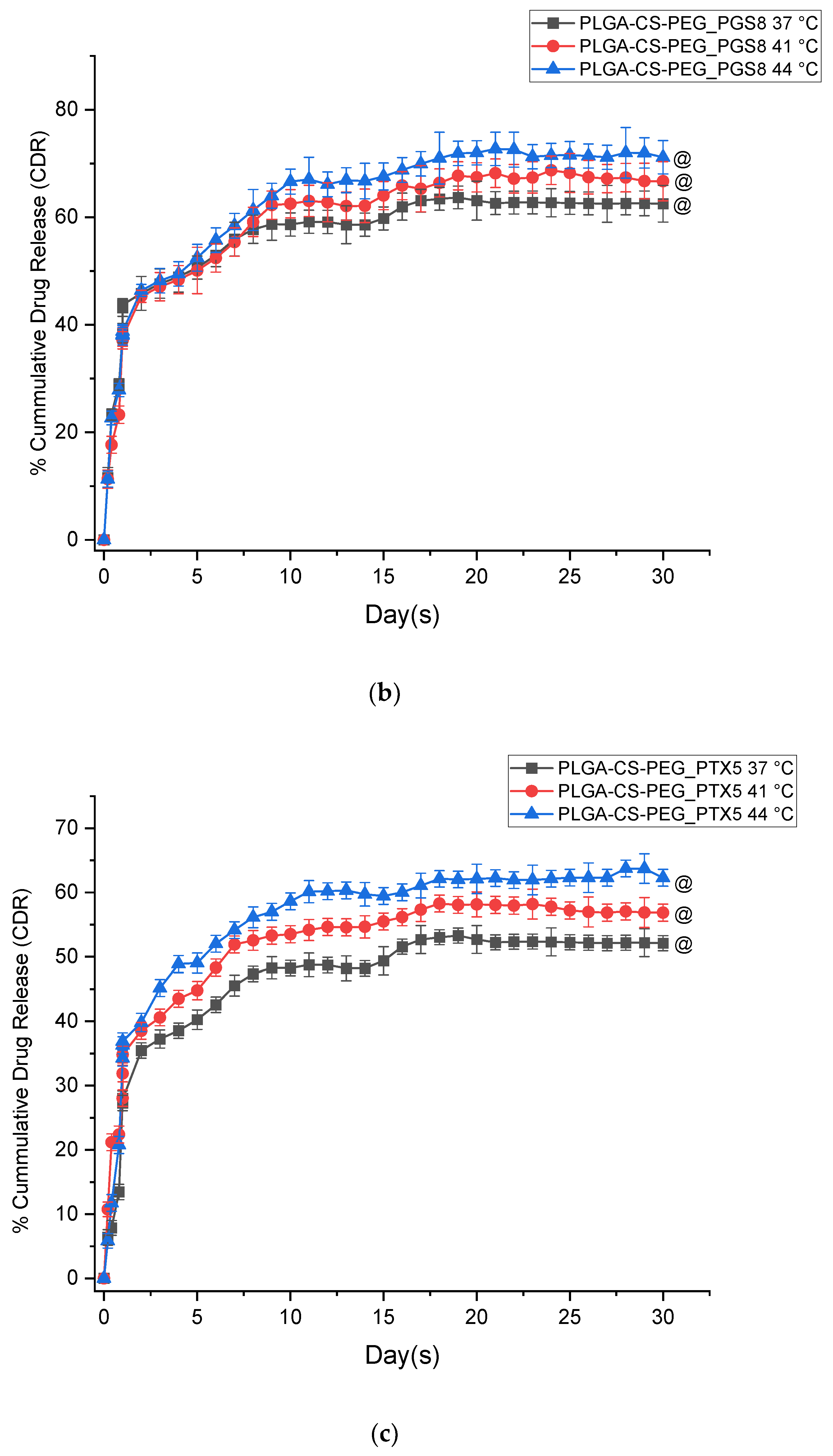
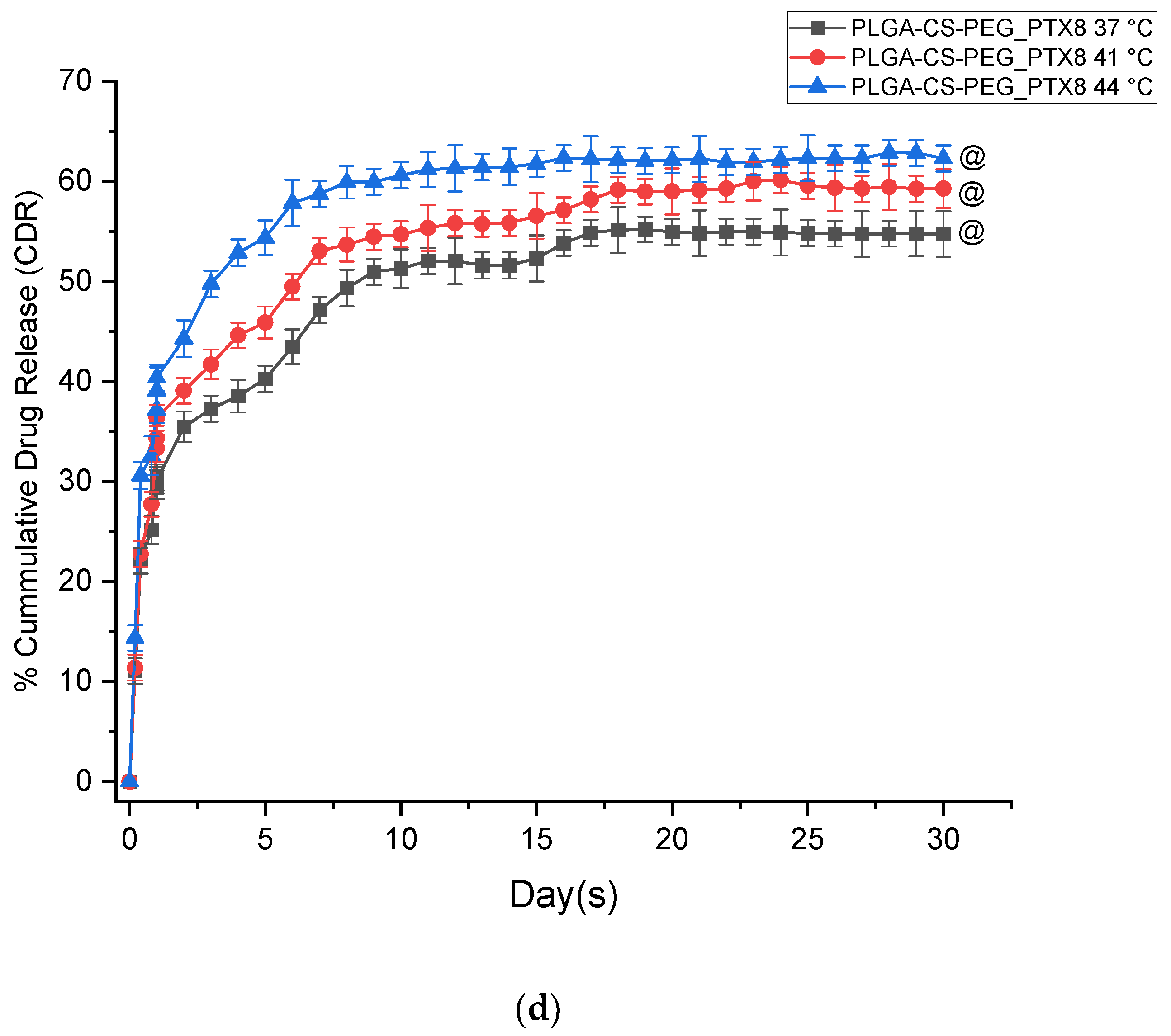
3.1. In Vitro Drug Release Kinetics
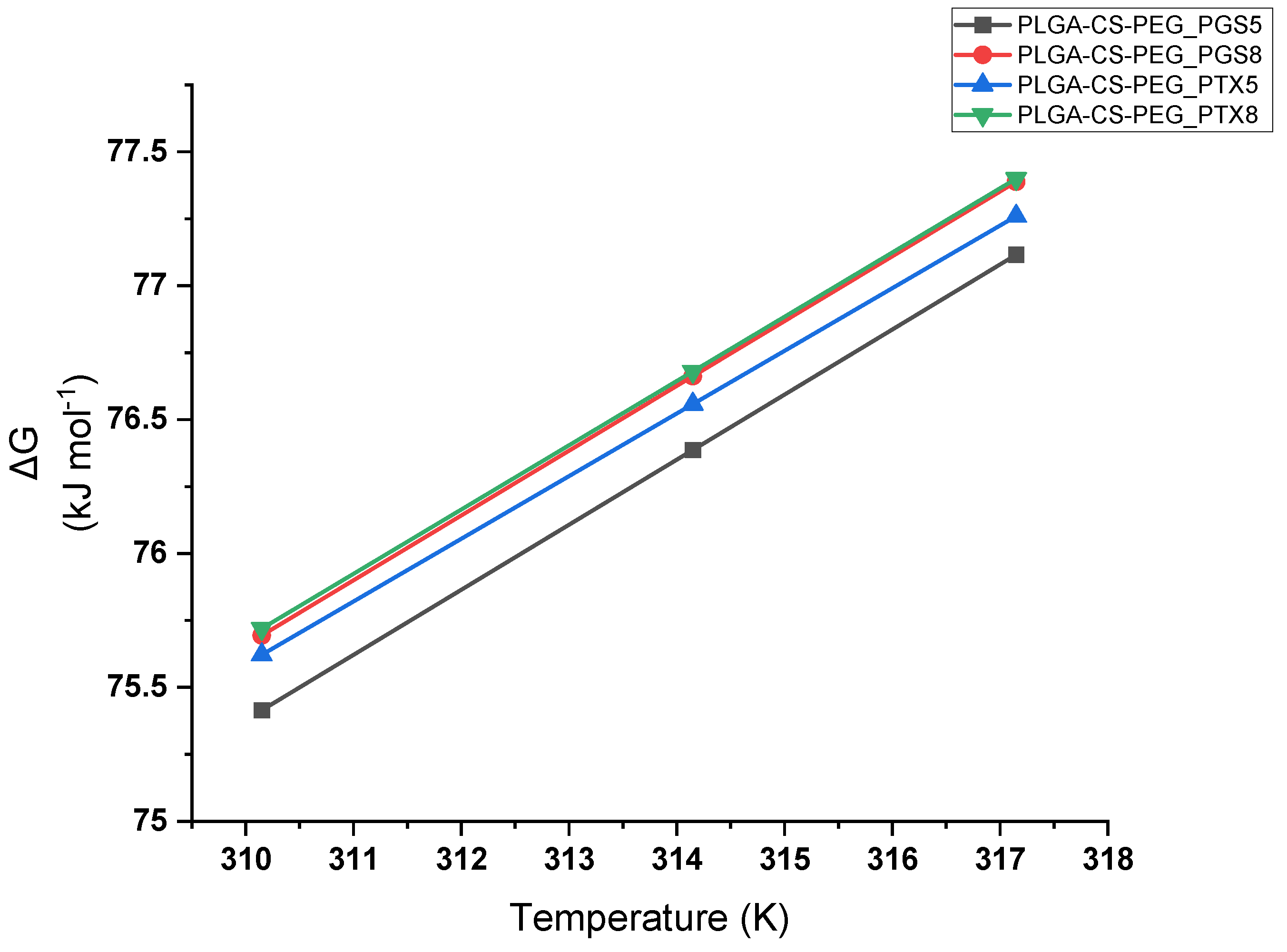
3.2. In Vitro Drug Release Thermodynamics
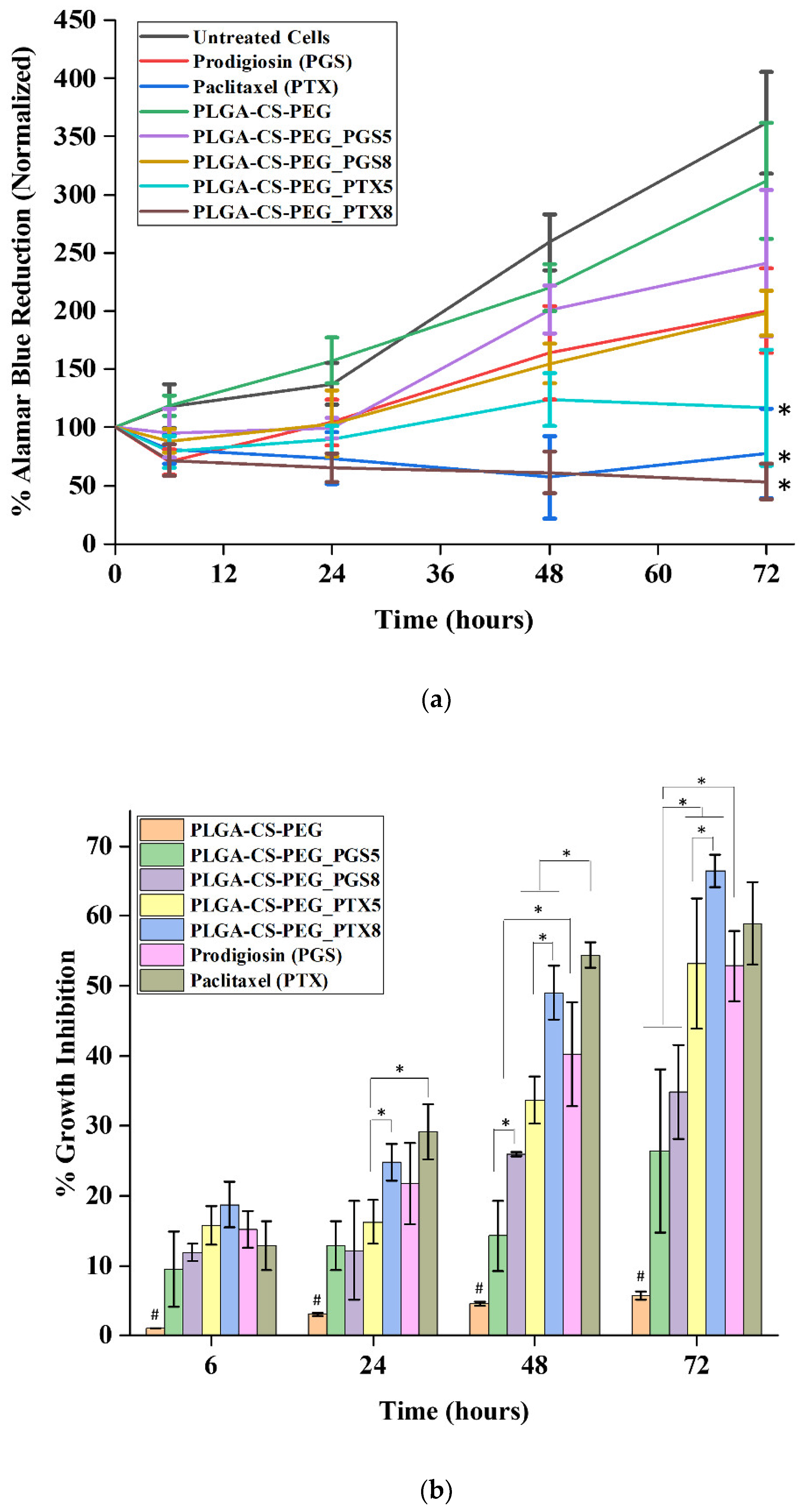
3.3. In Vitro Cell Viability and Cytotoxicity
4. Discussion
5. Conclusions
- The PGS-loaded microspheres have lower burst release profiles than the PTX-loaded microspheres. The kinetics of drug release from both types of microspheres are also well characterized by the Korsmeyer–Peppas model, with release exponents n within the range of 0.45 < n < 0.89. This range of n corresponds to drug release occurring by anomalous non-Fickian release and is associated with drug diffusion and the relaxation of the polymer chains between the networks.
- The in vitro drug release profiles obtained in this study exhibit a biphasic-controlled release of anticancer drugs (paclitaxel and prodigiosin) from drug-loaded PLGA-CS-PEG microspheres with >50% drug released at day 30. This controlled release is attributed to the presence of CS and PEG blends that limit the diffusion and erosion processes associated with drug transport through the blended microspheres.
- The thermodynamic analysis of in vitro drug release from PLGA-CS-PEG microspheres loaded with drugs revealed positive values of ΔG and ΔH, and negative values of ΔS at set temperatures of 37, 41, and 44 °C. This is compatible with endothermic, nonspontaneous, ordered release of anticancer drugs.
- Controlled in vitro drug release from cancer drug (paclitaxel and prodigiosin)-loaded PLGA-CS-PEG microspheres reduces the viability of MDA-MB-231 cancer cells.
