
Whole-Genome Sequencing and Comparative Genomic Analysis of Enterococcus spp. Isolated from Dairy Products: Genomic Diversity, Functional Characteristics, and Pathogenic Potential


Abstract
:1. Introduction
2. Materials and Methods
2.1. Microbial Strains and Culture Conditions
2.2. Whole Genome Sequencing, Assembly, and Quality Control
2.3. In Silico Typing and Characterization
2.4. Phylogenetic Analysis and Comparative Genomics
2.5. Statistical Analysis
3. Results
3.1. Species Identification, Assembly Statistics and Subsystem Analysis
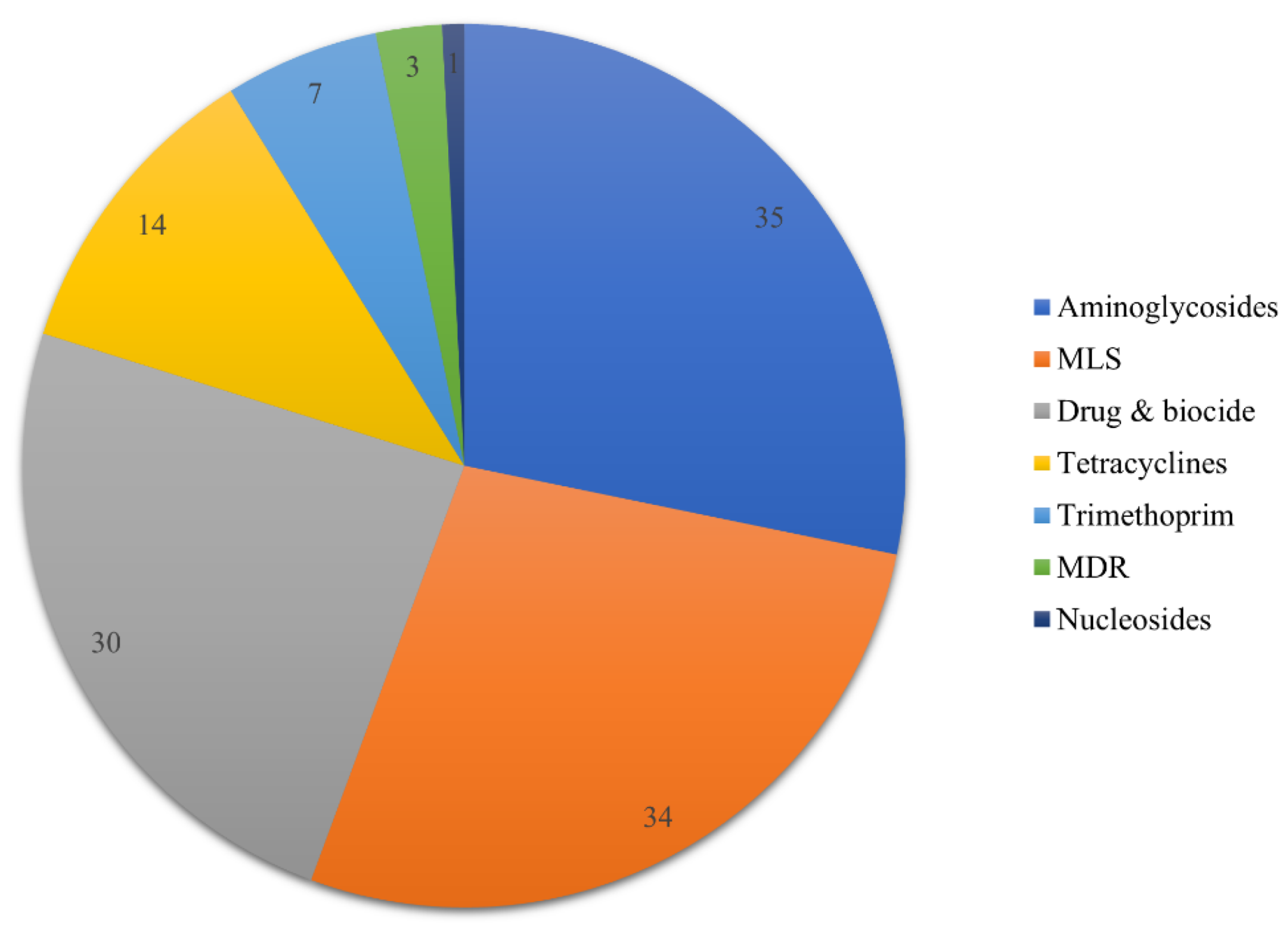
3.2. Presence of Resistance Genes
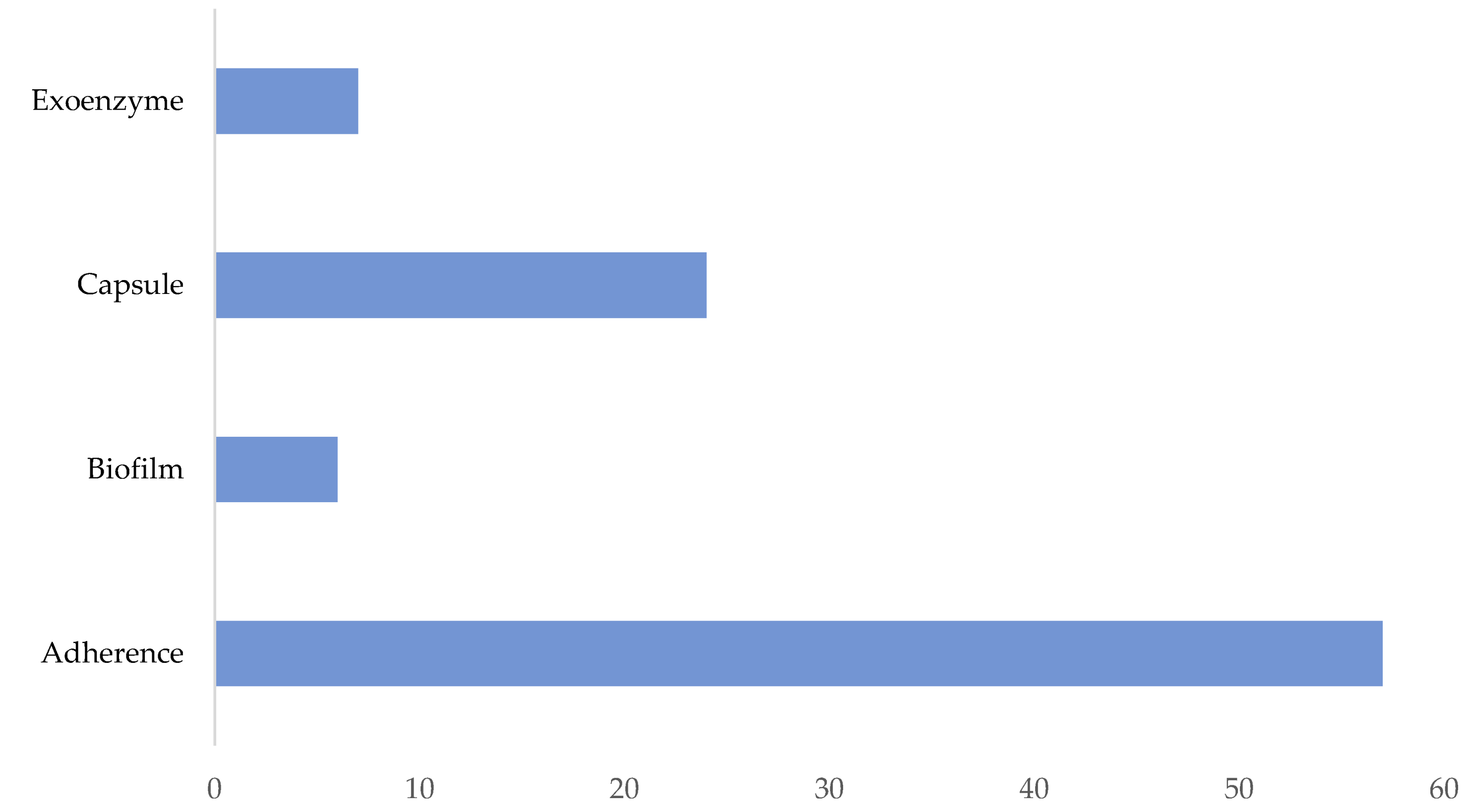
3.3. Presence of Virulence Genes
3.4. Other Genomic Features
3.4.1. Bacteriocins, Prophages and CRISPR-Cas
3.4.2. Plasmids and other MGEs
3.5. Phylogenetic Analysis and Comparative Genomics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Różańska, H.; Lewtak-Piłat, A.; Kubajka, M.; Weiner, M. Occurrence of Enterococci in Mastitic Cow’s Milk and Their Antimicrobial Resistance. J. Vet. Res. 2019, 63, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Jan, O.; Jaśkowski, J.M. Mastitis in Small Ruminants. Med. Weter. 2014, 70, 67–72. [Google Scholar] [CrossRef]
- Bergonier, D.; de Crémoux, R.; Rupp, R.; Lagriffoul, G.; Berthelot, X. Mastitis of Dairy Small Ruminants. Vet. Res. 2003, 34, 689–716. [Google Scholar] [CrossRef] [PubMed]
