
Metformin Serves as a Novel Drug Treatment for Arterial Thrombosis: Inhibitory Mechanisms on Collagen-Induced Human Platelet Activation

 , , and
, , and 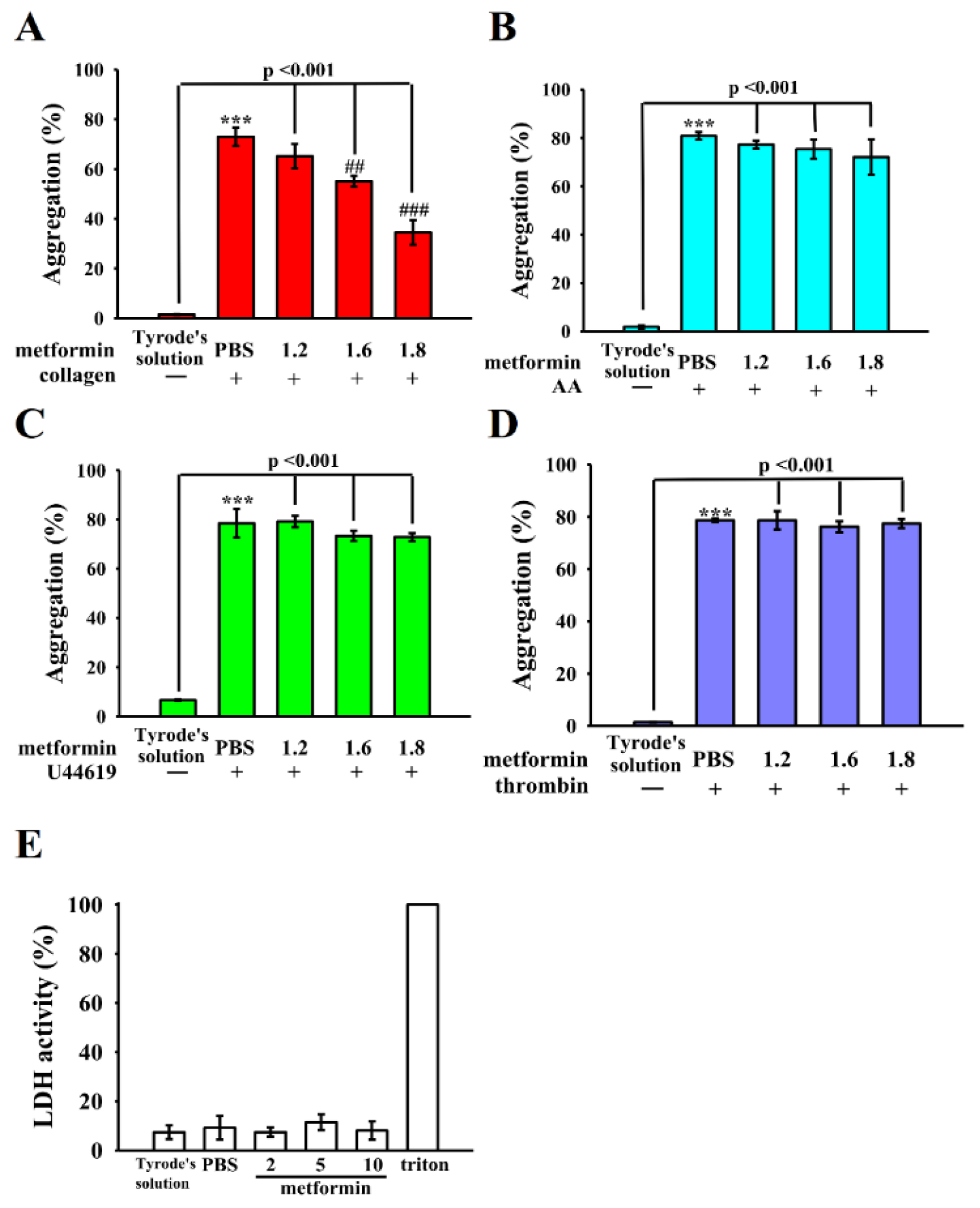{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
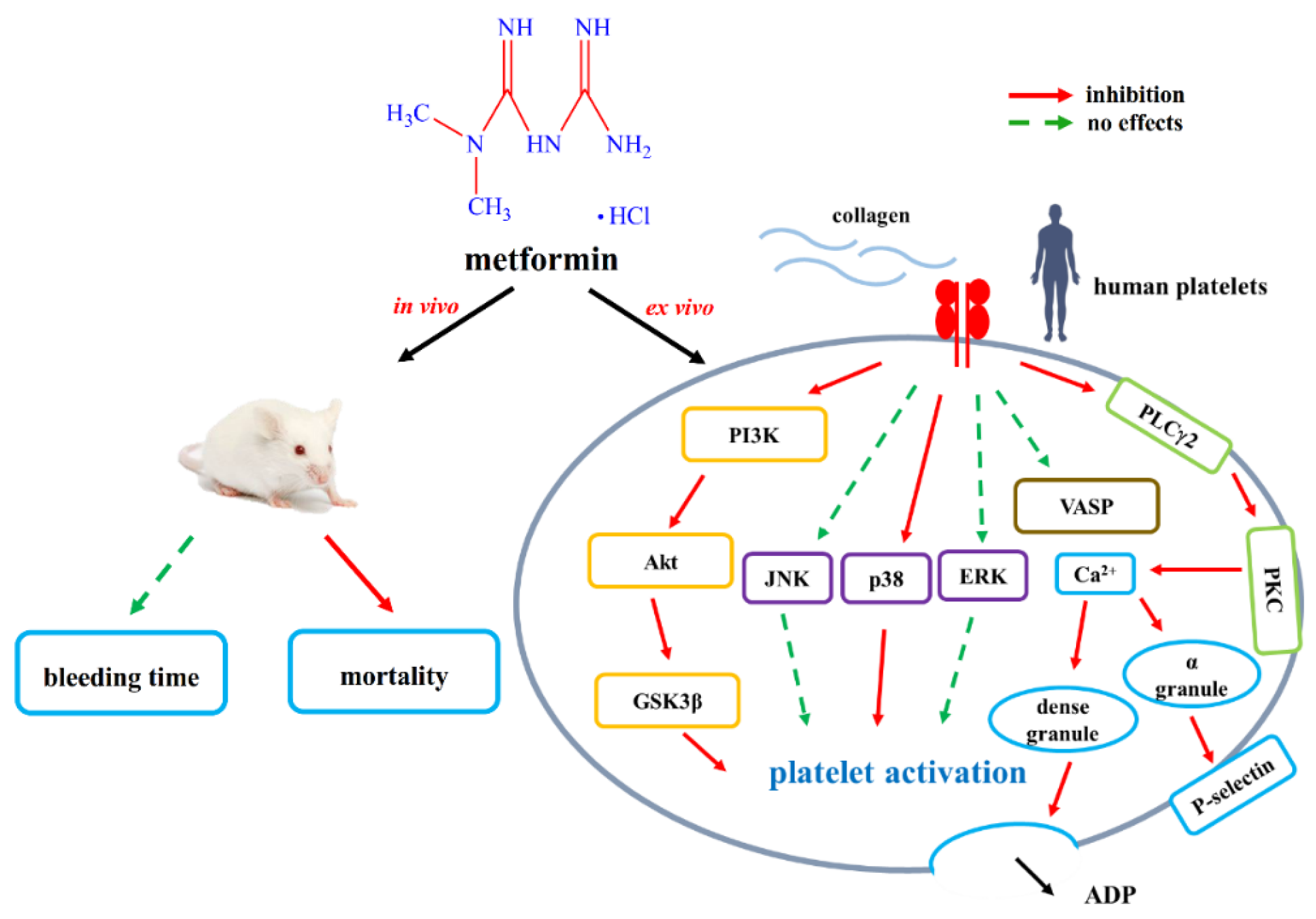{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Platelet Aggregation, ATP Release Reaction, and Lactate Dehydrogenase Assay
2.3. Intracellular [Ca2+]i Mobilization and FITC-P-Selectin Expression in Human Platelets
2.4. Immunoblotting
2.5. Immunofluorescence Staining Assay
2.6. Measurement of Pulmonary Microvascular Thrombosis in Mice
2.7. Mouse Tail Bleeding Assay
2.8. Statistical Analysis
3. Results
3.1. Mitigation of Human Platelet Aggregation by Metformin
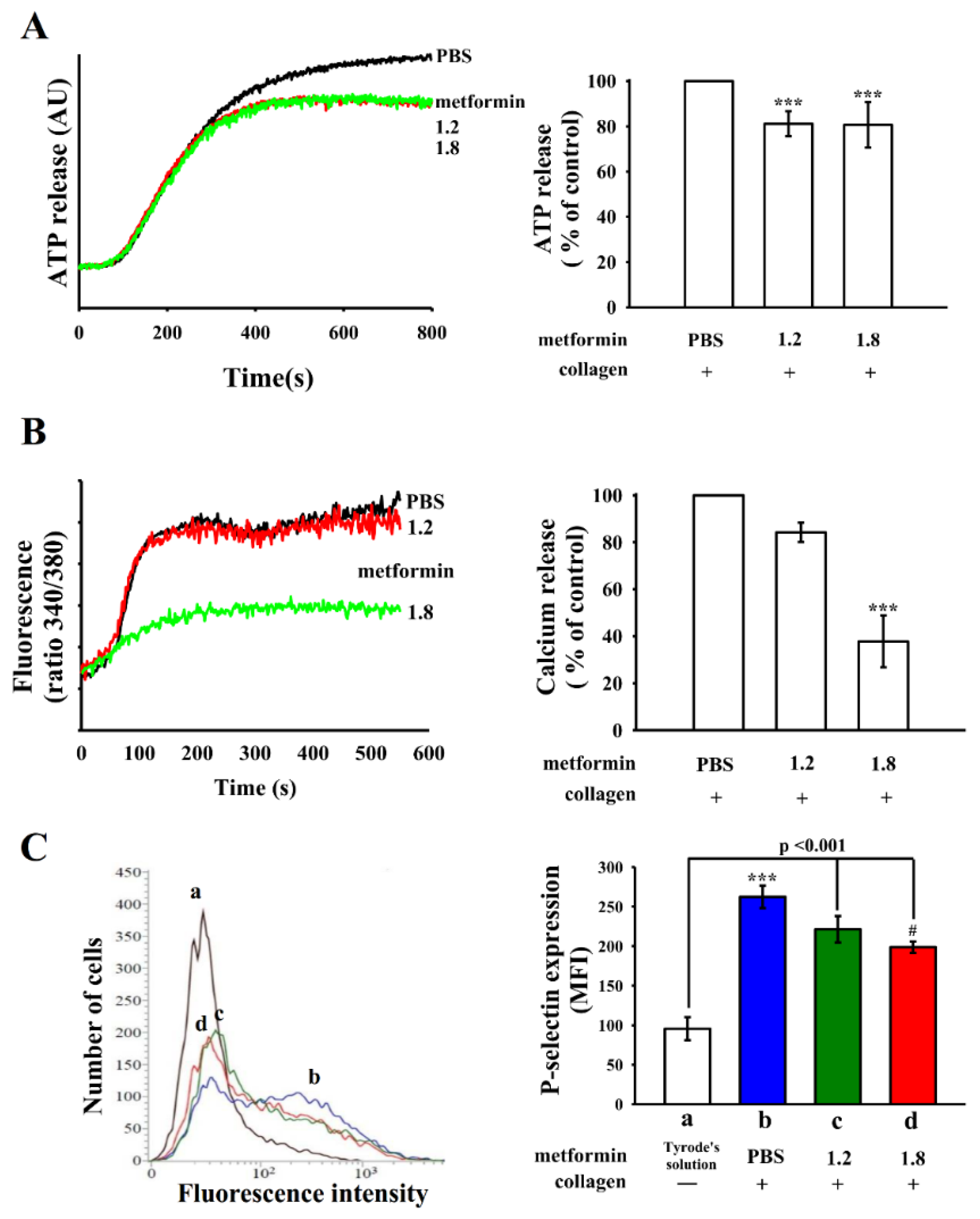
3.2. Effects of Metformin on ATP Release, Relative [Ca2+]i Mobilization, and Surface P-Selectin Expression
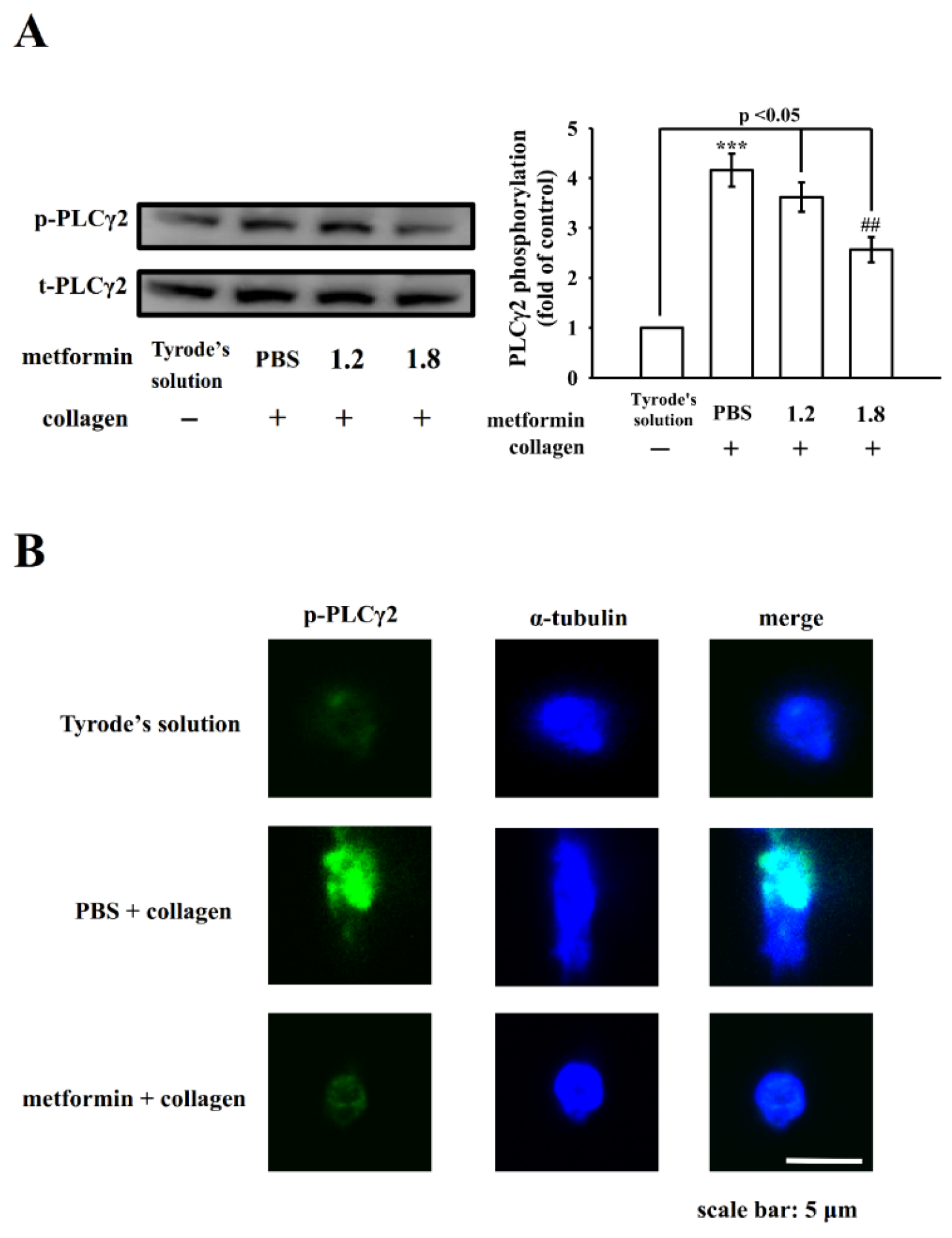
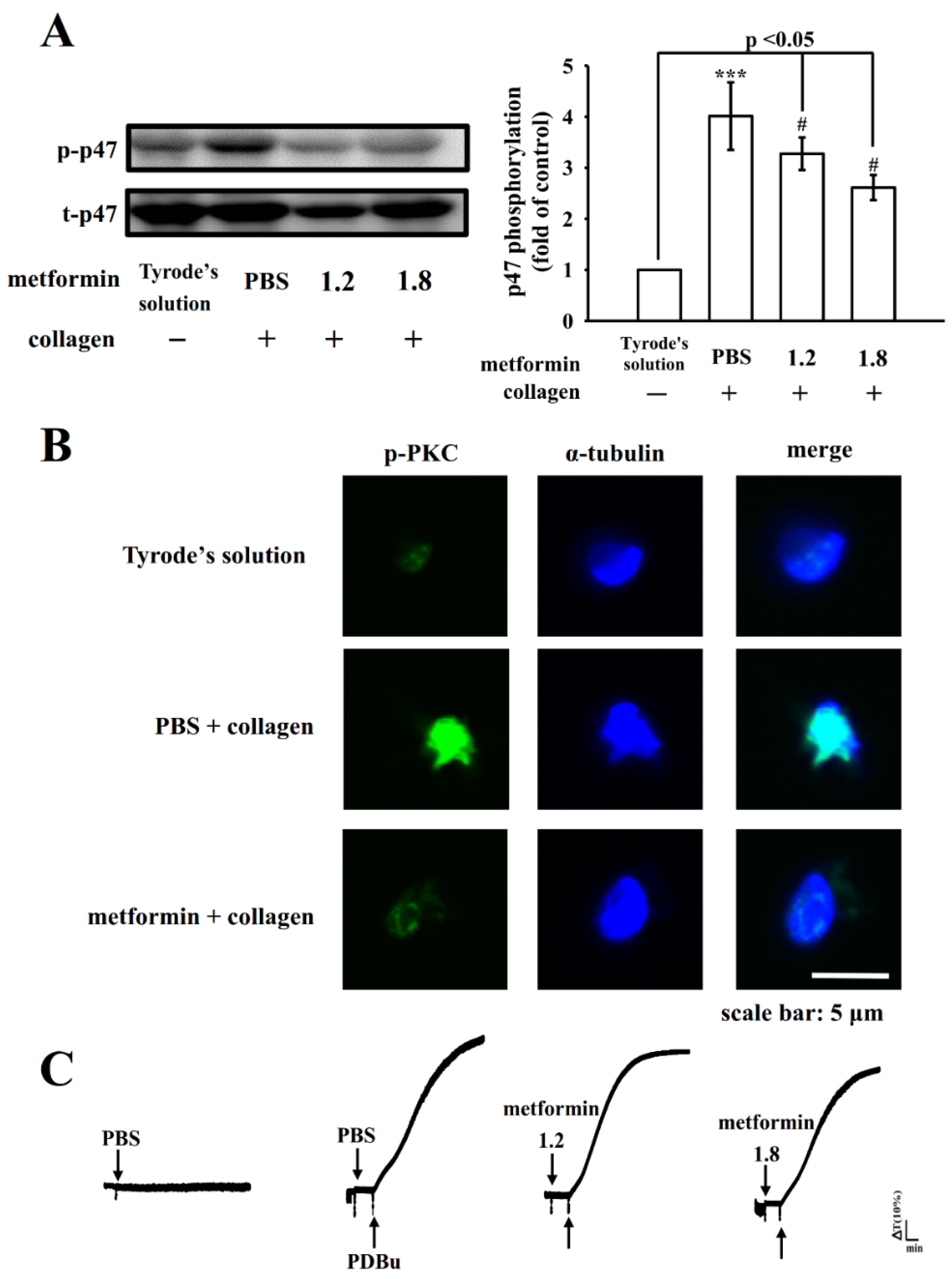
3.3. Influence of Metformin in PLCγ2-Mediated Downstream Signals
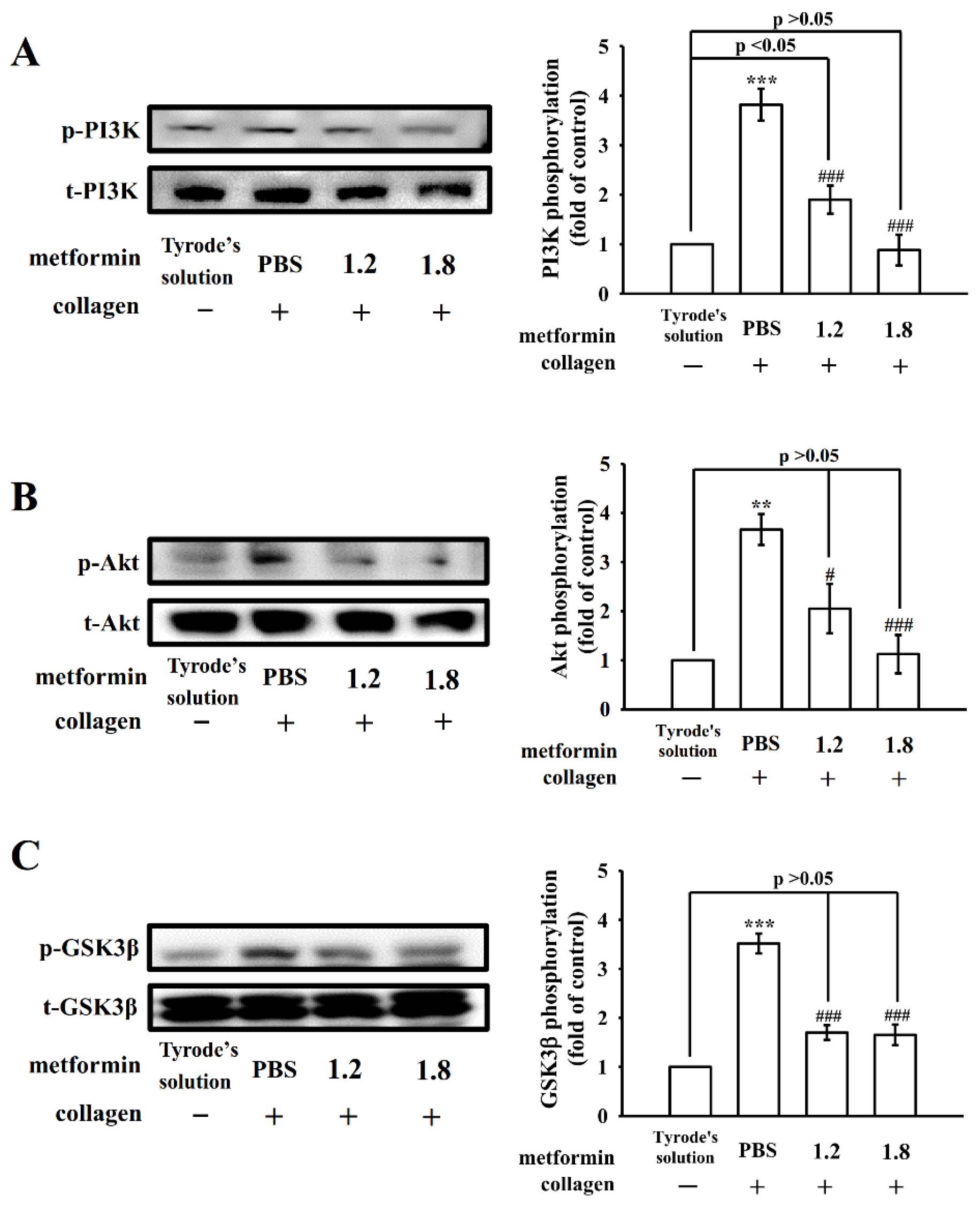
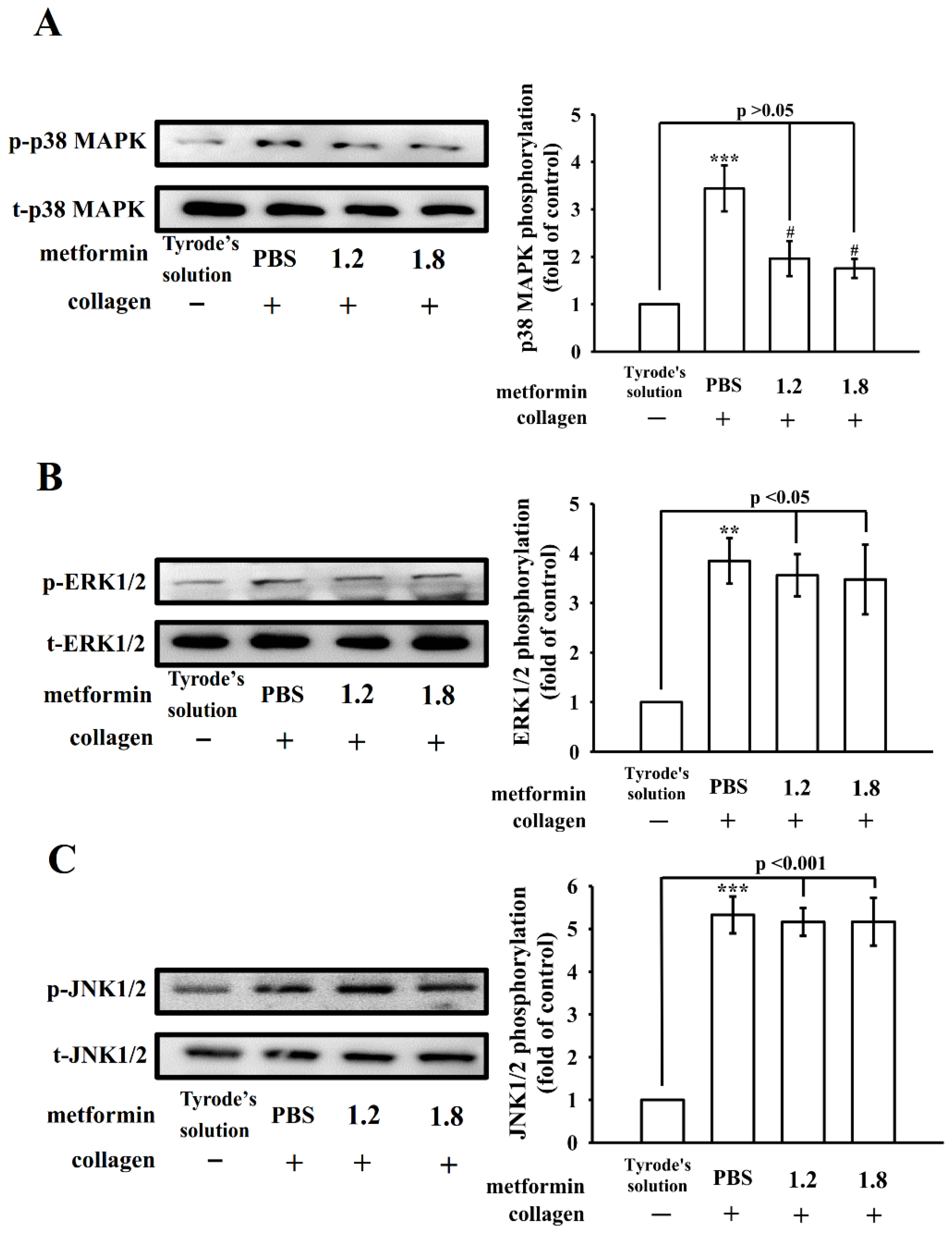
3.4. Regulatory Activity of PI3K–Akt–GSK3β and MAPK Activation by Metformin
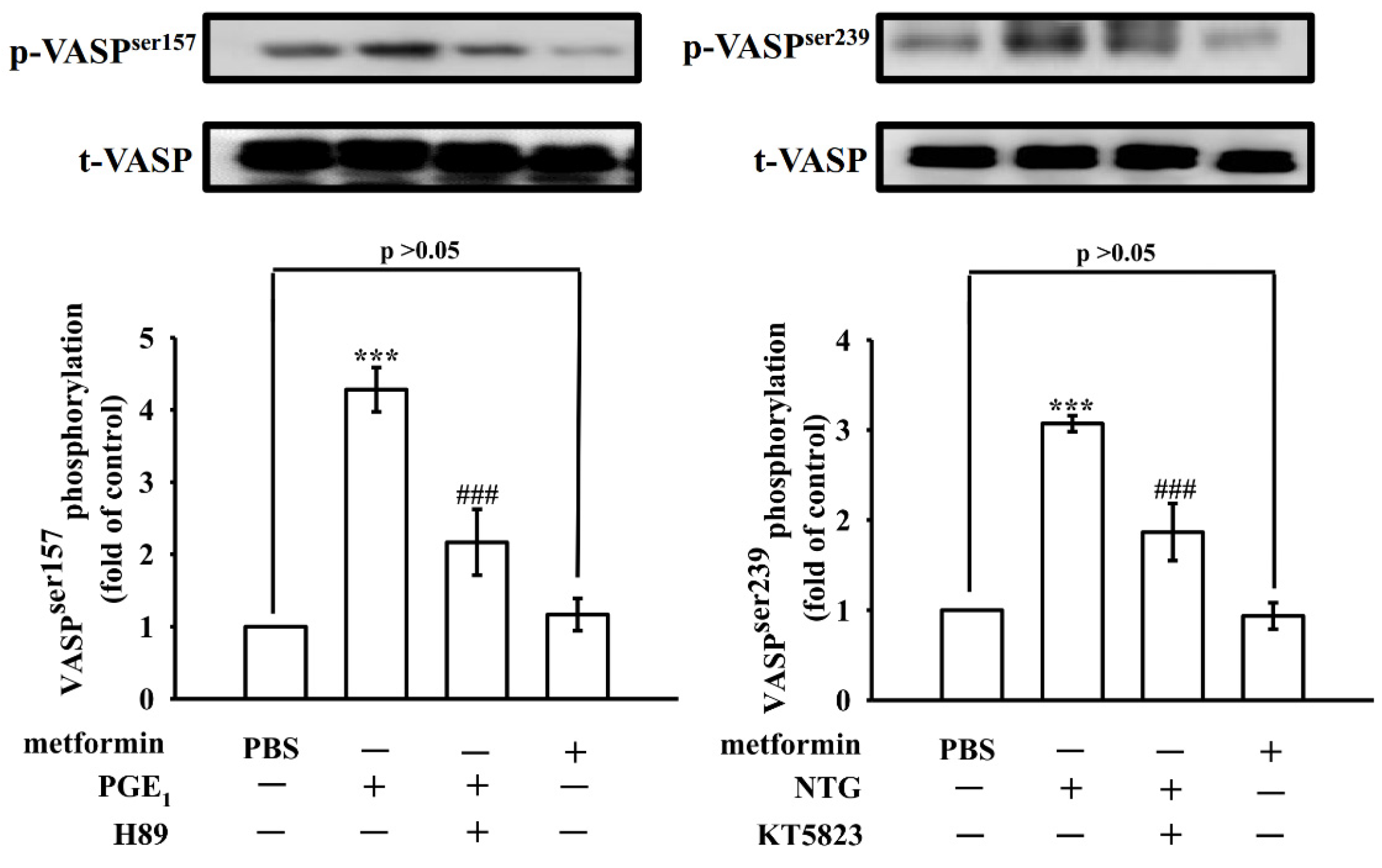
3.5. Effect of Metformin in Phosphorylation of VASP
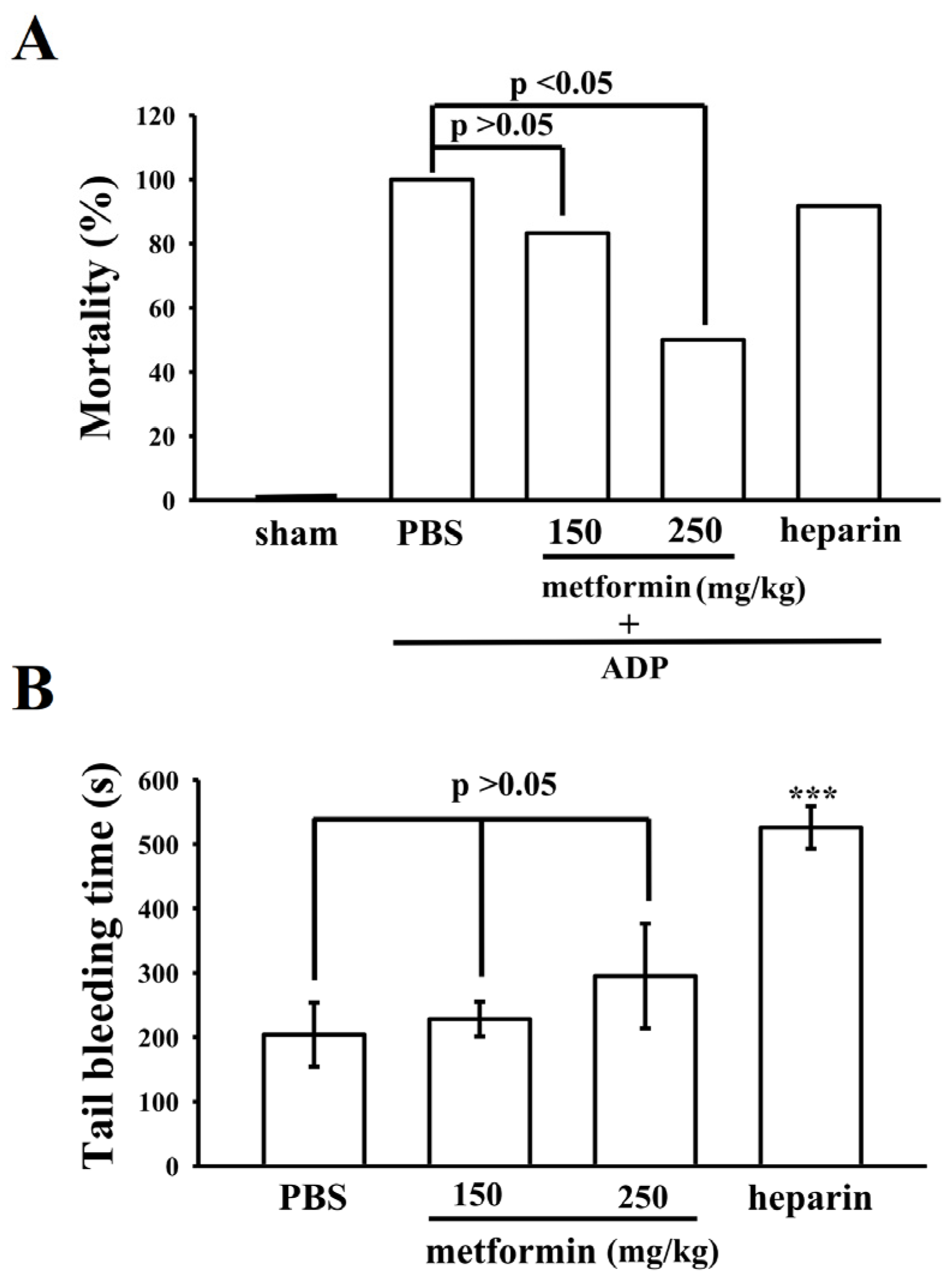
