In Situ Hyperspectral Raman Imaging: A New Method to Investigate Sintering Processes of Ceramic Material at High-temperature


Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Method
2.1. Analytical Details
2.2. Experimental Series
2.3. Sample Preparation
2.4. Heating Stage and Temperature Calibration
2.5. Map Programming
2.6. Data reduction, Quantification, and Visualization
2.6.1. Data Reduction
2.6.2. Quantification and Image Visualization
2.6.3. Factors Affecting Phase Quantification
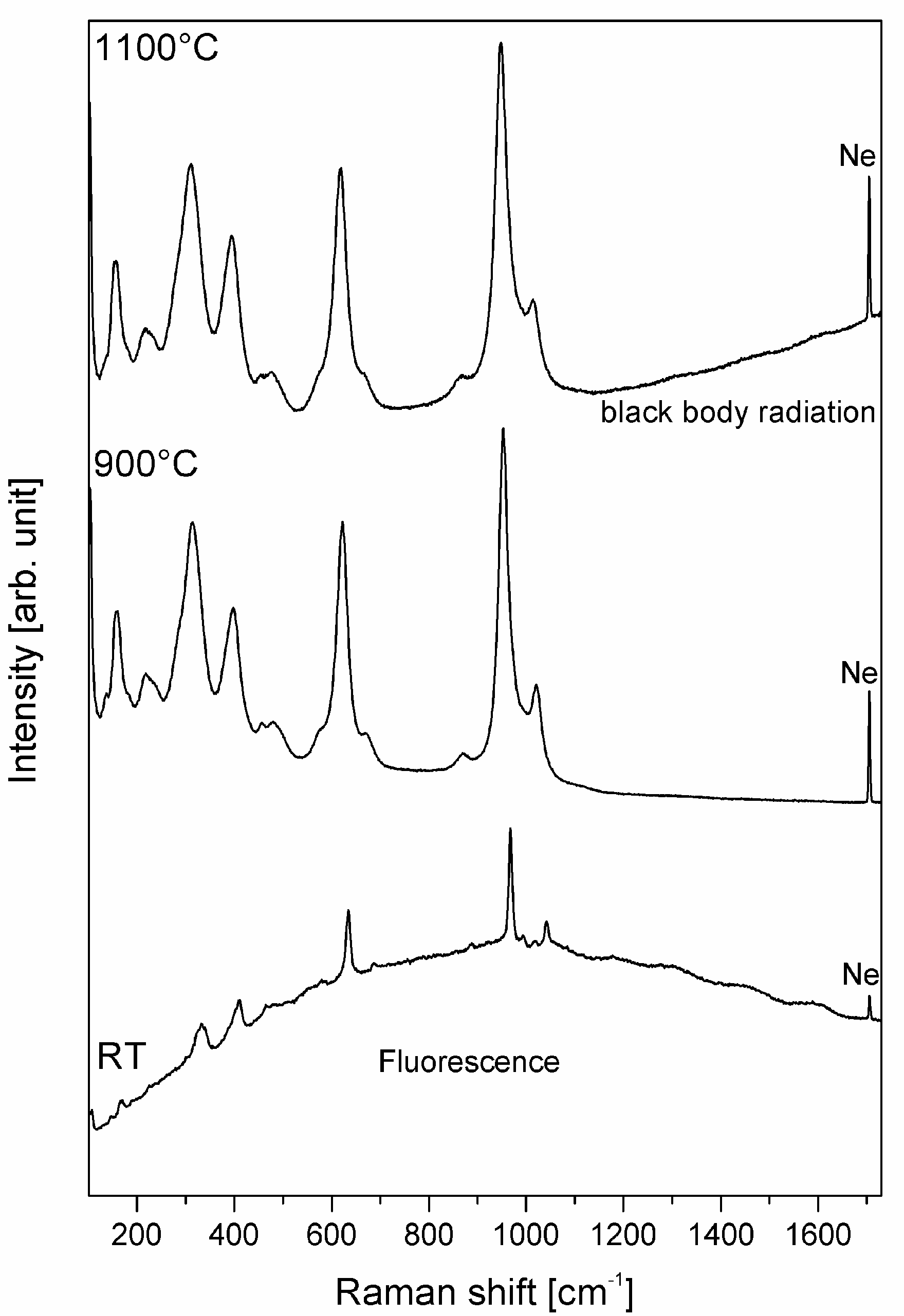
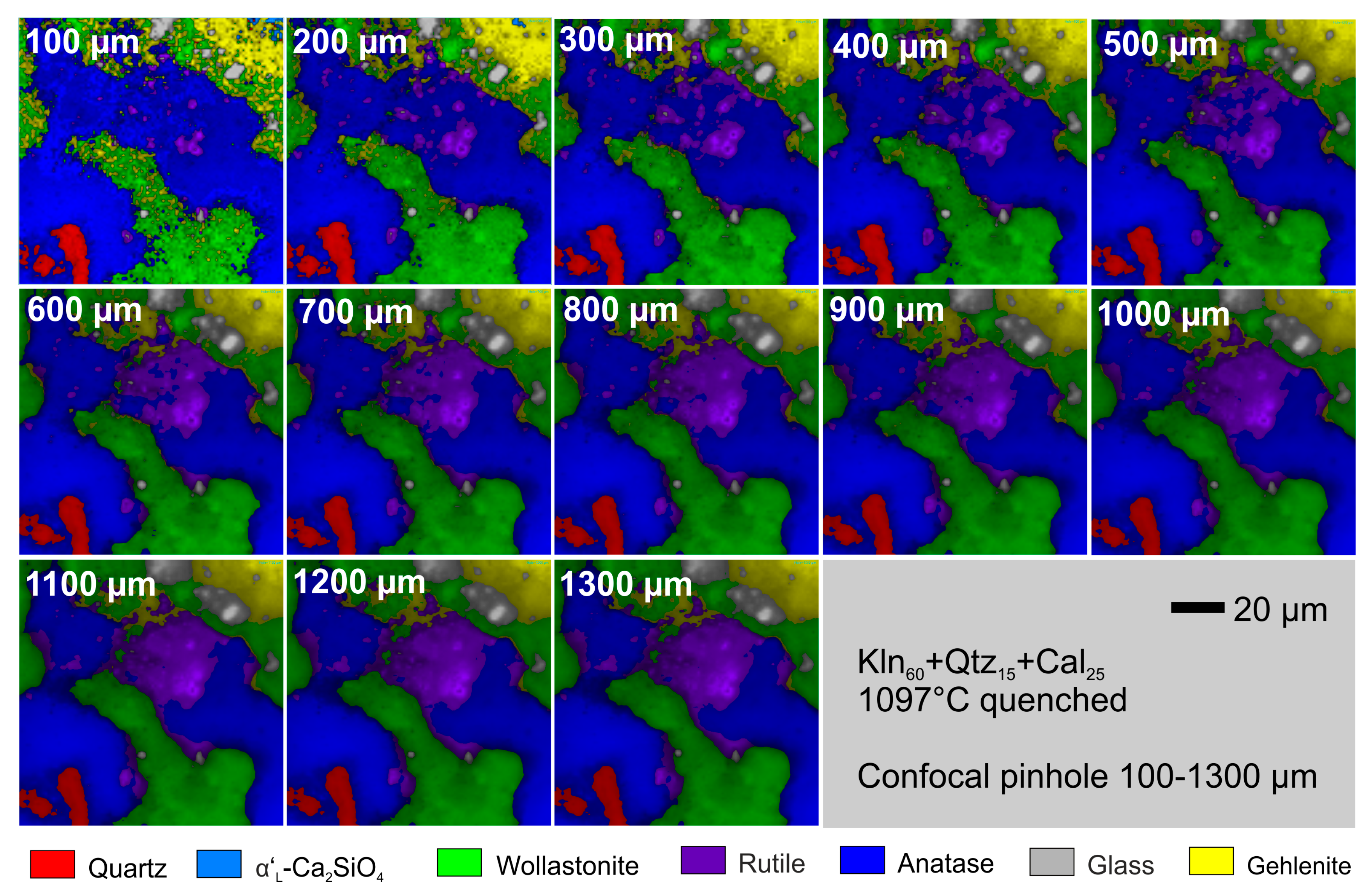
2.7. Lateral and Axial Resolution
3. Applications
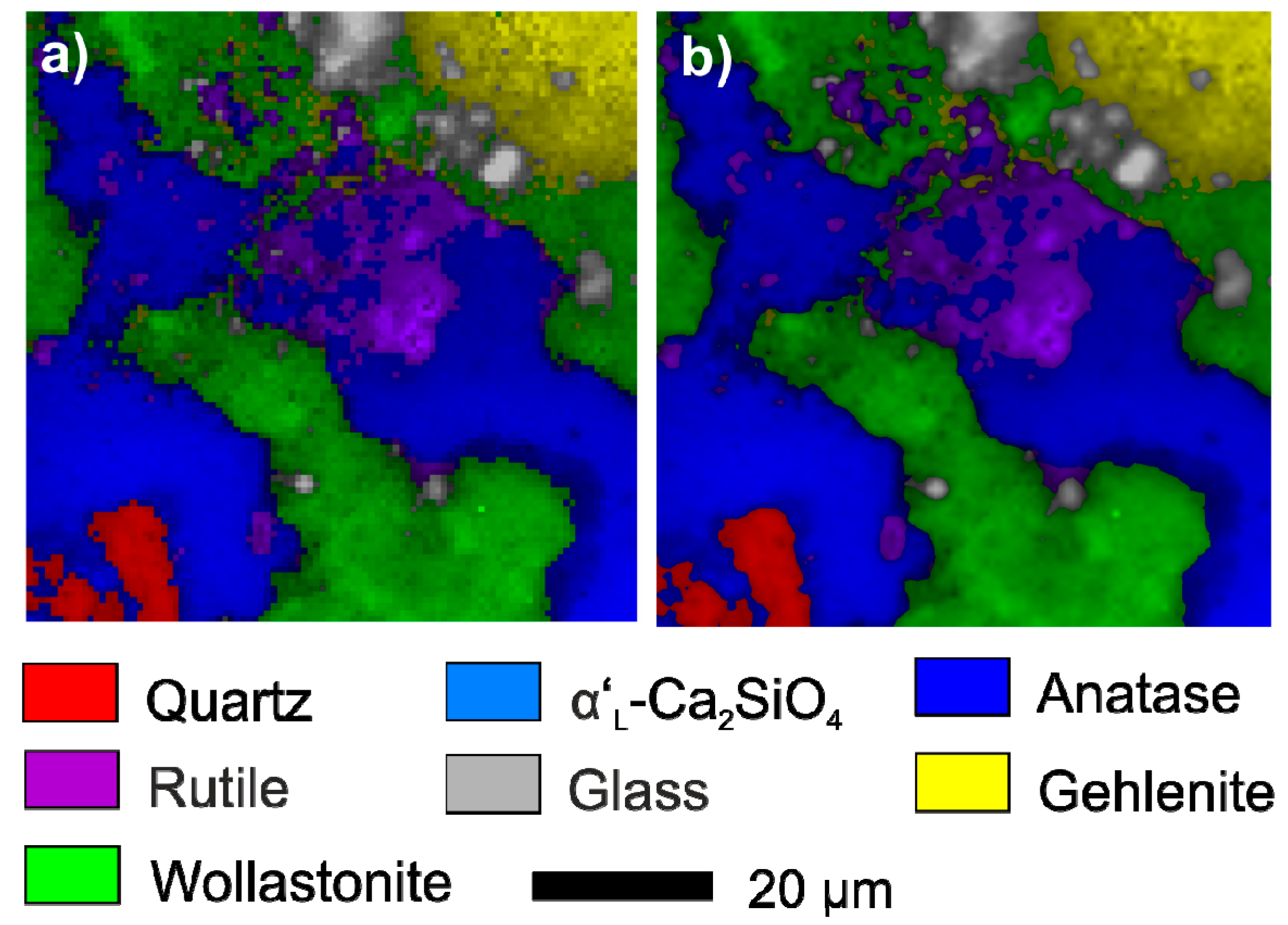
3.1. Mineral Reactions During Firing and Cooling
3.2. Isothermal Mineral Reactions and Grain Growth
3.3. In Situ Observations of the Migration of Solid-Solid Reaction Fronts
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Becker, M.; Sarau, G.; Strunk, H.P.; Christiansen, S. Raman imaging of grain orientation, strain, crystallinity and doping levels in solar silicon. In Raman Imaging: Techniques and Applications; Zoubir, A., Ed.; Springer Series in Optical Sciences; Springer: Berlin, Germany, 2012; pp. 256–299. [Google Scholar]
- Stewart, S.; Priore, R.J.; Nelson, M.P.; Treado, P.J. Raman imaging. Annu. Rev. Anal. Chem. 2012, 5, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, T.; Sun, D.; Cheng, J. Raman imaging for food quality and safety evaluation: Fundamentals and applications. Trends Food Sci. Technol. 2017, 62, 177–189. [Google Scholar] [CrossRef]
