
Dynamics of Planktonic Microbial Community Associated with Saccharina japonica Seedling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Environmental Characterization
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Data Analysis
2.4. Statistical Analysis
2.5. Network Analysis
3. Results
3.1. Physicochemical Parameters of Seawater Samples
3.2. Microbial Alpha and Beta Diversities
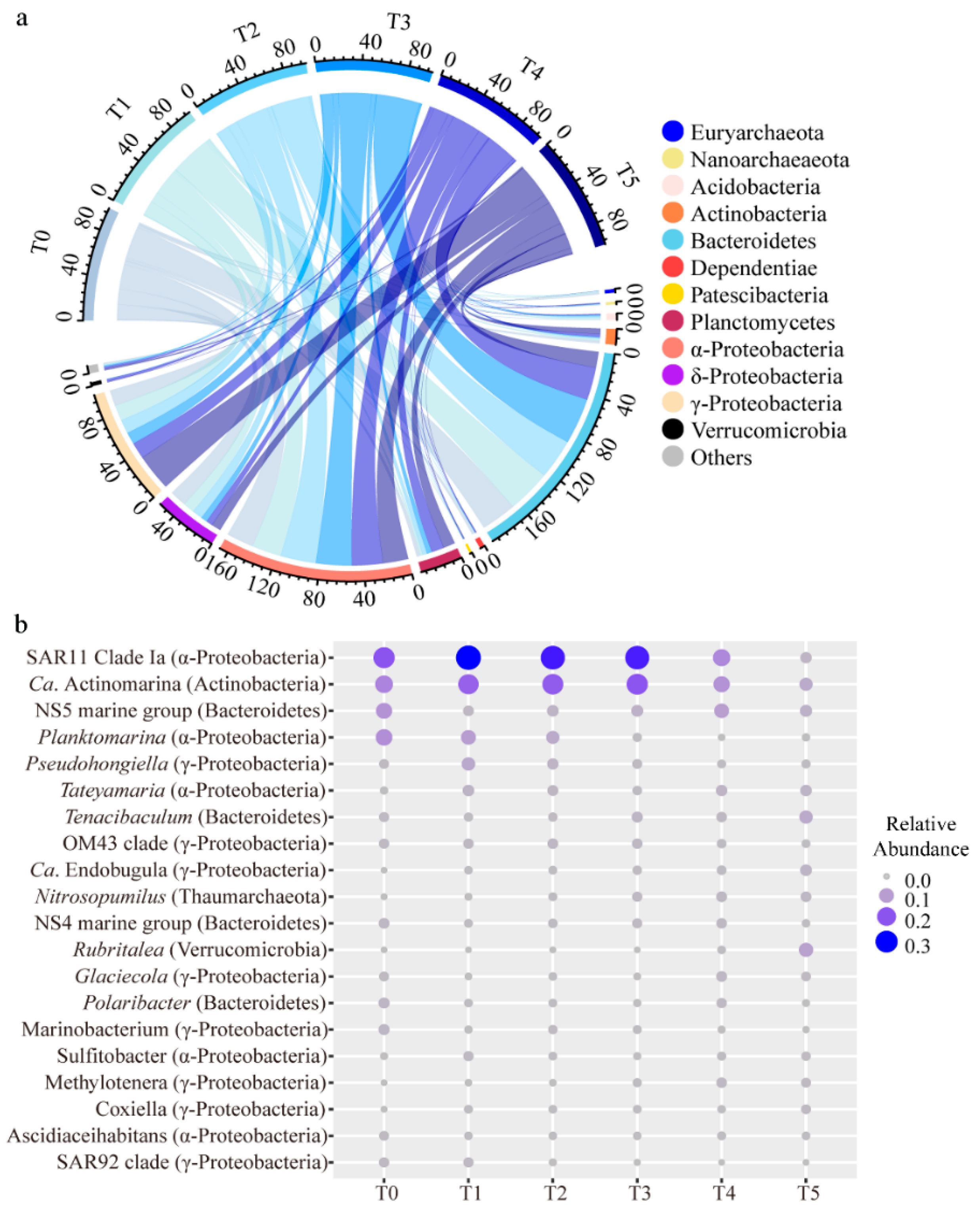
3.3. Distribution of Microbial Taxa
3.4. Environmental Variables and Microbial Communities
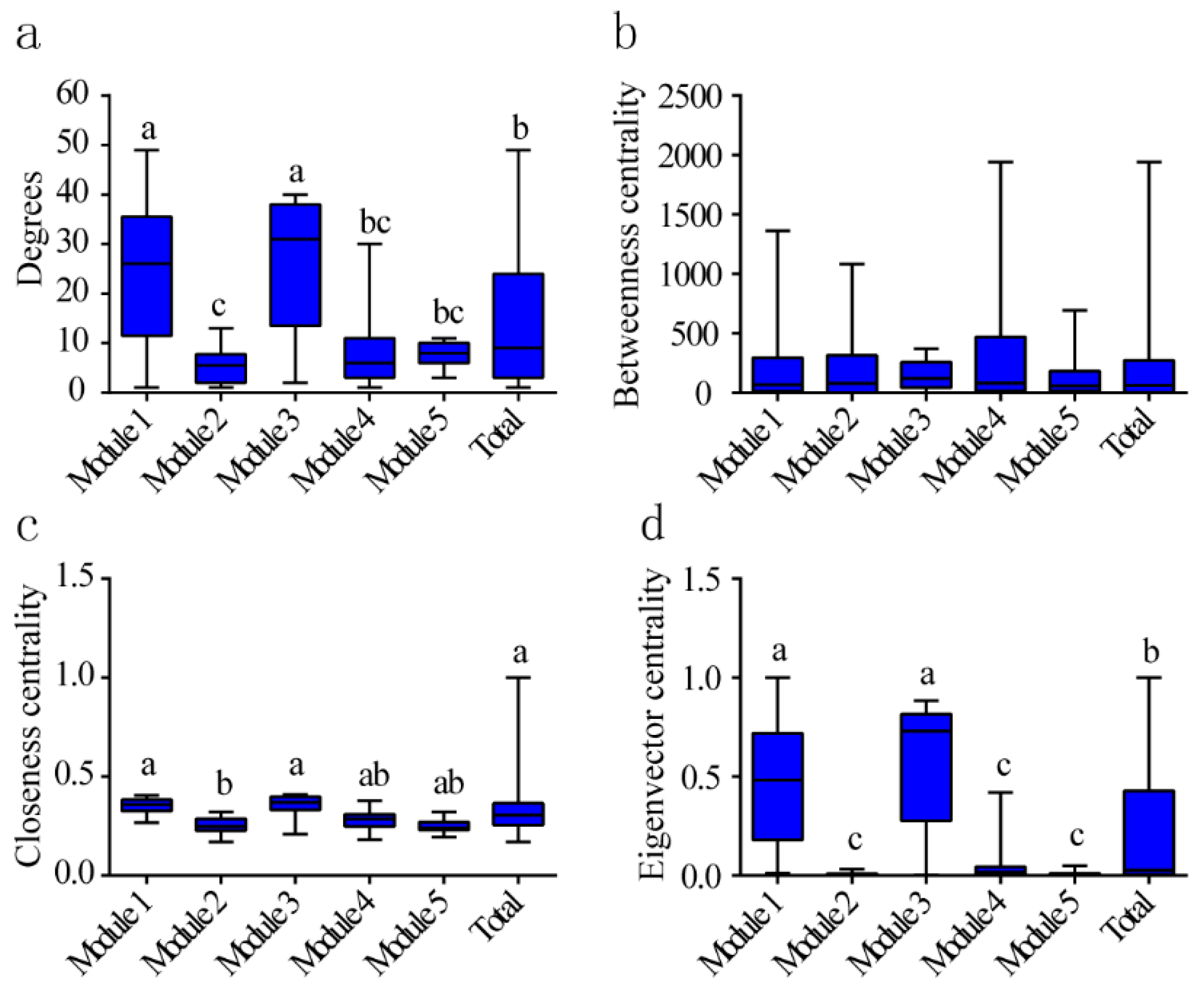
3.5. Microbial Community Co-Occurrence Network
4. Discussion
4.1. Microbial Diversities during the Seedling Production Stage
4.2. The Dominant Microbial Groups and Keystone Taxa
4.3. Co-Occurrence Patterns of Microbial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tseng, C.K. Algal biotechnology industries and research activities in China. J. Appl. Phycol. 2001, 13, 375–380. [Google Scholar] [CrossRef]
- Bocanegra, A.; Bastida, S.; Benedí, J.; Ródenas, S.; Sánchez-Muniz, F.J. Characteristics and nutritional and cardiovascular-health properties of seaweeds. J. Med. Food 2009, 12, 236–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, D.; Kim, Y.; Hwang, H.; Nam, T.; Kong, I. An improved method of protein isolation and proteome analysis with Saccharina japonica (Laminariales) incubated under different pH conditions. J. Appl. Phycol. 2011, 23, 123–130. [Google Scholar] [CrossRef]
- Zhang, J.; Fang, J.; Wang, W.; Du, M.; Gao, Y.; Zhang, M. Growth and loss of mariculture kelp Saccharina japonica in Sungo Bay, China. J. Appl. Phycol. 2012, 24, 1209–1216. [Google Scholar] [CrossRef]
