
Bioinformatics Characterization of Candidate Genes Associated with Gene Network and miRNA Regulation in Esophageal Squamous Cell Carcinoma Patients

 , ,
, ,
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Affymetrix Microarray Data
2.2. DEG’s Processing Using GEO2R
2.3. PPI Network Construction to Identify Hub Genes
2.4. Analysis and Validation of HGsT10 Expression and Its Various ESCC Stages
2.5. The Overall Survivability and Promoter Methylation
2.6. Comparison of HGsT10 Gene Expression and Somatic Mutation Identification
2.7. HGsT10 Interaction with Other Genes and miRNA
2.8. Functional and Pathway Enrichment Analysis for the Predicted HGsT10 Genes
3. Results
3.1. The GEO Dataset Validation
3.2. Identification of DEGs and HGsT10 and PPI Network Construction for ESCC Tumor and Normal Tissues
3.3. Verification of the HGsT10 Expression Patterns
3.4. The Comparison of Expression of HGsT10 in the ESCC Patients with Other Types of EC
3.5. Correlation between HGST10 Genes Expression and Tumor Stage in Patients with ESCC Carcinoma
3.6. The Validation of HGsT10 Expression by Promoter Methylation in ESCC Patients
3.7. The Predicted HmiRT10 from the Constructed miRNA-HGsT10 Regulatory Network
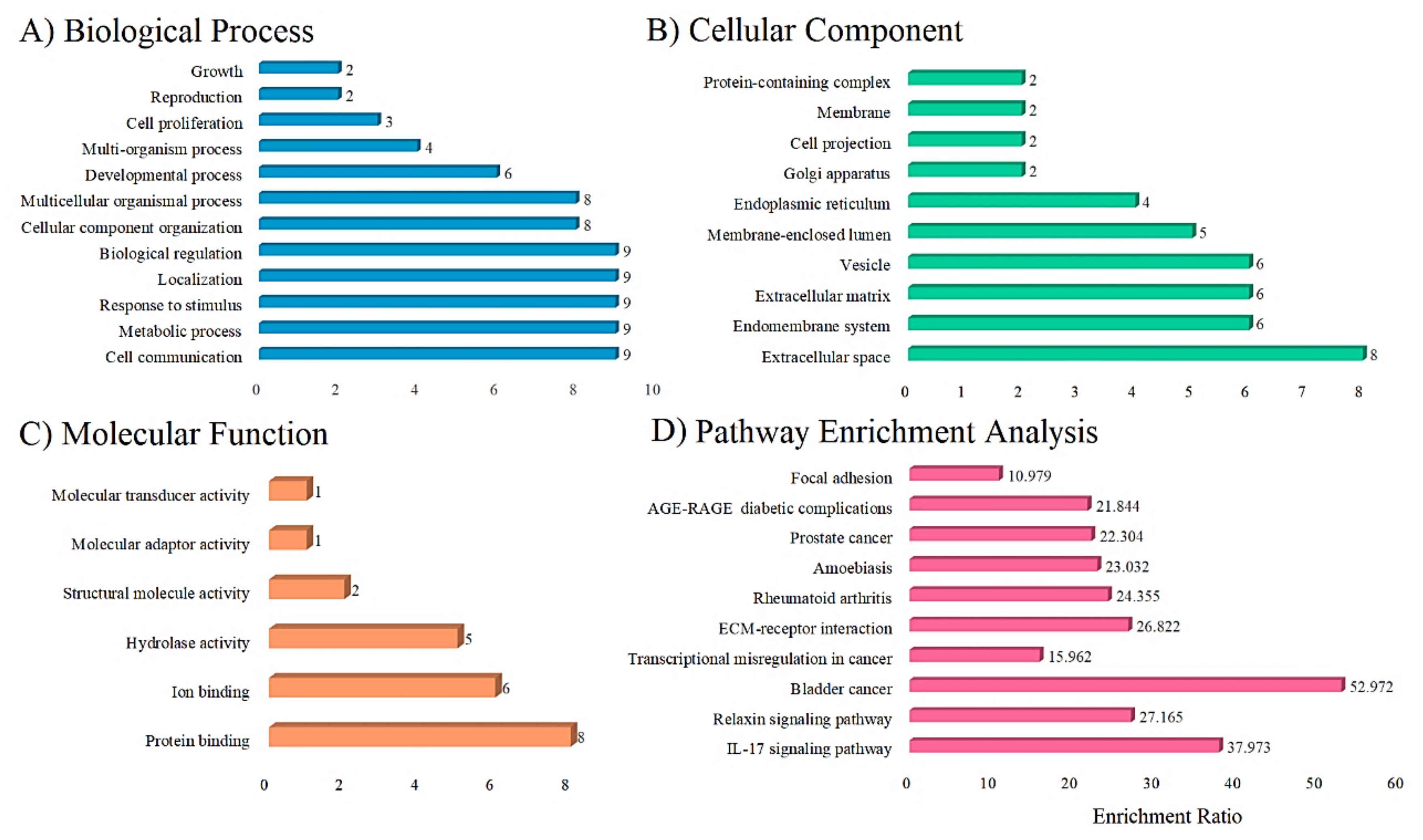
3.8. Functional and Pathway Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Dong, K.; Guo, P.; Tan, Z.; Zhang, F.; Tian, Y.; Lv, H. Identification of Autophagy-Related Genes and Small-Molecule Drugs in Esophageal Carcinoma. Med. Sci. Monit. 2020, 16, e921855. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Zhou, Z.; Wang, X.; Hua, X.; Zou, M.; Zhang, X. SHCBP1 Promotes the Progression of Esophageal Squamous Cell Carcinoma Via the TGFβ Pathway. Appl. Immunohistochem. Mol. Morphol. 2021, 29, 136–143. [Google Scholar] [CrossRef]
- Chen, Y.; Bieerkehazhi, S.; Li, X.; Ma, L.; Yibulayin, W.; Ran, J. Survivin Regulates Bad Gene Expression by Binding to Its Promoter and Modulates Cell Cycle and Apoptosis in Esophageal Carcinoma Cell. J. Oncol. 2021, 2021, 1384289. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jiang, Y.G. Current gene expression studies in esophageal carcinoma. Curr. Genom. 2009, 10, 534–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Xu, R.; Li, Q.; Zhao, J.Y.; Peng, B.; Zhang, H.; Guo, J.D.; Zhang, S.Q.; Li, H.W.; Wang, J.; et al. Systematic profiling of ferroptosis gene signatures predicts prognostic factors in esophageal squamous cell carcinoma. Mol. Ther. Oncolytics 2021, 21, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Liu, Z.; Shang, L.; Cai, H.Q.; Zhang, Y.; Cai, Y.; Xu, X.; Hao, J.J.; Wang, M.R. Deletion and downregulation of MTAP contribute to the motility of esophageal squamous carcinoma cells. Onco Targets Ther. 2017, 10, 5855–5862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Shan, Y.; Zhou, X.; Zhang, C.; Zhao, R.; Zhao, L.; Shan, B. MicroRNA-485-5p suppresses the progression of esophageal squamous cell carcinoma by targeting flotillin-1 and inhibits the epithelial-mesenchymal transition. Oncol. Rep. 2021, 45, 93. [Google Scholar] [CrossRef]