- The controlled release of cancer drugs (paclitaxel and prodigiosin) from drug-loaded PLGA-CS-PEG microspheres occurs at slower rates than the controlled release of cancer drugs from PLGA-PEG microspheres during the first 30 days of controlled cancer drug (paclitaxel and prodigiosin) release.
- Prior to in vivo studies, further work is required to provide more insight into the nonspecific toxicity to noncancer cells. There is also a need for further studies of biodegradability.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, P.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. 2018 GLOBOCAN stats. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. World Health Organization—Key Facts on Cancer. Available online: https://www.who.int/cancer/resources/keyfacts/en/ (accessed on 9 September 2019).
- Boyle, J. Molecular biology of the cell, 5th edition by B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts, and P. Walter. Biochem. Mol. Biol. Educ. 2008, 36, 317–318. [Google Scholar] [CrossRef]
- Abbas, Z.; Rehman, S. An Overview of Cancer Treatment Modalities. In Neoplasm; IntechOpen: London, UK, 2018; Volume 1, pp. 139–157. [Google Scholar]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef]
- Mitra, A.; Dey, B. Chitosan microspheres in novel drug delivery systems. Indian J. Pharm. Sci. 2011, 73, 355. [Google Scholar] [PubMed]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef]
- Gao, N.; Li, X.J. Controlled drug delivery using microfluidic devices. In Microfluidic Devices for Biomedical Applications, 1st ed.; Li, X., Zhou, Y., Eds.; Woodhead Publishing: Sawston, UK, 2013; pp. 167–185e. ISBN 9780857096975. [Google Scholar]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Hans, M.L.; Lowman, A.M. Lowman Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci. 2002, 6, 319–327. [Google Scholar] [CrossRef]
- Obayemi, J.D.; Jusu, S.M.; Salifu, A.A.; Ghahremani, S.; Tadesse, M.; Uzonwanne, V.O.; Soboyejo, W.O. Degradable porous drug-loaded polymer scaffolds for localized cancer drug delivery and breast cell/tissue growth. Mater. Sci. Eng. C 2020, 112, 110794. [Google Scholar] [CrossRef]
- Jusu, S.M.; Obayemi, J.D.; Salifu, A.A.; Nwazojie, C.C.; Uzonwanne, V.; Odusanya, O.S.; Soboyejo, W.O. Drug-encapsulated blend of PLGA-PEG microspheres: In vitro and in vivo study of the effects of localized/targeted drug delivery on the treatment of triple-negative breast cancer. Sci. Rep. 2020, 10, 14188. [Google Scholar] [CrossRef]
- Obayemi, J.D.; Danyuo, Y.; Dozie-Nwachukwu, S.; Odusanya, O.S.; Anuku, N.; Malatesta, K.; Yu, W.; Uhrich, K.E.; Soboyejo, W.O. PLGA-based microparticles loaded with bacterial-synthesized prodigiosin for anticancer drug release: Effects of particle size on drug release kinetics and cell viability. Mater. Sci. Eng. C 2016, 66, 51–65. [Google Scholar] [CrossRef]
- Mir, M.; Ahmed, N.; ur Rehman, A. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanafari, A.; Assadi, M.M.; Fakhr, F.A. Review of Prodigiosin, Pigmentation in Serratia marcescens. Online J. Biol. Sci. 2006, 6, 1–13. [Google Scholar] [CrossRef]
- Pérez-Tomás, R.; Montaner, B.; Llagostera, E.; Soto-Cerrato, V. The prodigiosins, proapoptotic drugs with anticancer properties. Biochem. Pharmacol. 2003, 66, 1447–1452. [Google Scholar] [CrossRef]