3.6. Effect of Metformin on Acute Pulmonary Thrombosis and Bleeding Time in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, J.; Bhutani, S. Worldwide Burden of Diabetes. Indian. J. Endocrinol. Metab. 2014, 18, 868–870. [Google Scholar] [CrossRef]
- Bai, B.; Chen, H. Metformin: A Novel Weapon Against Inflammation. Front. Pharmacol. 2021, 12, 622262. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.I.; Sang, Y.; Chang, A.R.; Dunning, S.C.; Coresh, J.; Inker, L.A.; Selvin, E.; Ballew, S.H.; Grams, M.E. The FDA Metformin Label Change and Racial and Sex Disparities in Metformin Prescription Among Patients with CKD. J. Am. Soc. Nephrol. 2020, 31, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Ala, M. Metformin for Cardiovascular Protection, Inflammatory Bowel Disease, Osteoporosis, Periodontitis, Polycystic Ovarian Syndrome, Neurodegeneration, Cancer, Inflammation and Senescence: What Is Next? ACS Pharmacol. Transl. Sci. 2021, 4, 1747–1770. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Yen, M.H.; Hung, W.C.; Lee, Y.M.; Su, C.H.; Huang, T.F. Triflavin Inhibits Platelet-Induced Vasoconstriction in de-Endothelialized Aorta. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3461–3468. [Google Scholar] [CrossRef]
- Stegner, D.; Nieswandt, B. Platelet Receptor Signaling in Thrombus Formation. J. Mol. Med. 2011, 89, 109–121. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef]
- Anfossi, G.; Russo, I.; Bonomo, K.; Trovati, M. The Cardiovascular Effects of Metformin: Further Reasons to Consider an Old Drug as a Cornerstone in the Therapy of Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2010, 8, 327–337. [Google Scholar] [CrossRef]