- Abramczyk, H.; Brozek-pluska, B. Raman imaging in biochemical and biomedical applications. Diagnosis and treatment of breast cancer. Chem. Rev. 2012, 113, 5766–5781. [Google Scholar] [CrossRef]
- Everall, N. Depth profiling with confocal Raman microscopy, part I. Spectroscopy 2004, 19, 22–28. [Google Scholar]
- Toporski, J. Confocal Raman Microscopy; Toporski, J., Dieing, T., Hollrichter, O., Eds.; Springer Series in Surface Sciences; Springer Verlag: Heidelberg, Germany, 2018; Volume 66, ISBN 978-3-642-12521-8. [Google Scholar]
- Geisler, T.; Burakov, B.E.; Zirlin, V.; Nikolaeva, L.; Pöml, P. A Raman spectroscopic study of high-uranium zircon from the Chernobyl “lava”. Eur. J. Mineral. 2005, 17, 883–894. [Google Scholar] [CrossRef]
- Geisler, T.; Popa, K.; Konings, R.J.M. Evidence for lattice strain and non-ideal behavior in the (La1−xEux)PO4 solid solution from X-ray diffraction and vibrational spectroscopy. Front. Earth Sci. 2016, 4. [Google Scholar] [CrossRef]
- King, H.; Geisler, T. Tracing mineral reactions using confocal Raman spectroscopy. Minerals 2018, 8, 158. [Google Scholar] [CrossRef]
- Geisler, T.; Dohmen, L.; Lenting, C.; Fritzsche, M.B.K. Real time in situ observations of reaction and transport phenomena during silicate glass corrosion by fluid-cell Raman spectroscopy. Nat. Mater. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.S.; Bursill, L.A.; Prawer, S.; Beserman, R. Temperature dependence of the first-order Raman phonon line of diamond. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 61, 3391–3395. [Google Scholar] [CrossRef]