- Predescu, D.; Gheorghe, M.; Boeriu, M.; Constantin, A.; Iosif, C.; Anghel, R.; Constantinoiu, S. Molecular factors and criteria for predicting the response to neoadjuvant treatment in patients with esophageal squamous cell carcinoma (ESCC)—Responder/non-responder. Chirurgia 2012, 107, 583–590. [Google Scholar]
- Napier, K.J.; Scheerer, M.; Misra, S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J. Gastrointest. Oncol. 2014, 6, 112–120. [Google Scholar] [CrossRef]
- Businello, G.; Parente, P.; Mastracci, L.; Pennelli, G.; Traverso, G.; Milione, M.; Bellan, E.; Michelotto, M.; Kotsafti, A.; Grillo, F.; et al. The Pathologic and Molecular Landscape of Esophageal Squamous Cell Carcinogenesis. Cancers 2020, 12, 2160. [Google Scholar] [CrossRef]
- Redondo, J.A.; Bibes, R.; Vercauteren Drubbel, A.; Dassy, B.; Bisteau, X.; Maury, E.; Beck, B. PER2 Circadian Oscillation Sensitizes Esophageal Cancer Cells to Chemotherapy. Biology 2021, 10, 266. [Google Scholar] [CrossRef]
- Lee, T.G.; Jeong, E.H.; Kim, S.Y.; Kim, H.R.; Kim, H.; Kim, C.H. Fhit, a tumor suppressor protein, induces autophagy via 14–3-3τ in non-small cell lung cancer cells. Oncotarget 2017, 8, 31923–31937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, C.W.; Wu, M.T.; Chen, Y.K.; Wu, C.C.; Chen, W.C.; Li, H.P.; Chou, S.H.; Wu, D.C.; Wu, I.C. Identification of biomarkers for esophageal squamous cell carcinoma using feature selection and decision tree methods. Sci. World J. 2013, 2013, 782031. [Google Scholar] [CrossRef]
- Couch, G.; Redman, J.E.; Wernisch, L.; Newton, R.; Malhotra, S.; Dawsey, S.M.; Lao-Sirieix, P.; Fitzgerald, R.C. The Discovery and Validation of Biomarkers for the Diagnosis of Esophageal Squamous Dysplasia and Squamous Cell Carcinoma. Cancer Prev. Res. 2016, 9, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sario, A. Clinical and molecular overview of inherited disorders resulting from epigenomic dysregulation. Eur. J. Med. Genet. 2009, 52, 363–372. [Google Scholar] [CrossRef]
- Fontes, L.P.; Jimenez, Q.P.; Iriarte, M.M. Epigenetics and epilepsy. Neurologia 2015, 30, 111–118. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Das, R.; Weidman, J.R.; Jirtle, R.L. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr. Res. 2007, 61, 30R–37R. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.P.; Fahimipour, F.; Tarran, R.; Kim, S.; Scarel-Caminaga, R.M.; Justice, A.; North, K. Epigenetic reprogramming in periodontal disease: Dynamic crosstalk with potential impact in oncogenesis. Periodontology 2000, 82, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Joehanes, R.; Wilson, R.; Tanaka, T.; Ferrucci, L.; Kretschmer, A.; Prokisch, H.; Schramm, K.; Gieger, C.; Peters, A.; et al. Epigenome-wide association study of whole blood gene expression in Framingham Heart Study participants provides molecular insight into the potential role of CHRNA5 in cigarette smoking-related lung diseases. Clin. Epigenet. 