- Manderville, R.A. Synthesis, proton-affinity and anti-cancer properties of the prodigiosin-group natural products. Curr. Med. Chem. Anticancer Agents 2001, 1, 195–218. [Google Scholar] [CrossRef]
- Darshan, N.; Manonmani, H.K. Prodigiosin and its potential applications. J. Food Sci. Technol. 2015, 52, 5393–5407. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.M.; Shalaby, M.; Embaby, A.M.; Saeed, H.; Agwa, M.M.; Hussein, A. Prodigiosin/PU-H71 as a novel potential combined therapy for triple negative breast cancer (TNBC): Preclinical insights. Sci. Rep. 2020, 10, 14706. [Google Scholar] [CrossRef]
- Sang, B.H.; Se, H.P.; Young, J.J.; Young, K.K.; Hwan, M.K.; Kyu, H.Y. Prodigiosin blocks T cell activation by inhibiting interleukin-2Rα expression and delays progression of autoimmune diabetes and collagen-induced arthritis. J. Pharmacol. Exp. Ther. 2001, 299, 415–425. [Google Scholar]
- Venil, C.K.; Zakaria, Z.A.; Ahmad, W.A. Bacterial pigments and their applications. Process Biochem. 2013, 48, 1065–1079. [Google Scholar] [CrossRef]
- Singh, B.; Vishwakarma, R.; Bharate, S. QSAR and Pharmacophore Modeling of Natural and Synthetic Antimalarial Prodiginines. Curr. Comput. Aided-Drug Des. 2013, 9, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, D.; Nazari, T.F.; Kassim, J.; Lim, S.H. Prodigiosin—An antibacterial red pigment produced by Serratia marcescens IBRL USM 84 associated with a marine sponge Xestospongia testudinaria. J. Appl. Pharm. Sci. 2014, 4, 1–6. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; Mcphail, A.T. Plant Antitumor Agents. VI. The Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Renneberg, R. Biotech History: Yew trees, paclitaxel synthesis and fungi. Biotechnol. J. Healthc. Nutr. Technol. 2007, 2, 1207–1209. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. Drug Res. 2010, 67, 217–223. [Google Scholar]
- Freire, M.C.L.C.; Alexandrino, F.; Marcelino, H.R.; Picciani, P.H.d.S.; Silva, K.G.d.H.; Genre, J.; de Oliveira, A.G.; do Egito, E.S.T. Understanding drug release data through thermodynamic analysis. Materials 2017, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Maria, G.; Luta, I. Precautions in using global kinetic and thermodynamic models for characterization of drug release from multivalent supports. Chem. Pap. 2011, 65, 542–552. [Google Scholar] [CrossRef]
- Wang, J.; Shi, A.; Agyei, D.; Wang, Q. Formulation of water-in-oil-in-water (W/O/W) emulsions containing trans-resveratrol. RSC Adv. 2017, 7, 35917–35927. [Google Scholar] [CrossRef] [Green Version]
- Suphiya Parveen, S.K.S. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur. J. Pharmacol. 2011, 670, 372–383. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Katrin, K.; Richard, H.; Dagmar, F.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Al Rubeaan, K.; Rafiullah, M.; Jayavanth, S. Oral insulin delivery systems using chitosan-based formulation: A review. Expert Opin. Drug Deliv. 2016, 13, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Abd-Rabou, A.A.; Ahmed, H.H. CS-PEG decorated PLGA nano-prototype for delivery of bioactive compounds: A novel approach for induction of apoptosis in HepG2 cell line. Adv. Med. Sci. 2017, 62, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Sadoughi, F.; Mansournia, M.A.; Mirhashemi, S.M. The potential role of chitosan-based nanoparticles as drug delivery systems in pancreatic cancer. IUBMB Life 2020, 72, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Tekade, R.K.; Jain, N.K. Spectrophotometric estimation of paclitaxel. Int. J. Adv. Pharm. Sci. 2011, 2, 29–32. [Google Scholar]