- Menéndez, J.; Cardona, M. Temperature dependence of the first-order Raman scattering by phonons in Si, Ge, and -Sn: Anharmonic effects. Phys. Rev. B 1984, 29, 2051–2059. [Google Scholar] [CrossRef]
- Klemens, P.G. Anharmonic decay of optical phonon in diamond. Phys. Rev. B 1975, 11, 3206–3207. [Google Scholar] [CrossRef]
- Huang, F.; Yue, K.T.; Tan, P.; Zhang, S.-L.; Shi, Z.; Zhou, X.; Gu, Z. Temperature dependence of the Raman spectra of carbon nanotubes. J. Appl. Phys. 1998, 84, 4022–4024. [Google Scholar] [CrossRef]
- Bouhifd, M.A.; Gruener, G.; Mysen, B.O.; Richet, P. Premelting and calcium mobility in gehlenite (Ca2Al2SiO7) and pseudowollastonite (CaSiO3). Phys. Chem. Miner. 2002, 29, 655–662. [Google Scholar] [CrossRef]
- Richet, P.; Ingrin, J.; Mysen, B.O.; Courtial, P.; Gillet, P. Premelting effects in minerals: An experimental study. Earth Planet. Sci. Lett. 1994, 121, 589–600. [Google Scholar] [CrossRef]
- Malfait, W.J.; Halter, W.E. Structural relaxation in silicate glasses and melts: High-temperature Raman spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 1–6. [Google Scholar] [CrossRef]
- Neuville, D.R.; Mysen, B.O. Role of aluminium in the silicate network: In situ, high-temperature study of glasses and melts on the join SiO2-NaAlO2. Geochim. Cosmochim. Acta 1996, 60, 1727–1737. [Google Scholar] [CrossRef]