2021, 13, 60. [Google Scholar] [CrossRef]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 629780. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenet. Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Eladl, M.A.; Khoder, G. Helicobacter pylori-induced DNA Methylation as an Epigenetic Modulator of Gastric Cancer: Recent Outcomes and Future Direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Wang, Q.; Dai, Y.; Wang, H.; Wang, M.; Wang, J.; Zhang, D.; Sun, P.; Qi, T.; Jin, X.; et al. Hypomethylation of PlncRNA-1 promoter enhances bladder cancer progression through the miR-136-5p/Smad3 axis. Cell Death Dis. 2020, 11, 1038. [Google Scholar] [CrossRef]
- Wang, X.; Shi, D.; Zhao, D.; Hu, D. Aberrant Methylation and Differential Expression of SLC2A1, TNS4, GAPDH, ATP8A2, and CASZ1 Are Associated with the Prognosis of Lung Adenocarcinoma. Biomed Res. Int. 2020, 2020, 1807089. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhuang, X.; Xia, X.; Li, X.; Xiao, K.; Liu, X. Correlation Between Promoter Hypomethylation and Increased Expression of Syncytin-1 in Non-Small Cell Lung Cancer. Int. J. Gen. Med. 2021, 14, 957–965. [Google Scholar] [CrossRef]
- Liu, G.M.; Zeng, H.D.; Zhang, C.Y.; Xu, J.W. Identification of a six-gene signature predicting overall survival for hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yi, Y.; Wu, W.; Wu, K.; Zhang, W. Bioinformatics prediction and analysis of hub genes and pathways of three types of gynecological cancer. Oncol. Lett. 2019, 18, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Gu, Z.Y.; Li, Q.; Liu, Q.H.; Yang, X.Y.; Zhang, J.J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef]
- Wang, M.; Zhong, B.; Li, M.; Wang, Y.; Yang, H.; Du, K. Identification of potential core genes and pathways predicting pathogenesis in head and neck squamous cell carcinoma. Biosci. Rep. 2021, 41, BSR20204148. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Jin, D.; Wang, B.; Gou, Y.J.; Dong, X.C. Identification of miRNAs as prognostic factors for esophageal squamous cell carcinoma. Math. Biosci. Eng. 2020, 17, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zhao, C.H.; Li, P.S.; Liu, R.R.; Zhang, Y.; Chen, H.E.; Chang, L.P.; Gong, Y.H.; Guan, Y.F.; Yi, X.; et al. Post-radiation circulating tumor DNA as a prognostic factor in locally advanced esophageal squamous cell carcinoma. Oncol. Lett. 2021, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The gene expression omnibus database. Methods Mol. Biol. 2016, 14, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Li, R.; Li, P.; Xing, W. Expression Data from Esophageal Squamous Cell Carcinoma Patients (GSE161533). 2020. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/geo/ (accessed on 17 December 2021).