- Hammerum, A.M. Enterococci of Animal Origin and Their Significance for Public Health. Clin. Microbiol. Infect. 2012, 18, 619–625. [Google Scholar] [CrossRef]
- Markwart, R.; Willrich, N.; Haller, S.; Noll, I.; Koppe, U.; Werner, G.; Eckmanns, T.; Reuss, A. The Rise in Vancomy-Cin-Resistant Enterococcus faecium in Germany: Data from the German Antimicrobial Resistance Surveillance (ARS). Antimicrob. Resist. Infect. Control. 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Hammerum, A.M.; Lester, C.H.; Heuer, O.E. Antimicrobial-Resistant Enterococci in Animals and Meat: A Human Health Hazard? Foodborne Pathog. Dis. 2010, 7, 1137–1146. [Google Scholar] [CrossRef]
- Dapkevicius, M.; Sgardioli, B.; Câmara, S.; Poeta, P.; Malcata, F. Current Trends of Enterococci in Dairy Products: A Comprehensive Review of Their Multiple Roles. Foods 2021, 10, 821. [Google Scholar] [CrossRef]
- Ghattargi, V.C.; Gaikwad, M.A.; Meti, B.S.; Nimonkar, Y.S.; Dixit, K.; Prakash, O.; Shouche, Y.S.; Pawar, S.P.; Dhotre, D.P. Comparative Genome Analysis Reveals Key Genetic Factors Associated with Probiotic Property in Enterococcus faecium Strains. BMC Genom. 2018, 19, 652. [Google Scholar] [CrossRef]