- Ye, N.; Zhang, X.; Miao, M.; Fan, X.; Zheng, Y.; Xu, D.; Wang, J.; Zhou, L.; Wang, D.; Gao, Y.; et al. Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 2015, 6, 6986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabu, H. Alternation of chromosomes in the life history of Laminaria japonica Aresch. Bull. Fac. Fish. Hokkaido Univ. 1973, 23, 171–176. [Google Scholar]
- Wang, G.; Lu, B.; Shuai, L.; Li, D.; Zhang, R. Microbial diseases of nursery and field-cultivated Saccharina japonica (Phaeophyta) in China. Algol. Stud. 2014, 145, 39–51. [Google Scholar] [CrossRef]
- Peng, Y.; Li, W. A bacterial pathogen infecting gametophytes of Saccharina japonica (Laminariales, Phaeophyceae). Chin. J. Oceanol. Limn. 2013, 31, 366–373. [Google Scholar] [CrossRef]
- Gai, C.; Xu, L.; Diao, J.; Yang, X.; Liu, W.; Ye, H. Establishment and utilization of quick retrieval table of kelp common disease. Chin. Fish. Qual. Stand. 2017, 7, 51–57. [Google Scholar]
- Li, J.; Pang, S.; Shan, T.; Su, L. Changes of microbial community structures associated with seedlings of Saccharina japonica at early stage of outbreak of green rotten disease. J. Appl. Phycol. 2020, 62, 191. [Google Scholar] [CrossRef]
- Zhang, R.; Chang, L.; Xiao, L.; Zhang, X.; Han, Q.; Li, N.; Egan, S.; Wang, G. Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. J. Appl. Phycol. 2020, 32, 2071–2080. [Google Scholar] [CrossRef]
- Han, Q.; Zhang, X.; Chang, L.; Xiao, L.; Ahmad, R.; Saha, M.; Wu, H.; Wang, G. Dynamic shift of the epibacterial communities on commercially cultivated Saccharina japonica from mature sporophytes to sporelings and juvenile sporophytes. J. Appl. Phycol. 2021, 33, 1171–1179. [Google Scholar] [CrossRef]
- Burke, C.; Thomas, T.; Lewis, M.; Steinberg, P.; Kjelleberg, S. Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 2011, 5, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 2011, 13, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Reddy, C.R.K. Seaweed-microbial interactions: Key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 2014, 88, 213–230. [Google Scholar] [CrossRef]