- Xin, G.; Wei, Z.; Ji, C.; Zheng, H.; Gu, J.; Ma, L.; Huang, W.; Morris-Natschke, S.L.; Yeh, J.L.; Zhang, R.; et al. Metformin Uniquely Prevents Thrombosis by Inhibiting Platelet Activation and mtDNA Release. Sci. Rep. 2016, 6, 36222. [Google Scholar] [CrossRef] [Green Version]
- Gin, H.; Freyburger, G.; Boisseau, M.; Aubertin, J. Study of the Effect of Metformin on Platelet Aggregation in Insulin-Dependent Diabetics. Diabetes Res. Clin. Pract. 1989, 6, 61–67. [Google Scholar] [CrossRef]
- De Caterina, R.; Marchetti, P.; Bernini, W.; Giannarelli, R.; Giannessi, D.; Navalesi, R. The Direct Effects of Metformin on Platelet Function In Vitro. Eur. J. Clin. Pharmacol. 1989, 37, 211–213. [Google Scholar] [CrossRef]
- Gargiulo, P.; Caccese, D.; Pignatelli, P.; Brufani, C.; De Vito, F.; Marino, R.; Lauro, R.; Violi, F.; Di Mario, U.; Sanguigni, V. Metformin Decreases Platelet Superoxide Anion Production in Diabetic Patients. Diabetes Metab. Res. Rev. 2002, 18, 156–159. [Google Scholar] [CrossRef]
- Formoso, G.; De Filippis, E.A.; Michetti, N.; Di Fulvio, P.; Pandolfi, A.; Bucciarelli, T.; Ciabattoni, G.; Nicolucci, A.; Davì, G.; Consoli, A. Decreased In Vivo Oxidative Stress and Decreased Platelet Activation Following Metformin Treatment in Newly Diagnosed Type 2 Diabetic Subjects. Diabetes Metab. Res. Rev. 2008, 24, 231–237. [Google Scholar] [CrossRef]
