
Treatment of Pulmonary Disease of Cystic Fibrosis: A Comprehensive Review

 and
and
Abstract
:1. Introduction
2. Obstruction Treatment
2.1. Physiotherapy
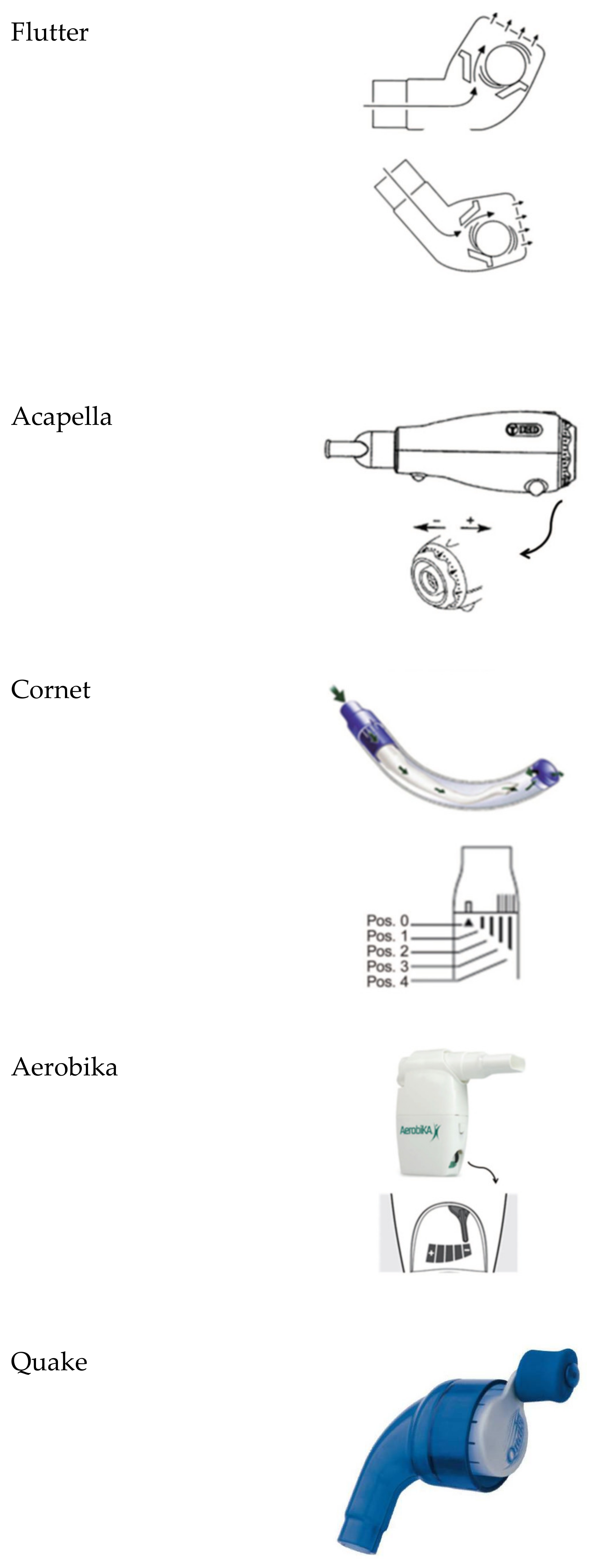
- Flutter®: It is a small plastic device containing a large ball bearing, which repeatedly interrupts the outward flow of air.
- Acapella®: It is a flow-operated oscillating PEP device, which generates oscillating resistance using a plug and magnet counterweight.
- Cornet®: A horn-shaped tube, which houses a rubber inner tube. The degree of rotation of this inner tube reflects the resistance generated.
- Quake®: This device oscillates a column of air in both inspiration and suction. A manually rotated cylinder that fits inside another cylinder is used. Airflow occurs only when the grooves inside the two cylinders are aligned. Therefore, the airflow is interrupted at regular intervals as the user rotates the crank.
- Aerobika®: Exhaled gas passes through a one-way valve housed within a chamber, creating airflow oscillations and PEP as the valve chatters.
- Intrapulmonary percussive ventilation (IPV): This provides continuous oscillation to the airways via the mouth.
- Extra-thoracic oscillations (HFCWO): Extra-thoracic oscillations are generated by forces external to the respiratory system, e.g., high-frequency chest wall oscillation. This type of device is also known as the Vest® or Hayek Oscillator.