- Mesquita, M.M.F.; Crapez, M.A.C.; Teixeira, V.L.; Cavalcanti, D.N. Potential interactions bacteria-brown algae. J. Appl. Phycol. 2019, 31, 867–883. [Google Scholar] [CrossRef]
- Egan, S.; Fernandes, N.D.; Kumar, V.; Gardiner, M.; Thomas, T. Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ. Microbiol. 2014, 16, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Gardiner, M. Microbial dysbiosis: Rethinking disease in marine ecosystems. Front. Microbiol. 2016, 7, 991. [Google Scholar] [CrossRef]
- Clasen, J.L.; Shurin, J.B. Kelp forest size alters microbial community structure and function on Vancouver Island, Canada. Ecology 2015, 96, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; He, Z.; Hu, X.; Yin, H.; Liu, X.; Yang, Y. Large-scale seaweed cultivation diverges water and sediment microbial communities in the coast of Nan’ao Island, South China Sea. Sci. Total Environ. 2017, 598, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Dahms, H.U.; Dobretsov, S. Antifouling compounds from marine macroalgae. Mar. Drugs 2017, 15, 265. [Google Scholar] [CrossRef]
- Steinberg, P.D.; Rice, S.A.; Campbell, A.H.; Mcdougald, D.; Harder, T. Interfaces between bacterial and eukaryotic “neuroecology”. Integr. Comp. Biol. 2011, 51, 794–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.Y.; Parfrey, L.W. Incubation with macroalgae induces large shifts in water column microbiota, but minor changes to the epibiota of co-occurring macroalgae. Mol. Ecol. 2018, 27, 1966–1979. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, L.; Xu, K.; Xu, Y.; Ji, D.; Chen, C.; Xie, C. The cultivation of Pyropia haitanensis has important impacts on the seawater microbial community. J. Appl. Phycol. 