- Voron’ko, Y.K.; Sobol’, A.A.; Ushakov, S.N.; Jiang, G.; You, J. Phase transformations and melt structure of calcium metasilicate. Inorg. Mater. 2002, 38, 825–830. [Google Scholar]
- Gillet, P. Raman spectroscopy at high pressure and high-temperature. Phase transitions and thermodynamic properties of minerals. Phys. Chem. Miner. 1996, 23, 263–275. [Google Scholar] [CrossRef]
- Osipov, A.A.; Osipova, L.M. Raman scattering study of barium borate glasses and melts. J. Phys. Chem. Solids 2013, 74, 971–978. [Google Scholar] [CrossRef]
- McKeown, D.A. Raman spectroscopy and vibrational analyses of albite: From 25 °C through the melting temperature. Am. Mineral. 2005, 90, 1506–1517. [Google Scholar] [CrossRef]
- Remy, C.; Reynard, B.; Madon, M. Raman Spectroscopic Investigations of Dicalcium Silicate: Polymorphs and High-Temperature Phase Transformations. J. Am. Ceram. Soc. 2005, 80, 413–423. [Google Scholar] [CrossRef]
- Stange, K.; Lenting, C.; Geisler, T. Insights into the evolution of carbonate-bearing kaolin during sintering revealed by in situ hyperspectral Raman imaging. J. Am. Ceram. Soc. 2018, 101, 897–910. [Google Scholar] [CrossRef]
- Seryotkin, Y.V.; Sokol, E.V.; Kokh, S.N. Natural pseudowollastonite: Crystal structure, associated minerals, and geological context. Lithos 2012, 134–135, 75–90. [Google Scholar] [CrossRef]