- The VibraLung®: It is an acoustic percussor, where sound waves are applied directly to the tracheobronchial tract at frequencies that cover the range of resonant frequencies of the human tracheobronchial tract (5 to 1200 Hz).
- Metaneb®: It is a pneumatic compressor system, which delivers continuous high-frequency oscillation and continuous positive expiratory pressure.
2.2. Bronchodilators
2.2.1. β2-Adrenergic Receptor Agonists
2.2.2. Inhaled Corticosteroids
2.3. Mucolytic
rhDNase
2.4. Hypertonic Substances
2.4.1. Hypertonic Saline
2.4.2. Mannitol
2.4.3. Other Investigation Substances
- Inhibition of the ENaC [38]: ENaC has been proposed as a therapeutic target to ameliorate airway surface liquid dehydration and improve mucus transport. To date, no one therapy inhibiting ENaC has successfully translated to clinical efficacy, in part due to concerns regarding off-target effects, systemic exposure, durability of effect, and adverse effects.
- ○
- ○
- SPX-101. A phase II study to test the safety and effectiveness of it in people with CF is finished, and no further development in CF is planned at this time. Discontinued due to lack of efficacy (NCT03229252).
- ○
- AZD5634. A Phase Ib study to test the safety and effectiveness of it in adults with CF did not have a significant impact on mucociliary clearance when compared with placebo. At this moment, it is discontinued. (NCT02950805).
- ○
- IONIS-ENaC-2.5Rx. A Phase 1/2a study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple doses of IONIS-ENaCRx in healthy volunteers and CF patients is underway. Data collection is finalized for the primary outcome measure (NCT03647228).
- ○
- AROENaC1001. A Phase 1/2 dose-escalating study to evaluate the safety, tolerability, and pharmacokinetic effects of ARO-ENaC in healthy volunteers and patients with CF is underway (NCT04375514).
Additionally, there are other preclinical models [41], such as:- ○
- NVP-QBE 170. It is an inhaled ENaC blocker effective in airways with a reduced risk of hyperkalemia.
- ○
- QUB-TL1. It is designed to inhibit ENaC signaling in CF airways and restores ASL volume and mucociliary function.
- ○
- MK 104. Its mode of action is a channel-activating protease inhibitor.
- Modulators of SLC26A9. They contribute to the secretion of anions and fluids in the airway epithelium. SLC26A9 transports chloride ions through both CFTR-dependent and -independent mechanisms, and positive and negative regulators of SLC26A9 function are required to treat mucus obstruction, although its function is not yet understood [42].
- Modulation of the airway epithelial calcium-activated chloride channel (CaCC), TMEM16A. Positive modulation of TMEM16A favors mucosal hydration in CF. Pre-clinical data with the TMEM16A potentiator ETX001 show that it can increase fluid into the airway mucosa and ccelerate mucus clearance in vivo [43,44]. ETD002 is a compound designed to increase the activity of TMEM16A. A Phase 1 study to test the safety of ETD002 in healthy participants is underway.
- SNSP113. A new class of glycopolymers includes polycationic poly-N (acetyl, arginyl) glucosamine (PAAG), which is a polycationic biopolymer suitable for human use. SNSP113 could separate mucus from individuals with CF by chelation of calcium without any harmful effect on tissue. It can be exploited to treat mucus stagnation and can help clarify secretions more easily [45]. SNSP113 may also help improve the effectiveness of some antibiotics and it is under development as an alternative to treat methicillin-resistant staphylococcus aureus (MRSA) infections [46]. It is administered using a dry-powder inhaler and was developed as a liquid for use with a nebulizer. A recent study demonstrated the potential use of SNSP113 as a molecular agent that could benefit patients with a broad array of mucus diseases [47].