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006, 34, D322–D326. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Fau, C.C.; Chen, S.-H. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.Y.; Peng, Y.H.; Weng, X.F.; Xie, J.J.; Xu, Y.W. Blood-based biomarkers for early detection of esophageal squamous cell carcinoma. World J. Gastroenterol. 2020, 26, 1708–1725. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Cao, S.; Dong, X.; Rao, S.; Cai, K. Understanding Esophageal Cancer: The Challenges and Opportunities for the Next Decade. Front. Oncol. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hu, Y.; Zhang, M.F.; Luo, K.J.; Xie, X.Y.; Wen, J.; Fu, J.H.; Yang, H. MMP1 promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2016, 377, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, J.; Guo, Q.; Duan, F.; Tang, F.; Zheng, P.; Zhao, Z.; Lu, G. Overexpression of MMP-2 and MMP-9 in esophageal squamous cell carcinoma. Dis. Esophagus 2009, 22, 664–667. [Google Scholar] [CrossRef]
- Song, Y.; Wang, X.; Wang, F.; Peng, X.; Li, P.; Liu, S.; Zhang, D. Identification of four genes and biological characteristics of esophageal squamous cell carcinoma by integrated bioinformatics analysis. Cancer Cell Int. 2021, 21, 123. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Z.; Hu, X.; Luo, M.; Sun, Z.; Li, J.; Shi, S.; Feng, X.; Zhou, C.; Li, Z.; et al. AJUBA promotes the migration and invasion of esophageal squamous cell carcinoma cells through upregulation of MMP10 and MMP13 expression. Oncotarget 2016, 7, 36407–36418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Huang, M.; Chen, Z.; Chen, J.; Chao, Q.; Yin, X.; Quan, M. FTO promotes cell proliferation and migration in esophageal squamous cell carcinoma through up-regulation of MMP13. Exp. Cell Res. 2020, 389, 111894. [Google Scholar] [CrossRef]
- Mahmoudian, R.A.; Gharaie, M.L.; Abbaszadegan, M.R.; Alasti, A.; Forghanifard, M.M.; Mansouri, A.; Gholamin, M. Crosstalk between MMP-13, CD44, and TWIST1 and its role in regulation of EMT in patients with esophageal squamous cell carcinoma. Mol. Cell. Biochem. 2021, 476, 2465–2478. [Google Scholar] [CrossRef]
- Jiao, X.L.; Chen, D.; Wang, J.G.; Zhang, K.J. Clinical significance of serum matrix metalloproteinase-13 levels in patients with esophageal squamous cell carcinoma (ESCC). Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 509–515. [Google Scholar]
- Sedighi, M.; Aledavood, S.A.; Abbaszadegan, M.; Memar, B.; Montazer, M.; Rajabian, M.; Gholamin, M. Matrix Metalloproteinase-13—A Potential Biomarker for Detection and Prognostic Assessment of Patients with Esophageal Squamous Cell Carcinoma. Asian Pac. J. Cancer Prev. 2016, 17, 2781–2785. [Google Scholar]