- Saloman, E.B. Wavelengths, energy level classifications, and energy levels for the spectrum of neutral mercury. J. Phys. Chem. Ref. Data 2006, 35, 1519–1548. [Google Scholar] [CrossRef]
- von Salje, E. Experimentelle Untersuchung der Ramanstreuung an Kristallpulvern (experimental investigation of Raman scattering of crystall powder). J. Appl. Cryst. 1973, 6, 442–446. [Google Scholar] [CrossRef]
- Linkam Scientific. Linkam Scientific Instruments TS1500, TS1200 and TS1000 Systems 2018. Available online: https://static1.squarespace.com/static/556d800ae4b0e8f91507450c/t/56d07c8f2fe13114fc6d5287/1456503961692/TS1000_1200_1500_systems_v1.4.pdf (accessed on 3 October 2018).
- Neuville, D.R.; de Ligny, D.; Henderson, G.S. Advances in Raman spectroscopy applied to earth and material sciences. Rev. Mineral. Geochem. 2014, 78, 509–541. [Google Scholar] [CrossRef]
- Vajna, B.; Patyi, G.; Nagy, Z.; Bódis, A.; Farkas, A.; Marosi, G. Comparison of chemometric methods in the analysis of pharmaceuticals with hyperspectral Raman imaging. J. Raman Spectrosc. 2011, 42, 1977–1986. [Google Scholar] [CrossRef]
- Nasdala, L.; Beyssac, O.; Schopf, J.W.; Bleisteiner, B. Application of Raman-based images in the Earth Sciences. In Raman Imaging: Techniques and Applications; Springer Series in Optical Sciences; Springer: Berlin, Germany, 2012; pp. 145–189. [Google Scholar]
- Kramida, A.; Ralchenko, Y.; Reader, J. NIST: Atomic Spectra Database—Version History; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2016.