- Graham, K.; Stack, H.; Rea, R. Safety, Beneficial and Technological Properties of Enterococci for Use in Functional Food Applications—A Review. Crit. Rev. Food Sci. Nutr. 2020, 60, 3836–3861. [Google Scholar] [CrossRef]
- Tsigkrimani, M.; Bakogianni, M.; Paramithiotis, S.; Bosnea, L.; Pappa, E.; Drosinos, E.H.; Skandamis, P.N.; Mataragas, M. Microbial Ecology of Artisanal Feta and Kefalograviera Cheeses, Part I: Bacterial Community and Its Functional Characteristics with Focus on Lactic Acid Bacteria as Determined by Culture-Dependent Methods and Phenotype Microarrays. Microorganisms 2022, 10, 161. [Google Scholar] [CrossRef]
- Tsigkrimani, M.; Panagiotarea, K.; Paramithiotis, S.; Bosnea, L.; Pappa, E.; Drosinos, E.H.; Skandamis, P.N.; Mataragas, M. Microbial Ecology of Sheep Milk, Artisanal Feta, and Kefalograviera Cheeses. Part II: Technological, Safety, and Probiotic Attributes of Lactic Acid Bacteria Isolates. Foods 2022, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Syrokou, M.K.; Themeli, C.; Paramithiotis, S.; Mataragas, M.; Bosnea, L.; Argyri, A.A.; Chorianopoulos, N.G.; Skandamis, P.N.; Drosinos, E.H. Microbial Ecology of Greek Wheat Sourdoughs, Identified by a Culture-Dependent and a Culture-Independent Approach. Foods 2020, 9, 1603. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2019. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 11 May 2022).