- Chen, W.F.; Lee, J.J.; Chang, C.C.; Lin, K.H.; Wang, S.H.; Sheu, J.R. Platelet Protease-Activated Receptor (PAR)4, but not PAR1, Associated with Neutral Sphingomyelinase Responsible for Thrombin-Stimulated Ceramide-NF-κB Signaling in Human Platelets. Haematologica 2013, 98, 793–801. [Google Scholar] [CrossRef]
- Sheu, J.R.; Lee, C.R.; Lin, C.H.; Hsiao, G.; Ko, W.C.; Chen, Y.C.; Yen, M.H. Mechanisms Involved in the Antiplatelet Activity of Staphylococcus Aureus Lipoteichoic Acid in Human Platelets. Thromb. Haemost. 2000, 83, 777–784. [Google Scholar] [CrossRef]
- Sheu, J.R.; Hung, W.C.; Wu, C.H.; Lee, Y.M.; Yen, M.H. Antithrombotic Effect of Rutaecarpine, an Alkaloid Isolated from Evodia Rutaecarpa, on Platelet Plug Formation in vivo Experiments. Br. J. Haematol. 2000, 110, 110–115. [Google Scholar] [CrossRef]
- Merten, M.; Thiagarajan, P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation 2000, 102, 1931–1936. [Google Scholar] [CrossRef]
- Moroi, M.; Jung, S.M. Platelet Glycoprotein VI: Its Structure and Function. Thromb. Res. 2004, 114, 221–233. [Google Scholar] [CrossRef]
- Laurent, P.A.; Severin, S.; Gratacap, M.P.; Payrastre, B. Class I PI 3-Kinases Signaling in Platelet Activation and Thrombosis: PDK1/Akt/GSK3 Axis and Impact of PTEN and SHIP1. Adv. Biol. Regul. 2014, 54, 162–174. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Inzucchi, S.E. Metformin and Heart Failure: Innocent Until Proven Guilty. Diabetes Care 2005, 28, 2585–2587. [Google Scholar] [CrossRef] [Green Version]