- Tian, H.; Wachs, I.E.; Briand, L.E. Comparison of UV and visible Raman spectroscopy of bulk metal molybdate and metal vanadate catalysts. J. Phys. Chem. B 2005, 109, 23491–23499. [Google Scholar] [CrossRef]
- Zouboulis, E.; Renusch, D.; Grimsditch, M. Advantages of ultraviolet Raman scattering for high-temperature investigations. Appl. Phys. Lett. 1998, 72, 1–3. [Google Scholar] [CrossRef]
- Swamy, V.; Dubrovinsky, L.S.; Tutti, F. Thermal Expansion of Wollastonite. J. Am. Ceram. Soc. 1997, 80, 2237–2247. [Google Scholar] [CrossRef]
- Hope, G.A.; Woods, R.; Munce, C.G. Raman microprobe mineral identification. Miner. Eng. 2001, 14, 1565–1577. [Google Scholar] [CrossRef]
- Hopkins, J.B.; Farrow, L.A. Raman microprobe determination of local crystal orientation. J. Appl. Phys. 1986, 59, 1103–1110. [Google Scholar] [CrossRef]
- Chaigneau, M.; Picardi, G.; Girard, H.A.; Arnault, J.C.; Ossikovski, R. Laser heating versus phonon confinement effect in the Raman spectra of diamond nanoparticles. J. Nanopart. Res. 2012, 14, 955. [Google Scholar] [CrossRef]
- Bersani, D.; Lottici, P.P.; Ding, X. Phonon confinement effects in the Raman scattering by TiO2 nanocrystals. Appl. Phys. Lett. 1998, 72, 72–75. [Google Scholar] [CrossRef]
- Swamy, V.; Kuznetsov, A.; Dubrovinsky, L.S.; Caruso, R.A.; Shchukin, D.G.; Muddle, B.C. Finite-size and pressure effects on the Raman spectrum of nanocrystalline anatase TiO2. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 15–17. [Google Scholar] [CrossRef]
- Swamy, V. Size-dependent modifications of the first-order Raman spectra of nanostructured rutile TiO2. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 15–18. [Google Scholar] [CrossRef]
- Namba, Y.; Heidarpour, E.; Nakayama, M. Size effects appearing in the Raman spectra of polycrystalline diamonds Size effects appearing in the Raman spectra of polycrystalline. J. Appl. Phys. 2012, 1748, 1–5. [Google Scholar]
- Yoshikawa, M.; Mori, Y.; Obata, H.; Maegawa, M.; Katagiri, G.; Ishida, H.; Ishitani, A.; Yoshikawa, M.; Mori, Y.; Obata, H.; et al. Raman scattering from nanometer-sized diamond Raman scattering from nanometer-sized diamond. Appl. Phys. Lett. 2008, 694, 5–8. [Google Scholar]
- Zhao, X.Z.; Cherian, K.A.; Roy, R.; White, W.B. Downshift of Raman peak in diamond powders. J. Mater. Res. 1998, 13, 1974–1976. [Google Scholar] [CrossRef]
- Richet, P.; Mysen, B.O.; Ingrin, J. High-temperature X-ray diffraction and Raman spectroscopy of diopside and pseudowollastonite. Phys. Chem. Miner. 1998, 25, 401–414. [Google Scholar] [CrossRef]
- Hibbert, R.; Price, M.C.; Kinnear, T.M.; Cole, M.J.; Burchell, M.J. The effects of temperature on the Raman spectrum of high purity quartz crystals. In Proceedings of the 46th Lunar and Planetary Science Conference, The Woodlands, TX, USA, 16–20 March 2015; pp. 2–3. [Google Scholar]
- Polsky, C.H.; Smith, K.H.; Wolf, G.H. Effect of pressure on the absolute Raman scattering cross section of SiO2 and GeO2 glasses. J. Non-Cryst. Solids 1999, 248, 159–168. [Google Scholar] [CrossRef]
- Quittet, A.M.; Lambert, M. Temperature dependence of the Raman cross section and light absorption in cubic BaTiO3. Solid State Commun. 1973, 12, 1053–1055. [Google Scholar] [CrossRef]
- Demos, S.G.; Raman, R.N.; Yang, S.T.; Negres, R.A.; Schaffers, K.I.; Henesian, M.A. Measurement of the Raman scattering cross section of the breathing mode in KDP and DKDP crystals. Opt. Express 2011, 19, 21050. [Google Scholar] [CrossRef]
- Rappoport, D. Berechnung von Raman-Intensitäten mit zeitabhängiger Dichte-Funktionaltheorie; Universitätsverlag Karlsruhe: Karlsruhe, Germany, 2007. [Google Scholar]