- Narasimhan, B.; Mallapragada, S.K.; Peppas, N.A. Release kinetics, data interpretation. Encycl. Control. Drug Deliv. 1999, 2, 921–935. [Google Scholar]
- Shaikh, H.K.; Kshirsagar, R.V.; Patil, S.G. Mathematical models for drug release characterization: A review. World J. Pharm. Pharm. Sci. 2015, 4, 324–338. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Sawston, UK, 2015; ISBN 9780081001127. [Google Scholar]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Smith, J.M. Introduction to chemical engineering thermodynamics. J. Chem. Educ. 1950. [Google Scholar] [CrossRef] [Green Version]
- Sari, A.; Tuzen, M.; Citak, D.; Soylak, M. Equilibrium, kinetic and thermodynamic studies of adsorption of Pb(II) from aqueous solution onto Turkish kaolinite clay. J. Hazard. Mater. 2007, 149, 283–291. [Google Scholar] [CrossRef]
- Obayemi, J.D.; Salifu, A.A.; Eluu, S.C.; Uzonwanne, V.O.; Jusu, S.M.; Nwazojie, C.C.; Onyekanne, C.E.; Ojelabi, O.; Payne, L.; Moore, C.M.; et al. LHRH-Conjugated Drugs as Targeted Therapeutic Agents for the Specific Targeting and Localized Treatment of Triple Negative Breast Cancer. Sci. Rep. 2020, 10, 8212. [Google Scholar] [CrossRef]
- Danyuo, Y.; Ani, C.J.; Salifu, A.A.; Obayemi, J.D.; Dozie-Nwachukwu, S.; Obanawu, V.O.; Akpan, U.M.; Odusanya, O.S.; Abade-Abugre, M.; McBagonluri, F.; et al. Anomalous Release Kinetics of Prodigiosin from Poly-N-Isopropyl-Acrylamid based Hydrogels for The Treatment of Triple Negative Breast Cancer. Sci. Rep. 2019, 9, 3862. [Google Scholar] [CrossRef] [Green Version]
- Hassani Najafabadi, A.; Abdouss, M.; Faghihi, S. Synthesis and evaluation of PEG-O-chitosan nanoparticles for delivery of poor water soluble drugs: Ibuprofen. Mater. Sci. Eng. C 2014, 41, 91–99. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Egea, M.A.; Cano, A.; Espina, M.; Calpena, A.C.; Ettcheto, M.; Camins, A.; Souto, E.B.; Silva, A.M.; García, M.L. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen-in vitro, ex vivo and in vivo characterization. Colloids Surf. B Biointerfaces 2016, 147, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Rahman, M.H.; Rahman, M.M. Biphasic oral solid drug delivery system: A review. Int. J. Pharm. Sci. Res. 2011, 2, 1108. [Google Scholar]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]








| Serial Number | Name of Equation | Mathematical Expression |
|---|---|---|
| 1 | Arrhenius (1st form) | |
| 2 | Arrhenius (2nd form) | |
| 3 | Eyring | |
| 4 | Change in Gibbs free energy |
| Formulations | Drug Release Temperature (°C) | Percentage Burst Release after 48 h | Encapsulation Efficiency (%) | Percentage Cumulative Drug Release for 30 Days |
|---|---|---|---|---|
| PLGA-CS-PEG_PGS5 | 37 | 27.290 ± 2.074 | 56.5 | 52.117 ± 2.506 |
| PLGA-CS-PEG_PGS5 | 41 | 27.290 ± 1.031 | 54.471 ± 1.095 | |
| PLGA-CS-PEG_PGS5 | 44 | 27.130 ± 1.031 | 59.151 ± 2.084 | |
| PLGA-CS-PEG_PGS8 | 37 | 28.910 ± 1.082 | 58.5 | 62.502 ± 3.412 |
| PLGA-CS-PEG_PGS8 | 41 | 23.277 ± 1.230 | 66.700 ± 3.641 | |
| PLGA-CS-PEG_PGS8 | 44 | 27.872 ± 1.807 | 71.153 ± 3.103 | |
| PLGA-CS-PEG_PTX5 | 37 | 28.055 ± 1.157 | 57.5 | 52.117 ± 1.172 |