- Sato, F.; Shimada, Y.; Watanabe, G.; Uchida, S.; Makino, T.; Imamura, M. Expression of vascular endothelial growth factor, matrix metalloproteinase-9 and E-cadherin in the process of lymph node metastasis in oesophageal cancer. Br. J. Cancer 1999, 80, 1366–1372. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.D.; Li, J.Y.; Li, M.; Gu, J.; Shi, X.T.; Ke, Y.; Chen, K.N. Matrix metalloproteinases expression correlates with survival in patients with esophageal squamous cell carcinoma. Am. J. Gastroenterol. 2005, 100, 1835–1843. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.S.; Ma, X.Y.; Chai, D.M.; Ma, L.; Feng, Z.Z.; Cheng, Z.N.; Lai, M.D. Overexpression of MMP-1 and VEGF-C is associated with a less favorable prognosis in esophageal squamous cell carcinoma. Onkologie 2012, 35, 651–656. [Google Scholar] [CrossRef]
- Hosono, M.; Koma, Y.I.; Takase, N.; Urakawa, N.; Higashino, N.; Suemune, K.; Kodaira, H.; Nishio, M.; Shigeoka, M.; Kakeji, Y.; et al. CXCL8 derived from tumor-associated macrophages and esophageal squamous cell carcinomas contributes to tumor progression by promoting migration and invasion of cancer cells. Oncotarget 2017, 8, 106071–106088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, J.L.; Zhang, C.Y.; Ma, Y.F.; Zhao, R.; Wang, Y.Y. The prognostic role of FZD6 in esophageal squamous cell carcinoma patients. Clin. Transl. Oncol. 2020, 22, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Liu, M. Chemokines and their receptors as biomarkers in esophageal cancer. Esophagus 2020, 17, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Liu, S.; Zhang, T.; Wang, Y.; Qin, G.; Chen, X.; Zhang, H.; Wang, D.; Huang, D.; Wang, F.; et al. NEDD9 promotes cancer stemness by recruiting myeloid-derived suppressor cells via CXCL8 in esophageal squamous cell carcinoma. Cancer Biol. Med. 2021, 18, 705. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Che, Y.; Zhang, C.; Huang, J.; Lei, Y.; Lu, Z.; Sun, N.; He, J. PLAU. directs conversion of fibroblasts to inflammatory cancer-associated fibroblasts, promoting esophageal squamous cell carcinoma progression via uPAR/Akt/NF-κB/IL8 pathway. Cell Death Discov. 2021, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zheng, K.; Liu, Y.; Li, J.; Wang, S.; Liu, K.; Song, X.; Li, N.; Xie, S.; et al. The clinical significance of collagen family gene expression in esophageal squamous cell carcinoma. PeerJ 2019, 7, e7705. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Jia, E.; Ren, N.; Xin, H. Identification of prognostic miRNA biomarkers for esophageal cancer based on The Cancer Genome Atlas and Gene Expression Omnibus. Medicine 2021, 100, e24832. [Google Scholar] [CrossRef]
- Booka, E.; Tsubosa, Y.; Yokota, T.; Mayanagi, S.; Ishii, K.; Urakami, K.; Ohshima, K.; Ohnami, S.; Nagashima, T.; Yamaguchi, K. Whole exome sequencing and deep sequencing of esophageal squamous cell carcinoma and adenocarcinoma in Japanese patients using the Japanese version of the Genome Atlas, JCGA. Esophagus 2021, 18, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Suo, J.; Shao, S.; Xue, L.; Chen, W.; Dong, L.; Shi, J.; Fu, M.; Lu, N.; Zhan, Q.; et al. Overexpression of OLC1 promotes tumorigenesis of human esophageal squamous cell carcinoma. PLoS ONE 2014, 9, e90958. [Google Scholar] [CrossRef]