- Neuville, D.R.; Cormier, L.; Massiot, D. Al coordination and speciation in calcium aluminosilicate glasses: Effects of composition determined by27Al MQ-MAS NMR and Raman spectroscopy. Chem. Geol. 2006, 229, 173–185. [Google Scholar] [CrossRef]
- Neuville, D.R. Viscosity, structure and mixing in (Ca, Na) silicate melts. Chem. Geol. 2006, 229, 28–41. [Google Scholar] [CrossRef]
- Guerette, M.; Huang, L. A simple and convenient set-up for high-temperature Brillouin light scattering. J. Phys. D Appl. Phys. 2012, 45, 1–7. [Google Scholar] [CrossRef]
- Behrens, H.; Roux, J.; Neuville, D.R.; Siemann, M. Quantification of dissolved H2O in silicate glasses using confocal microRaman spectroscopy. Chem. Geol. 2006, 229, 96–112. [Google Scholar] [CrossRef]
- Everall, N.J. Confocal Raman microscopy: Why the depth resolution and spatial accuracy can be much worse than you think. Appl. Spectrosc. 2000, 54, 1515–1520. [Google Scholar] [CrossRef]
- Everall, N.J. Depth Profiling with confocal Raman microscopy, part II. Spectroscopy 2004, 19, 16–27. [Google Scholar]
- Everall, N.J. Confocal Raman microscopy: Common errors and artefacts. Analyst 2010, 135, 2512. [Google Scholar] [CrossRef] [PubMed]
- Presser, V.; Keuper, M.; Berthold, C.; Nickel, K.G. Experimental determination of the Raman sampling depth in zirconia ceramics. Appl. Spectrosc. 2009, 63, 1288–1292. [Google Scholar] [CrossRef]
- Everall, N. Optimising image quality in 2D and 3D confocal Raman mapping. J. Raman Spectrosc. 2014, 45, 133–138. [Google Scholar] [CrossRef]
- Zoubir, A. Raman Imaging: Techniques and Applications; Zoubir, A., Ed.; Springer: Berlin, Germany, 2012; Volume 168, ISBN 9783642282515. [Google Scholar]
- Everall, N.J. Modeling and measuring the effect of refraction on the depth resolution of confocal Raman microscopy. Appl. Spectrosc. 2000, 54, 773–782. [Google Scholar] [CrossRef]
- Juang, C.B.; Finzi, L.; Bustamante, C.J. Design and application of a computer-controlled confocal scanning differential polarization microscope. Rev. Sci. Instrum. 1988, 59, 2399–2408. [Google Scholar] [CrossRef]
- Tschegg, C.; Ntaflos, T.; Hein, I. Thermally triggered two-stage reaction of carbonates and clay during ceramic firing—A case study on Bronze Age Cypriot ceramics. Appl. Clay Sci. 2009, 43, 69–78. [Google Scholar] [CrossRef]
- Nankervis, J.C.; Furlong, R.B. Phase changes in mineral matter of North Dakota lignites caused by heating to 1200 °C. Fuel 1980, 59, 425–430. [Google Scholar] [CrossRef]
- Schmida, T.; Dariz, P. Shedding light onto the spectra of lime: Raman and luminescence bands of CaO, Ca(OH)2 and CaCO3. J. Raman Spectrosc. 2014, 46, 141–146. [Google Scholar] [CrossRef]
- Holm, E.A.; Foiles, S.M. How grain growth stops: A mechanism for grain-growth stagnation in pure materials. Science 2010, 328, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhai, W.; Ni, S.; Chang, J.; Zeng, Y.; Qian, W. Study of the mechanical property and in vitro biocompatibility of CaSiO3ceramics. Ceram. Int. 2005, 31, 323–326. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I: General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Knudsen, F.P. Dependence of mechanical strength of brittle polycrystalline specimens on porosity and grain size. J. Am. Ceram. Soc. 1959, 42, 376–387. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Milke, R.; Heinrich, W. Diffusion-controlled growth of wollastonite rims between quartz and calcite: Comparison between nature and experiment. J. Metamorph. Geol. 2002, 20, 467–480. [Google Scholar] [CrossRef]