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.-F.; Lio, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. SkewIT: The Skew Index Test for Large-Scale GC Skew Analysis of Bacterial Genomes. PLoS Comput. Biol. 2020, 16, e1008439. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Smirnov, S.; Nikolskaya, A.N.; et al. The COG Database: An Updated Vesion Includes Eukaryotes. BMC Bioinform. 2003, 4, 1–14. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, An Update of CRISRFinder, Includes A Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for Finding Prophage in Bacterial Genomes. Briefings Bioinform. 2017, 20, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Abricate, Github; Github. 2020. Available online: https://github.com/tseemann/abricate (accessed on 11 May 2022).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis—10 Years on. Nucleic Acids Res. 2015, 44, D694–D697. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2020, 76, 101–109. [Google Scholar] [CrossRef]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A User-Friendly Web Server to Thoroughly Mine Ripps and Bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H.E. MOB-Suite: Software Tools for Clustering, Reconstruction and Typing of Plasmids from Draft Assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid Phylogenetic Analysis of Large Samples of Recombinant Bacterial Whole Genome Sequences Using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Connor, T.; Sirén, J.; Aanensen, D.M.; Corander, J. Hierarchical and Spatially Explicit Clustering of DNA Sequences with BAPS Software. Mol. Biol. Evol. 2013, 30, 1224–1228. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Weimann, A.; Mooren, K.; Frank, J.; Pope, P.B.; Bremges, A.; McHardy, A.C. From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer. mSystems 2016, 1, e00101-16. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 11 May 2022).
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid Scoring of Genes in Microbial Pan-Genome-Wide Association Studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.W. Aminoglycoside Resistance in Enterococci. Clin. Infect. Dis. 2000, 31, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Park, C.H.; Hegde, S.S.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Mechanistic and Structural Analysis of Aminoglycoside N-Acetyltransferase AAC(6′)-Ib and Its Bifunctional, Fluoroquinolone-Active AAC(6′)-Ib-cr Variant. Biochemistry 2008, 47, 9825–9835. [Google Scholar] [CrossRef] [PubMed]
- Salah, A.N.; Elleboudy, N.S.; El-Housseiny, G.S.; Yassien, M.A. Cloning and Sequencing of Lsae Efflux Pump Gene from MDR Enterococci and Its Role in Erythromycin Resistance. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 94, 105010. [Google Scholar] [CrossRef] [PubMed]