| PLGA-CS-PEG_PTX5 | 41 | 28.055 ± 1.270 | 56.862 ± 1.329 | |
| PLGA-CS-PEG_PTX5 | 44 | 34.275 ± 1.231 | 62.300 ± 1.309 | |
| PLGA-CS-PEG_PTX8 | 37 | 30.400 ± 1.310 | 56.0 | 54.728 ± 2.290 |
| PLGA-CS-PEG_PTX8 | 41 | 33.323 ± 1.346 | 59.287 ± 1.930 | |
| PLGA-CS-PEG_PTX8 | 44 | 37.176 ± 1.320 | 62.300 ± 1.300 |
| Formulations | Temperature °C | Zeroth Order | First Order | Higuchi | Korsmeyer–Peppas | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| K | R2 | K | R2 | K | R2 | K | R2 | n | ||
| PLGA-CS-PEG_PGS5 | 37 | 1.162 | 0.593 | 0.034 | 0.418 | 8.020 | 0.791 | 1.277 | 0.840 | 0.462 |
| PLGA-CS-PEG_PGS5 | 41 | 1.295 | 0.611 | 0.037 | 0.438 | 8.902 | 0.809 | 1.279 | 0.860 | 0.482 |
| PLGA-CS-PEG_PGS5 | 44 | 1.452 | 0.672 | 0.038 | 0.488 | 9.811 | 0.859 | 1.308 | 0.894 | 0.464 |
| PLGA-CS-PEG_PGS8 | 37 | 1.146 | 0.539 | 0.023 | 0.415 | 8.005 | 0.737 | 1.179 | 0.934 | 0.542 |
| PLGA-CS-PEG_PGS8 | 41 | 1.438 | 0.624 | 0.029 | 0.486 | 9.828 | 0.817 | 1.266 | 0.839 | 0.494 |
| PLGA-CS-PEG_PGS8 | 44 | 1.546 | 0.648 | 0.029 | 0.509 | 10.507 | 0.838 | 1.205 | 0.948 | 0.536 |
| PLGA-CS-PEG_PTX5 | 37 | 1.162 | 0.593 | 0.034 | 0.418 | 8.020 | 0.791 | 1.277 | 0.840 | 0.462 |
| PLGA-CS-PEG_PTX5 | 41 | 1.162 | 0.587 | 0.027 | 0.481 | 8.059 | 0.791 | 1.065 | 0.900 | 0.522 |
| PLGA-CS-PEG_PTX5 | 44 | 1.332 | 0.575 | 0.032 | 0.384 | 9.238 | 0.776 | 1.371 | 0.814 | 0.457 |
| PLGA-CS-PEG_PTX8 | 37 | 1.122 | 0.626 | 0.026 | 0.530 | 7.681 | 0.822 | 1.081 | 0.908 | 0.488 |
| PLGA-CS-PEG_PTX8 | 41 | 1.169 | 0.601 | 0.025 | 0.489 | 8.053 | 0.799 | 1.126 | 0.884 | 0.483 |
| PLGA-CS-PEG_PTX8 | 44 | 1.067 | 0.494 | 0.020 | 0.418 | 7.620 | 0.706 | 1.115 | 0.913 | 0.591 |
| Formulations | Temperature °C/K | Ea (kJ mol−1) | ΔS (kJ mol−1 K−1) | ΔH (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|---|---|---|
| PLGA-CS-PEG_PGS5 | 37/310.15 | 2.656 | −0.243 | 0.050 | 75.415 |
| 41/314.15 | 76.387 | ||||
| 44/317.15 | 77.116 | ||||
| PLGA-CS-PEG_PGS8 | 37/310.15 | 3.245 | −0.242 | 0.638 | 75.694 |
| 41/314.15 | 76.662 | ||||
| 44/317.15 | 77.388 | ||||
| PLGA-CS-PEG_PTX5 | 37/310.15 | 5.653 | −0.234 | 3.047 | 75.622 |
| 41/314.15 | 76.558 | ||||
| 44/317.15 | 77.260 | ||||
| PLGA-CS--PEG_PTX8 | 37/310.15 | 3.890 | −0.240 | 1.284 | 75.720 |
| 41/314.15 | 76.680 | ||||
| 44/317.15 | 77.400 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jusu, S.M.; Obayemi, J.D.; Salifu, A.A.; Nwazojie, C.C.; Uzonwanne, V.O.; Odusanya, O.S.; Soboyejo, W.O. PLGA-CS-PEG Microparticles for Controlled Drug Delivery in the Treatment of Triple Negative Breast Cancer Cells. Appl. Sci. 2021, 11, 7112. https://0-doi-org.brum.beds.ac.uk/10.3390/app11157112
Jusu SM, Obayemi JD, Salifu AA, Nwazojie CC, Uzonwanne VO, Odusanya OS, Soboyejo WO. PLGA-CS-PEG Microparticles for Controlled Drug Delivery in the Treatment of Triple Negative Breast Cancer Cells. Applied Sciences. 2021; 11(15):7112. https://0-doi-org.brum.beds.ac.uk/10.3390/app11157112
Chicago/Turabian StyleJusu, Sandra Musu, John David Obayemi, Ali Azeko Salifu, Chukwudalu Clare Nwazojie, Vanessa Obiageli Uzonwanne, Olushola Segun Odusanya, and Winnston Oluwole Soboyejo. 2021. "PLGA-CS-PEG Microparticles for Controlled Drug Delivery in the Treatment of Triple Negative Breast Cancer Cells" Applied Sciences 11, no. 15: 7112. https://0-doi-org.brum.beds.ac.uk/10.3390/app11157112