3. Inflammation Treatment
3.1. Azithromycin
3.2. Anti-Inflammatory
3.2.1. Ibuprofen
3.2.2. Acebilustat (CTX-4430)
3.2.3. Lenabasum (JBT-101)
3.2.4. Lau-7b
3.2.5. CB-280
3.2.6. PoL 6014 (Lonodelestat)
4. Infection Treatment
4.1. Antibiotics for Pseudomonas aeruginosa Eradication
4.2. Antibiotics for Exacerbations
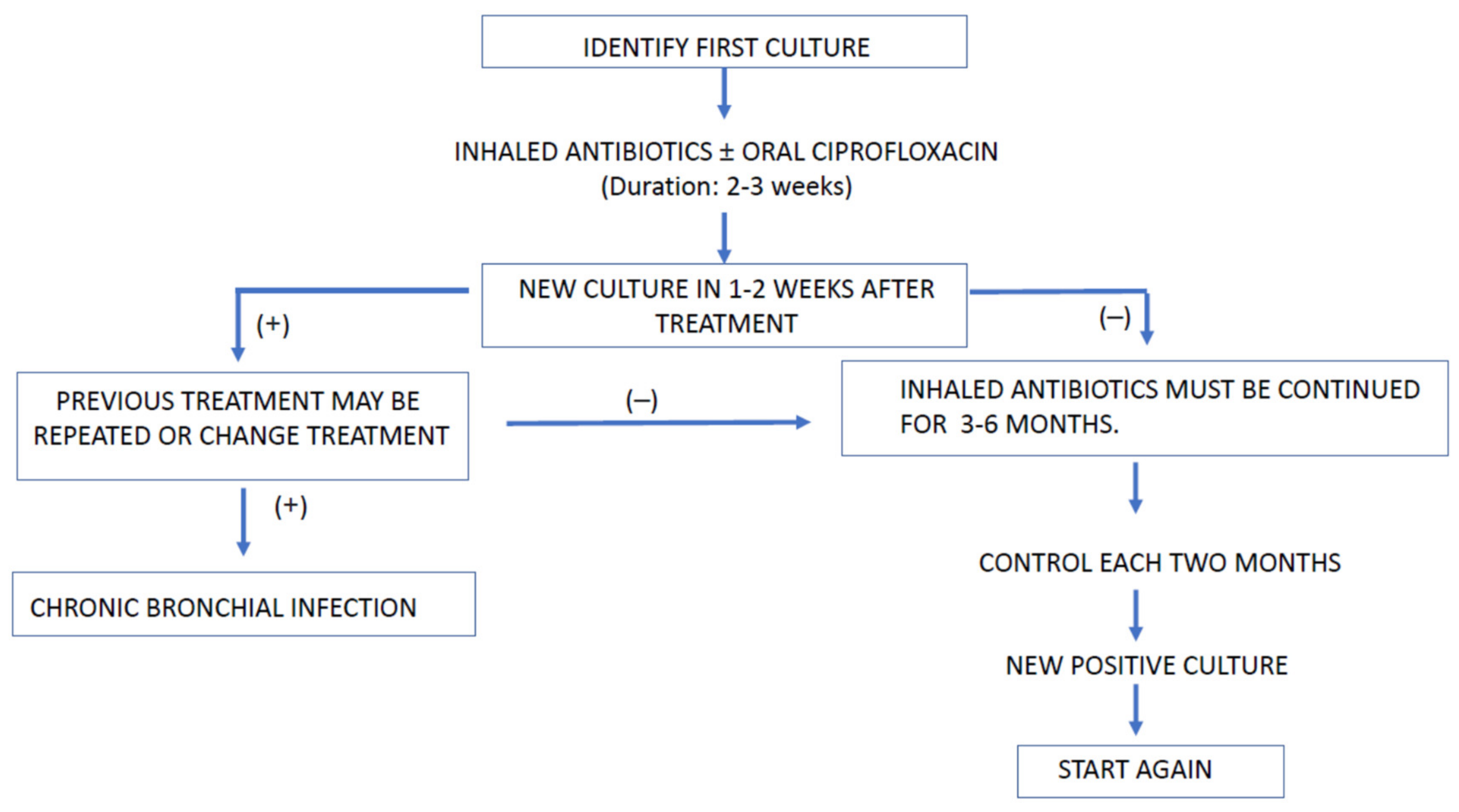
4.3. Duration of Antibiotic Therapy
4.4. Antibiotics for Bronchial Chronic Infection
5. Treatment of Chronic Respiratory Failure
- FEV1≤ or a rapid drop in FEV1 despite optimal treatment.
- 6-min march test < 400 m.
- Pulmonary hypertension in the absence of hypoxic exacerbation, pulmonary arterial pressure (PAP) <35 mmHg in echocardiogram or PAPm < 25 mmHg in catheterization.