- Gaidies, F.; Milke, R.; Heinrich, W.; Abart, R. Metamorphic mineral reactions: Porphyroblast, corona and symplectite growth. EMU Notes Mineral. 2017, 16, 469–540. [Google Scholar]
- Milke, R.; Wiedenbeck, M.; Heinrich, W. Grain boundary diffusion of Si, Mg, and O in enstatite reaction rims: A SIMS study using isotopically doped reactants. Contrib. to Mineral. Petrol. 2001, 142, 15–26. [Google Scholar] [CrossRef]
- Abart, R.; Kunze, K.; Milke, R.; Sperb, R.; Heinrich, W. Silicon and oxygen self diffusion in enstatite polycrystals: The Milke et al. (2001) rim growth experiments revisited. Contrib. Mineral. Petrol. 2004, 147, 633–646. [Google Scholar] [CrossRef]
- Diella, V.; Adamo, I.; Pagliari, L.; Pavese, A.; Francescon, F. Effects of particle size distribution and starting phase composition in Na-feldspar/kaolinite system at high-temperature. J. Eur. Ceram. Soc. 2015, 35, 1327–1335. [Google Scholar] [CrossRef]
- Freundlich, H. Kapillarchemie: Eine Darstellung der Chemie der Kolloide und verwandter Gebiete [Capillary Chemistry: A Presentation of Colloid Chemistry and Related Fields]; Akademische Verlagsgesellschaft: Leipzig, Germany, 1909. [Google Scholar]
- Fierens, P.; Picquet, P. Kinetic studies of the thermal synthesis of calcium silicates above 1400 °C: I, Dynamic Thermal Synthesis of Ca2SiO4. J. Am. Ceram. Soc. 1975, 58, 50–51. [Google Scholar] [CrossRef]
- Herzberg, G. Atomic Spectra and Atomic Structure; Dover puplications: New York, NY, USA, 1944. [Google Scholar]
- Geisler, T.; Pöml, P.; Stephan, T.; Janssen, A.; Putnis, A. Experimental observation of an interface-controlled pseudomorphic replacement reaction in a natural crystalline pyrochlore. Am. Mineral. 2005, 90, 1683–1687. [Google Scholar] [CrossRef]
- King, H.E.; Plümper, O.; Geisler, T.; Putnis, A. Experimental investigations into the silicification of olivine: Implications for the reaction mechanism and acid neutralization. Am. Mineral. 2011, 96, 1503–1511. [Google Scholar] [CrossRef]
- Hövelmann, J.; Putnis, A.; Geisler, T.; Schmidt, B.C.; Golla-Schindler, U. The replacement of plagioclase feldspars by albite: Observations from hydrothermal experiments. Contrib. Mineral. Petrol. 2010, 159, 43–59. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Company | Level of Purity | Particle Size |
|---|---|---|---|
| Quartz | Merck | 99.900% | 5–50 µm 1 |
| Calcite | Alfa Aesar | 99.950% | 2–20 µm 2 |
| CaO | Alfa Aesar | 99.995% | <10 µm |
| Amorphous SiO2 | Alfa Aesar | 99.900% | 2–20 µm 2 |
| Kaolinite China-Clay | Carl Jäger Tonindustriebedarf GmbH | - | <2 µm (58%) 8–53 µm (8%) >58 µm (0.05%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauke, K.; Kehren, J.; Böhme, N.; Zimmer, S.; Geisler, T. In Situ Hyperspectral Raman Imaging: A New Method to Investigate Sintering Processes of Ceramic Material at High-temperature. Appl. Sci. 2019, 9, 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/app9071310
Hauke K, Kehren J, Böhme N, Zimmer S, Geisler T. In Situ Hyperspectral Raman Imaging: A New Method to Investigate Sintering Processes of Ceramic Material at High-temperature. Applied Sciences. 2019; 9(7):1310. https://0-doi-org.brum.beds.ac.uk/10.3390/app9071310
Chicago/Turabian StyleHauke, Kerstin, Johannes Kehren, Nadine Böhme, Sinje Zimmer, and Thorsten Geisler. 2019. "In Situ Hyperspectral Raman Imaging: A New Method to Investigate Sintering Processes of Ceramic Material at High-temperature" Applied Sciences 9, no. 7: 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/app9071310