- Hancock, L.E.; Gilmore, M.S. The Capsular Polysaccharide of Enterococcus faecalis and Its Relationship to Other Polysaccharides in the Cell Wall. Proc. Natl. Acad. Sci. USA 2002, 99, 1574–1579. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Thomas, V.C.; Fleming, S.D.; Hancock, L.E. Enterococcus faecalis Capsular Polysaccharide Serotypes C and D and Their Contributions to Host Innate Immune Evasion. Infect. Immun. 2009, 77, 5551–5557. [Google Scholar] [CrossRef]
- Engelbert, M.; Mylonakis, E.; Ausubel, F.M.; Calderwood, S.B.; Gilmore, M.S. Contribution of Gelatinase, Serine Protease, and fsr to the Pathogenesis of Enterococcus faecalis Endophthalmitis. Infect. Immun. 2004, 72, 3628–3633. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Zhong, Z.; Kwok, L.-Y.; Hou, Q.; Sun, Y.; Li, W.; Zhang, H.; Sun, Z. Comparative Genomic Analysis Revealed Great Plasticity and Environmental Adaptation of the Genomes of Enterococcus faecium. BMC Genom. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Lebreton, F.; van Schaik, W.; McGuire, A.M.; Godfrey, P.; Griggs, A.; Mazumdar, V.; Corander, J.; Cheng, L.; Saif, S.; Young, S.; et al. Emergence of Epidemic Multidrug-Resistant Enterococcus faecium from Animal and Commensal Strains. mBio 2013, 4, e00534-13. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The Subsystems Approach to Genome Annotation and its Use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [Green Version]
- Henning, C.; Gautam, D.; Muriana, P. Identification of Multiple Bacteriocins in Enterococcus spp. Using an Enterococcus-Specific Bacteriocin PCR Array. Microorganisms 2015, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Hou, Q.; Wang, Y.; Li, J.; Li, W.; Kwok, L.-Y.; Sun, Z.; Zhang, H.; Zhong, Z. Comparative Genomic Analysis of Enterococcus faecalis: Insights into Their Environmental Adaptations. BMC Genom. 2018, 19, 527. [Google Scholar] [CrossRef] [PubMed]
- Kohler, V.; Vaishampayan, A.; Grohmann, E. Broad-Host-Range Inc18 Plasmids: Occurrence, Spread and Transfer Mechanisms. Plasmid 2018, 99, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Schwarz, S.; Li, Y.; Shen, Z.; Zhang, Q.; Wu, C.; Shen, J. Transferable Multiresistance Plasmids Carrying cfr in Enterococcus spp. from Swine and Farm Environment. Antimicrob. Agents Chemother. 2013, 57, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.M.; Halami, P.M. Detection of Mobile Genetic Elements in Pediocin PA-1 Like Producing Lactic Acid Bacteria. J. Basic Microbiol. 2013, 53, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Bringel, F. IS Lpl1 Is a Functional IS 30 -Related Insertion Element in Lactobacillus plantarum That Is Also Found in Other Lactic Acid Bacteria. Appl. Environ. Microbiol. 2003, 69, 6032–6040. [Google Scholar] [CrossRef]
- Hendrickx, A.P.A.; van Luit-Asbroek, M.; Schapendonk, C.M.E.; van Wamel, W.J.B.; Braat, J.C.; Wijnands, L.M.; Bonten, M.J.M.; Willems, R.J.L. SgrA, a Nidogen-Binding LPXTG Surface Adhesin Implicated in Biofilm Formation, and EcbA, a Collagen Binding MSCRAMM, Are Two Novel Adhesins of Hospital-Acquired Enterococcus faecium. Infect. Immun. 2009, 77, 5097–5106. [Google Scholar] [CrossRef]