2020, 461, 37. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Zhou, W.; Hu, C.; Zhu, M.; Ding, Y.; Gai, S.; Zheng, X.; Zhu, J.; Lu, Q. Bacterial community temporal dynamics and disease-related variations in the seawater of Pyropia (laver) seedling pools. J. Appl. Phycol. 2018, 30, 1217–1224. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, H.; Tang, L.; Li, J.; Mao, Y.; Mo, Z. Compositional shifts of bacterial communities associated with Pyropia yezoensis and surrounding seawater co-occurring with red rot disease. Front. Microbiol. 2019, 10, 1666. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A.; Steele, J.A. Community structure of marine bacterioplankton: Patterns, networks, and relationships to function. Aquat. Microb. Ecol. 2008, 53, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xu, Y.; Zhang, J.; Hao, X.; Lu, Y. Core microbiota in agricultural soils and their potential associations with nutrient cycling. mSystems 2019, 4, e00313-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín González, A.M.; Dalsgaard, B.; Olesen, J.M. Centrality measures and the importance of generalist species in pollination networks. Ecol. Complex. 2010, 7, 36–43. [Google Scholar] [CrossRef]
- Eaton, A.D.; Clesceri, L.S.; Rice, E.W.; Greenberg, A.F. Standard Methods for the Examination of Water and Wastewater; American Water Works Association: Washington, DC, USA, 2005. [Google Scholar]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.E.L.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Computing 2010, 14, 12–21. [Google Scholar]
- Faith, D.P.; Minchin, P.R.; Belbin, L. Compositional dissimilarity as a robust measure of ecological distance. Vegetatio 1987, 69, 57–68. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 January 2012).