- Roussel, R.; Travert, F.; Pasquet, B.; Wilson, P.W.; Smith, S.C., Jr.; Goto, S.; Ravaud, P.; Marre, M.; Porath, A.; Bhatt, D.L.; et al. Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. Metformin Use and Mortality Among Patients with Diabetes and Atherothrombosis. Arch. Intern. Med. 2010, 170, 1892–1899. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.Y.; Huang, C.C.; Huang, P.H.; Chung, C.M.; Lin, S.J.; Chen, J.W.; Chan, W.L.; Leu, H.B. Metformin Use in Patients with Type 2 Diabetes Mellitus is Associated with Reduced Risk of Deep Vein Thrombosis: A Non-Randomized, Pair-Matched Cohort Study. BMC Cardiovasc. Disord. 2014, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, T.; Ding, K.; Liu, Z.; Li, Y.; He, T.; Zhang, W.; Fan, Y.; Ma, W.; Cui, L.; et al. Phospholipase Cγ2 Signaling Cascade Contribute to the Antiplatelet Effect of Notoginsenoside Fc. Front. Pharmacol. 2018, 9, 1293. [Google Scholar] [CrossRef] [Green Version]
- Ragab, A.; Séverin, S.; Gratacap, M.P.; Aguado, E.; Malissen, M.; Jandrot-Perrus, M.; Malissen, B.; Ragab-Thomas, J.; Payrastre, B. Roles of the C-Terminal Tyrosine Residues of LAT in GPVI-Induced Platelet Activation: Insights into the Mechanism of PLC Gamma 2 Activation. Blood 2007, 110, 2466–2474. [Google Scholar] [CrossRef] [Green Version]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium Signaling in Platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Gratacap, M.P.; Guillermet-Guibert, J.; Martin, V.; Chicanne, G.; Tronchère, H.; Gaits-Iacovoni, F.; Payrastre, B. Regulation and Roles of PI3Kβ, a Major Actor in Platelet Signaling and Functions. Adv. Enzyme Regul. 2011, 51, 106–116. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-Kinase p110Beta: A New Target for Antithrombotic Therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, T.; Chen, W.F.; Lu, W.J.; Chou, D.S.; Hsiao, G.; Hsu, C.Y.; Sheu, J.R.; Hsieh, C.Y. A Novel Antithrombotic Effect of Sulforaphane via Activation of Platelet Adenylate cyclase: Ex vivo and in vivo Studies. J. Nutr. Biochem. 2013, 24, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, D.S. Akt Signaling in Platelets and Thrombosis. Expert Rev. Hematol. 2010, 2010 3, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; August, S.; Woulfe, D.S. GSK3 Beta is a Negative Regulator of Platelet Function and Thrombosis. Blood 2008, 111, 3522–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugaud, F.; Nadal-Wollbold, F.; Lévy-Toledano, S.; Rosa, J.P.; Bryckaert, M. Regulation of c-jun-NH2 Terminal Kinase and Extracellular-Signal Regulated Kinase in Human Platelets. Blood 1999, 94, 3800–3805. [Google Scholar] [CrossRef]
- Fan, X.; Wang, C.; Shi, P.; Gao, W.; Gu, J.; Geng, Y.; Yang, W.; Wu, N.; Wang, Y.; Xu, Y.; et al. Platelet MEKK3 Regulates Arterial Thrombosis and Myocardial Infarct Expansion in Mice. Blood Adv. 2018, 2, 1439–1448. [Google Scholar] [CrossRef]