- Margalho, L.P.; van Schalkwijk, S.; Bachmann, H.; Sant’Ana, A.S. Enterococcus spp. in Brazilian artisanal cheeses: Occurrence and Assessment of Phenotypic and Safety Properties of a Large Set of Strains Through the Use of High Throughput Tools Combined with Multivariate Statistics. Food Control 2020, 118, 107425. [Google Scholar] [CrossRef]
- Sun, Z.; Harris, H.M.B.; McCann, A.; Guo, C.; Argimón, S.; Zhang, W.; Yang, X.; Jeffery, I.; Cooney, J.; Kagawa, T.F.; et al. Expanding the Biotechnology Potential of Lactobacilli Through Comparative Genomics Of 213 Strains and Associated Genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef]
- Tarrah, A.; Pakroo, S.; Junior, W.J.F.L.; Guerra, A.F.; Corich, V.; Giacomini, A. Complete Genome Sequence and Carbohydrates-Active EnZymes (CAZymes) Analysis of Lactobacillus paracasei DTA72, a Potential Probiotic Strain with Strong Capability to Use Inulin. Curr. Microbiol. 2020, 77, 2867–2875. [Google Scholar] [CrossRef]
- Armenta, S.; Moreno, S.A.; Sánchez-Cuapio, Z.; Sánchez, S.; Rodríguez-Sanoja, R. Advances in Molecular Engineering of Carbohydrate-Binding Modules. Proteins Struct. Funct. Bioinform. 2017, 85, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Vaaje-Kolstad, G.; Ghosh, A.; Hurtado-Guerrero, R.; Konarev, P.V.; Ibrahim, A.F.M.; Svergun, D.I.; Eijsink, V.G.H.; Chatterjee, N.S.; Van Aalten, D.M.F. The Vibrio cholerae Colonization Factor GbpA Possesses a Modular Structure that Governs Binding to Different Host Surfaces. PLoS Pathog. 2012, 8, e1002373. [Google Scholar] [CrossRef]
- Kiousi, D.E.; Efstathiou, C.; Tegopoulos, K.; Mantzourani, I.; Alexopoulos, A.; Plessas, S.; Kolovos, P.; Koffa, M.; Galanis, A. Genomic Insight into Lacticaseibacillus paracasei SP5, Reveals Genes and Gene Clusters of Probiotic Interest and Biotechnological Potential. Front. Microbiol. 2022, 13, 2038. [Google Scholar] [CrossRef] [PubMed]
- Chilambi, G.S.; Nordstrom, H.R.; Evans, D.R.; Ferrolino, J.A.; Hayden, R.T.; Marón, G.M.; Vo, A.N.; Gilmore, M.S.; Wolf, J.; Rosch, J.W.; et al. Evolution of Vancomycin-Resistant Enterococcus faecium During Colonization and Infection in Immunocompromised Pediatric Patients. Proc. Natl. Acad. Sci. USA 2020, 117, 11703–11714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vrijenhoek, J.E.P.; Bonten, M.J.M.; Willems, R.J.L.; van Schaik, W. A Genetic Element Present on Megaplasmids Allows Enterococcus faecium to Use Raffinose as Carbon Source. Environ. Microbiol. 2011, 13, 518–528. [Google Scholar] [CrossRef]
- Saber, M.M.; Shapiro, B.J. Benchmarking Bacterial Genome-Wide Association Study Methods Using Simulated Genomes and Phenotypes. Microb. Genom. 2020, 6, e000337. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain ID | Genus & Species | Genome Size (Mb) | GC Content (%) | No of Scaffolds | N50 (Mb) | No of CDSs | Contamination (%) | Completeness (%) |
|---|---|---|---|---|---|---|---|---|
| ent_C1 | E. faecium | 3.12 | 37.69 | 89 | 1.68 | 3472 | 3.9 | 96.1 |
| ent_C11 | E. faecium | 2.74 | 37.94 | 5 | 2.28 | 2800 | 4.6 | 97.4 |
| ent_C111 | E. italicus | 2.33 | 39.25 | 28 | 2.21 | 2332 | 3.5 | 97.5 |