- Chambers, J.M.; Freeny, A.; Heiberger, R.M. Analysis of variance: Designed experiments. In Statistical Models; Chambers, J.M., Hastie, T.J., Eds.; Wadsworth & Brooks/Cole: Pacific Grove, CA, USA, 1992; Chapter 5. [Google Scholar]
- Legendre, P.; Legendre, L. Numerical Ecology, 3rd ed.; Elsevier Science BV: Amsterdam, The Netherlands, 2012; pp. 174–175. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the 3rd International Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Erdős, P.; Rényi, A. On the evolution of random graphs. Magyar Tud. Akad. Mat. Kutató Int. Közl. 1960, 5, 17–60. [Google Scholar]
- Vick-Majors, T.J.; Priscu, J.C.; Amaral-Zettler, L.A. Modular community structure suggests metabolic plasticity during the transition to polar night in ice-covered Antarctic lakes. ISME J. 2014, 8, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Baah-Acheamfour, M.; Carlyle, C.N.; Bissett, A.; Richardson, A.E.; Siddique, T.; Bork, E.W.; Chang, S.X. Determinants of bacterial communities in Canadian agroforestry systems. Environ. Microbiol. 2016, 18, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Heal, K.R.; Ramdasi, R.; Kobelt, J.N.; Martens-Habbena, W.; Bertagnolli, A.D.; Amin, S.A.; Walker, C.B.; Urakawa, H.; Könneke, M.; et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int. J. Syst. Evol. Microbiol. 2017, 67, 5067–5079. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.E.; Brady, A.L.; Sharp, G.H.; Grasby, S.E.; Stott, M.B.; Dunfield, P.F. Humboldt’s spa: Microbial diversity is controlled by temperature in geothermal environments. ISME J. 2014, 8, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Milici, M.; Tomasch, J.; Wos-Oxley, M.L.; Wang, H.; Jáuregui, R.; Camarinha-Silva, A.; Deng, Z.; Plumeier, I.; Giebel, H.; Wurst, M.; et al. Low diversity of planktonic bacteria in the tropical ocean. Sci. Rep. 2016, 6, 19054. [Google Scholar] [CrossRef]
- Yan, Q.; Stegen, J.C.; Yu, Y.; Deng, Y.; Li, X.; Wu, S.; Dai, L.; Zhang, X.; Li, J.; Wang, C.; et al. Nearly a decade-long repeatable seasonal diversity patterns of bacterioplankton communities in the eutrophic Lake Donghu (Wuhan, China). Mol. Ecol. 2017, 26, 3839–3850. [Google Scholar] [CrossRef]
- Mou, X.; Hodson, R.E.; Moran, M.A. Bacterioplankton assemblages transforming dissolved organic compounds in coastal seawater. Environ. Microbiol. 2007, 9, 2025–2037. [Google Scholar] [CrossRef]
- Mo, Y.; Zhang, W.; Yang, J.; Lin, Y.; Yu, Z.; Lin, S. Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J. 2018, 12, 2198–2210. [Google Scholar] [CrossRef] [Green Version]
- Rud, I.; Kolarevic, J.; Holan, A.B.; Berget, I.; Calabrese, S.; Terjesen, B.F. Deep-sequencing of the bacterial microbiota in commercial-scale recirculating and semi-closed aquaculture systems for Atlantic salmon post-smolt production. Aquacult. Eng. 2017, 78, 50–62. [Google Scholar] [CrossRef]
- Huang, X.; Du, S.; Zhang, H.; Chen, C.; Xiao, G.; Huang, L.; Zhang, D. Temporal patterns of bacterial communities in shrimp (Litopenaeus vannamei) culture systems loaded with oyster shells. Aquaculture 2020, 526, 735424. [Google Scholar] [CrossRef]