- Hughes, P.E.; Renshaw, M.W.; Pfaff, M.; Forsyth, J.; Keivens, V.M.; Schwartz, M.A.; Ginsberg, M.H. Suppression of Integrin Activation: A Novel Function of a Ras/Raf-Initiated MAP Kinase Pathway. Cell 1997, 88, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Saklatvala, J.; Rawlinson, L.; Waller, R.J.; Sarsfield, S.; Lee, J.C.; Morton, L.F.; Barnes, M.J.; Farndale, R.W. Role for p38 Mitogen-Activated Protein Kinase in Platelet Aggregation Caused by Collagen or a Thromboxane Analogue. J. Biol. Chem. 1996, 271, 6586–6589. [Google Scholar] [CrossRef] [Green Version]
- Adam, F.; Kauskot, A.; Rosa, J.P.; Bryckaert, M. Mitogen-Activated Protein Kinases in Hemostasis and Thrombosis. J. Thromb. Haemost. 2008, 6, 2007–2016. [Google Scholar] [CrossRef]
- Benz, P.M.; Laban, H.; Zink, J.; Günther, L.; Walter, U.; Gambaryan, S.; Dib, K. Vasodilator-Stimulated Phosphoprotein (VASP)-Dependent and -Independent Pathways Regulate Thrombin-Induced Activation of Rap1b in Platelets. Cell Commun. Signal. 2016, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Ke, J.; Liu, Y.; Yang, J.; Lu, R.; Tian, Q.; Hou, W.; Wang, G.; Wei, R.; Hong, T. Synergistic Effects of Metformin with Liraglutide Against Endothelial Dysfunction through GLP-1 Receptor and PKA Signalling Pathway. Sci. Rep. 2017, 7, 41085. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, L.; Kuijpers, M.J.; Gilio, K.; Hego, A.; Théâtre, E.; Maurissen, L.; Vandereyken, M.; Diogo, C.V.; Lecut, C.; Guilmain, W.; et al. Dual-Specificity Phosphatase 3 Deficiency or Inhibition Limits Platelet Activation and Arterial Thrombosis. Circulation 2015, 131, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen Peroxide Promotes Aging-Related Platelet Hyperactivation and Thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.; Huang, W.-C.; Hsu, C.-Y.; Hsia, C.-W.; Jayakumar, T.; Hsieh, C.-Y.; Lu, W.-J.; Chang, C.-C. Metformin Serves as a Novel Drug Treatment for Arterial Thrombosis: Inhibitory Mechanisms on Collagen-Induced Human Platelet Activation. Appl. Sci. 2022, 12, 7426. https://0-doi-org.brum.beds.ac.uk/10.3390/app12157426
Chang Y, Huang W-C, Hsu C-Y, Hsia C-W, Jayakumar T, Hsieh C-Y, Lu W-J, Chang C-C. Metformin Serves as a Novel Drug Treatment for Arterial Thrombosis: Inhibitory Mechanisms on Collagen-Induced Human Platelet Activation. Applied Sciences. 2022; 12(15):7426. https://0-doi-org.brum.beds.ac.uk/10.3390/app12157426
Chicago/Turabian StyleChang, Yi, Wei-Chieh Huang, Chia-Yuan Hsu, Chih-Wei Hsia, Thanasekaran Jayakumar, Cheng-Ying Hsieh, Wan-Jung Lu, and Chao-Chien Chang. 2022. "Metformin Serves as a Novel Drug Treatment for Arterial Thrombosis: Inhibitory Mechanisms on Collagen-Induced Human Platelet Activation" Applied Sciences 12, no. 15: 7426. https://0-doi-org.brum.beds.ac.uk/10.3390/app12157426