- Cui, L.; Feng, L.; Zhang, Z.H.; Jia, X.B. The anti-inflammation effect of baicalin on experimental colitis through inhibiting TLR4/NF-κB pathway activation. Int. Immunopharmacol. 2014, 23, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, T.; Sawada, G.; Amano, S.; Kume, K.; Ito, C.; Endo, F.; Konosu, M.; Shioi, Y.; Akiyama, Y.; Takahara, T.; et al. Downregulation of ST6GALNAC1 is associated with esophageal squamous cell carcinoma development. Int. J. Oncol. 2017, 50, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Lessi, F.; Aretini, P.; Rizzo, M.; Morelli, M.; Menicagli, M.; Franceschi, S.; Mazzanti, C.M. Analysis of exosome-derived microRNAs reveals insights of intercellular communication during invasion of breast, prostate and glioblastoma cancer cells. Cell Adhes. Migr. 2021, 15, 180–201. [Google Scholar] [CrossRef]
- Lorusso, C.; De Summa, S.; Pinto, R.; Danza, K.; Tommasi, S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells 2020, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- He, F.C.; Meng, W.W.; Qu, Y.H.; Zhou, M.X.; He, J.; Lv, P.; Ming, L. Expression of circulating microRNA-20a and let-7a in esophageal squamous cell carcinoma. World J. Gastroenterol. 2015, 21, 4660–4665. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Chu, Y.; Liu, N.; Wang, Q.; Yin, Z.; Lu, Y.; Chen, Y. Joint bioinformatics analysis of underlying potential functions of hsa-let-7b-5p and core genes in human glioma. J. Transl. Med. 2019, 17, 129. [Google Scholar] [CrossRef]
- Chirshev, E.; Oberg, K.C.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Aghdam, A.M.; Amiri, A.; Salarinia, R.; Masoudifar, A.; Ghasemi, F.; Mirzaei, H. MicroRNAs as Diagnostic, Prognostic, and Therapeutic Biomarkers in Prostate Cancer. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xiong, Y.; Liu, J.; Yang, X.; Wang, L.; Zhang, S.; Liu, M.; Wang, D. MMP-9-Related microRNAs as Prognostic Markers for Hemorrhagic Transformation in Cardioembolic Stroke Patients. Front. Neurol. 2019, 10, 945. [Google Scholar] [CrossRef]
- Jiang, T.; Ye, L.; Han, Z.; Liu, Y.; Yang, Y.; Peng, Z.; Fan, J. miR-19b-3p promotes colon cancer proliferation and oxaliplatin-based chemoresistance by targeting SMAD4: Validation by bioinformatics and experimental analyses. J. Exp. Clin. Cancer Res. 2017, 36, 131. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Huang, D.; Ma, K.; Deng, X.; Shao, Z. MiR-19a as a prognostic indicator for cancer patients: A meta-analysis. Biosci. Rep. 2019, 39, BSR20182370. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A., Jr.; Da Costa, N.M.; Pontes, B.; de Oliveira, F.L.; Codeço, M.L.; Pinto, L.F.R.; Nasciutti, L.E. Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix. Cells 2020, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.; Lee, S.; Park, S.K. Causal Inference between Rheumatoid Arthritis and Breast Cancer in East Asian and European Population: A Two-Sample Mendelian Randomization. Cancers 2020, 12, 3272. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Name of the Parameter | Top 250 Genes (GSE161533 Samples) | Predicted HGs T10 | miRNA Regulatory Network | Predicted HmiR T10 |