- Fernández-Gómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrós-Alió, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowell, S.M.; Abraham, P.E.; Shah, M.; Verberkmoes, N.C.; Smith, D.P.; Barofsky, D.F.; Giovannoni, S.J. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 2011, 5, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Voget, S.; Wemheuer, B.; Brinkhoff, T.; Vollmers, J.; Dietrich, S.; Giebel, H.; Beardsley, C.; Sardemann, C.; Bakenhus, I.; Billerbeck, S.; et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015, 9, 371–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zhou, P.; Wu, Y.; Xu, J.; Wu, Y.; Xu, X. Insight into adaptation mechanisms of marine bacterioplankton from comparative genomic analysis of the genus Pseudohongiella. Deep Sea Res. Part II 2019, 167, 62–69. [Google Scholar] [CrossRef]
- Ghai, R.; Mizuno, C.M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria. Sci. Rep. 2013, 3, 2471. [Google Scholar] [CrossRef] [Green Version]
- Signori, C.N.; Pellizari, V.H.; Enrich-Prast, A.; Sievert, S.M. Spatiotemporal dynamics of marine bacterial and archaeal communities in surface waters off the northern Antarctic Peninsula. Deep Sea Res. Part II 2018, 149, 150–160. [Google Scholar] [CrossRef]
- Halsey, K.H.; Carter, A.E.; Giovannoni, S.J. Synergistic metabolism of a broad range of C1 compounds in the marine methylotrophic bacterium HTCC2181. Environ. Microbiol. 2012, 14, 630–640. [Google Scholar] [CrossRef]
- Wong, S.; Yoshizawa, S.; Nakajima, Y.; Cuadra, M.J.; Nogi, Y.; Nakamura, K.; Takami, H.; Ogura, Y.; Hayashi, T.; Chiura, H.X.; et al. Amylibacter kogurei sp. nov., a novel marine alphaproteobacterium isolated from the coastal sea surface microlayer of a marine inlet. Int. J. Syst. Evol. Micr. 2018, 68, 2872–2877. [Google Scholar] [CrossRef]
- Choi, A.; Lee, K.; Oh, H.; Feng, J.; Cho, J. Litoricola marina sp. nov. Int. J. Syst. Evol. Micr. 2010, 60, 1303–1306. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Chen, H.; Hu, C.; Ye, X.; Kong, D.; Zhang, D. Evidence of bacterioplankton community adaptation in response to long-term mariculture disturbance. Sci. Rep. 2015, 5, 15274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Zheng, C.; Zheng, Z.; Wei, Y.; Lu, K.; Zhu, J. Nutrient enrichment during shrimp cultivation alters bacterioplankton assemblies and destroys community stability. Ecotox. Environ. Safe. 2018, 156, 366–374. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Collecting Date | Seedling Production Stage |
|---|---|---|
| T0 | 13 August | Hours after zoospore inoculated on collectors and transferal to culturing tanks; nutrients supplied 2 days after T0 |
| T1 | 4 September | Zygote formation |
| T2 | 10 September | 4–8 rows of cells |
| T3 | 20 September | 16–32 rows of cells |
| T4 | 5 October | 3–5 mm seedlings |
| T5 | 15 October | 1–2 cm seedlings |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Yan, Y.; Qian, H.; Li, J.; Liu, T.; Mo, Z. Dynamics of Planktonic Microbial Community Associated with Saccharina japonica Seedling. J. Mar. Sci. Eng. 2022, 10, 726. https://0-doi-org.brum.beds.ac.uk/10.3390/jmse10060726
Wang S, Yan Y, Qian H, Li J, Liu T, Mo Z. Dynamics of Planktonic Microbial Community Associated with Saccharina japonica Seedling. Journal of Marine Science and Engineering. 2022; 10(6):726. https://0-doi-org.brum.beds.ac.uk/10.3390/jmse10060726
Chicago/Turabian StyleWang, Shanshan, Yongwei Yan, Hao Qian, Jie Li, Tao Liu, and Zhaolan Mo. 2022. "Dynamics of Planktonic Microbial Community Associated with Saccharina japonica Seedling" Journal of Marine Science and Engineering 10, no. 6: 726. https://0-doi-org.brum.beds.ac.uk/10.3390/jmse10060726