| ent_C116 | E. faecium | 2.79 | 37.87 | 9 | 2.26 | 2863 | 3.5 | 98.2 |
| ent_C14 | E. faecium | 2.83 | 37.79 | 14 | 2.26 | 2874 | 3.2 | 98.7 |
| ent_C143 | E. faecium | 2.67 | 37.89 | 70 | 2.11 | 2811 | 3.3 | 98.3 |
| ent_C146 | E. faecium | 2.87 | 37.82 | 58 | 2.35 | 3010 | 3.4 | 95.0 |
| ent_C151 | E. faecium | 2.78 | 37.91 | 36 | 2.41 | 2919 | 2.0 | 98.8 |
| ent_C154 | E. faecium | 2.75 | 37.9 | 5 | 1.48 | 2793 | 3.9 | 96.7 |
| ent_C155 | E. faecium | 2.85 | 37.86 | 34 | 0.88 | 2993 | 3.7 | 99.0 |
| ent_C156 | E. faecium | 2.78 | 37.82 | 7 | 0.97 | 2840 | 3.0 | 96.1 |
| ent_C157 | E. faecium | 2.79 | 37.87 | 8 | 2.59 | 2858 | 3.7 | 96.5 |
| ent_C158 | E. faecalis | 2.90 | 37.45 | 13 | 2.85 | 2864 | 2.5 | 98.5 |
| ent_C159 | E. faecium | 2.76 | 37.88 | 7 | 2.56 | 2812 | 2.1 | 98.4 |
| ent_C179 | E. durans | 2.94 | 37.87 | 13 | 2.54 | 2933 | 4.9 | 96.9 |
| ent_C22 | E. faecium | 2.81 | 37.88 | 49 | 2.14 | 2951 | 3.6 | 97.2 |
| ent_C25 | E. faecium | 2.79 | 37.83 | 17 | 1.47 | 2832 | 4.6 | 95.4 |
| ent_C28 | E. faecalis | 2.95 | 37.42 | 18 | 2.86 | 2933 | 2.3 | 97.5 |
| ent_C29 | E. faecium | 2.77 | 37.86 | 13 | 1.64 | 2803 | 4.5 | 95.7 |
| ent_C3 | E. faecium | 2.76 | 37.88 | 8 | 2.57 | 2806 | 3.4 | 97.2 |
| ent_C4 | E. faecium | 2.79 | 37.86 | 11 | 1.20 | 2867 | 3.1 | 98.7 |
| ent_C5 | E. faecium | 2.75 | 37.9 | 6 | 1.48 | 2784 | 4.1 | 97.5 |
| ent_C57 | E. faecium | 2.75 | 37.9 | 4 | 2.41 | 2793 | 2.9 | 98.7 |
| ent_C6 | E. faecium | 2.79 | 37.87 | 7 | 2.59 | 2865 | 2.2 | 98.4 |
| ent_C62 | E. faecium | 2.85 | 37.83 | 38 | 2.42 | 2897 | 2.2 | 95.8 |
| ent_C7 | E. faecium | 2.79 | 37.87 | 12 | 1.63 | 2846 | 4.1 | 95.1 |
| ent_C71 | E. faecium | 2.76 | 37.87 | 15 | 2.08 | 2795 | 3.7 | 98.4 |
| ent_C74 | E. italicus | 2.39 | 39.36 | 29 | 1.43 | 2442 | 3.2 | 97.4 |
| ent_C78 | E. faecalis | 2.96 | 37.46 | 18 | 2.43 | 2918 | 2.8 | 98.0 |
| ent_C8 | E. faecium | 2.74 | 37.94 | 5 | 1.64 | 2798 | 3.2 | 98.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostolakos, I.; Tsigkrimani, M.; Paramithiotis, S.; Mataragas, M. Whole-Genome Sequencing and Comparative Genomic Analysis of Enterococcus spp. Isolated from Dairy Products: Genomic Diversity, Functional Characteristics, and Pathogenic Potential. Appl. Sci. 2022, 12, 9620. https://0-doi-org.brum.beds.ac.uk/10.3390/app12199620
Apostolakos I, Tsigkrimani M, Paramithiotis S, Mataragas M. Whole-Genome Sequencing and Comparative Genomic Analysis of Enterococcus spp. Isolated from Dairy Products: Genomic Diversity, Functional Characteristics, and Pathogenic Potential. Applied Sciences. 2022; 12(19):9620. https://0-doi-org.brum.beds.ac.uk/10.3390/app12199620
Chicago/Turabian StyleApostolakos, Ilias, Markella Tsigkrimani, Spiros Paramithiotis, and Marios Mataragas. 2022. "Whole-Genome Sequencing and Comparative Genomic Analysis of Enterococcus spp. Isolated from Dairy Products: Genomic Diversity, Functional Characteristics, and Pathogenic Potential" Applied Sciences 12, no. 19: 9620. https://0-doi-org.brum.beds.ac.uk/10.3390/app12199620