|---|---|---|---|---|---|
| 1. | Number of Nodes | 129 | 10 | 377 | 10 |
| 2. | Number of edges | 262 | 38 | 597 | 30 |
| 3. | Avg.number of neighbors | 4.339 | 7.600 | 3.167 | 3.333 |
| 4. | Network diameter | 13 | 2 | 4 | 4 |
| 5. | Network radius | 7 | 1 | 3 | 3 |
| 6. | Characteristic path length | 4.196 | 1.156 | 3.170 | 2.261 |
| 7. | Clustering coefficient | 0.215 | 0.898 | 0.000 | 0.000 |
| 8. | Network density | 0.037 | 0.844 | 0.008 | 0.196 |
| 9. | Network heterogeneity | 0.180 | 0.169 | 3.880 | 0.640 |
| Group | Pathway Enriched | Gene Symbol | FDR | p-Value | Expected Value | Enrichment Ratio |
|---|---|---|---|---|---|---|
| 1. | Bladder cancer | CXCL8, MMP1 and MMP9 | 0.0021221 | 0.000019649 | 0.056633 | 52.972 |
| 2. | IL-17 signaling pathway | CXCL8, MMP1, MMP13, MMP3, MMP9 | 0.000027535 | 8.4985 × 10−8 | 0.13167 | 37.973 |
| 3. | Relaxin signaling pathway | COL1A1, COL1A2, MMP1, MMP13, MMP9 | 0.000074171 | 4.5785 × 10−7 | 0.18406 | 27.165 |
| 4. | ECM-receptor interaction | COL1A1, COL1A2, SPP1 | 0.0098986 | 0.00015276 | 0.11185 | 26.822 |
| 5. | Rheumatoid arthritis | CXCL8, MMP1, MMP3 | 0.010116 | 0.00020353 | 0.12318 | 24.355 |
| 6, | Amoebiasis | COL1A1, COL1A2, CXCL8 | 0.010116 | 0.00024023 | 0.13026 | 23.032 |
| 7. | Prostate cancer | MMP3, MMP9, PLAU | 0.010116 | 0.00026419 | 0.13450 | 22.304 |
| 8. | AGE-RAGE signaling pathway in diabetic complications | COL1A1, COL1A2, CXCL8 | 0.010116 | 0.00028100 | 0.13734 | 21.844 |
| 9. | Transcriptional misregulation in cancer | CXCL8, MMP3, MMP9, PLAU | 0.0057635 | 0.000071154 | 0.25060 | 15.962 |
| 10. | Focal adhesion | COL1A1, COL1A2, SPP1 | 0.061616 | 0.0020919 | 0.27325 | 10.979 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muruganantham, B.; Sivamaruthi, B.S.; Kesika, P.; Thangaleela, S.; Chaiyasut, C. Bioinformatics Characterization of Candidate Genes Associated with Gene Network and miRNA Regulation in Esophageal Squamous Cell Carcinoma Patients. Appl. Sci. 2022, 12, 1083. https://0-doi-org.brum.beds.ac.uk/10.3390/app12031083
Muruganantham B, Sivamaruthi BS, Kesika P, Thangaleela S, Chaiyasut C. Bioinformatics Characterization of Candidate Genes Associated with Gene Network and miRNA Regulation in Esophageal Squamous Cell Carcinoma Patients. Applied Sciences. 2022; 12(3):1083. https://0-doi-org.brum.beds.ac.uk/10.3390/app12031083
Chicago/Turabian StyleMuruganantham, Bharathi, Bhagavathi Sundaram Sivamaruthi, Periyanaina Kesika, Subramanian Thangaleela, and Chaiyavat Chaiyasut. 2022. "Bioinformatics Characterization of Candidate Genes Associated with Gene Network and miRNA Regulation in Esophageal Squamous Cell Carcinoma Patients" Applied Sciences 12, no. 3: 1083. https://0-doi-org.brum.beds.ac.uk/10.3390/app12031083