- Clinical impairment with increased number of exacerbations associated with an exacerbation with respiratory failure, requiring noninvasive ventilation.
- Increased antibiotic resistance and worse recovery from sharpening.
- Worsening status to nutritional supplements.
- Relapsing pneumothorax.
- Frequent massive hemoptysis.
6. Treatment of Non-Infectious Respiratory Complications
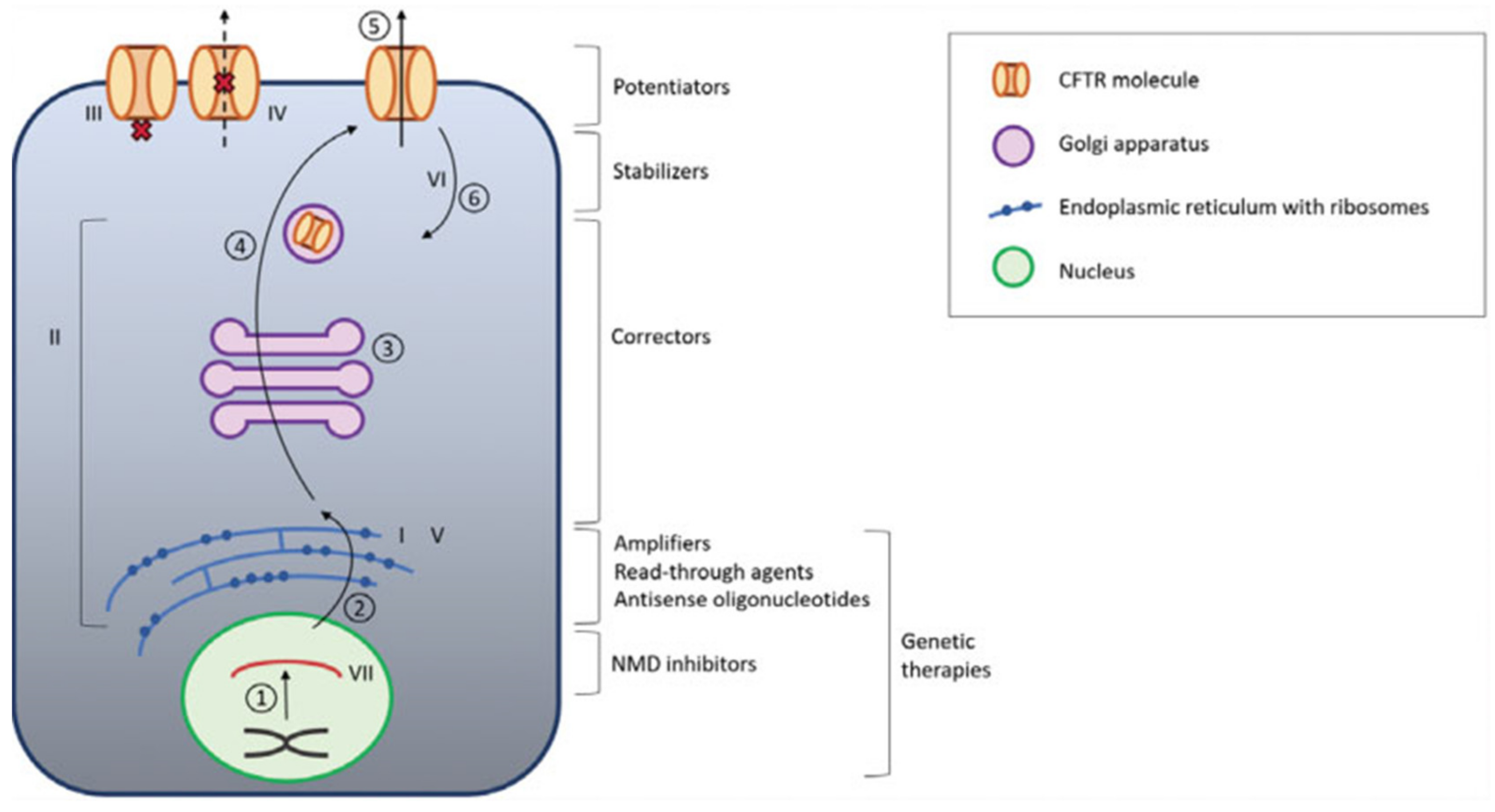
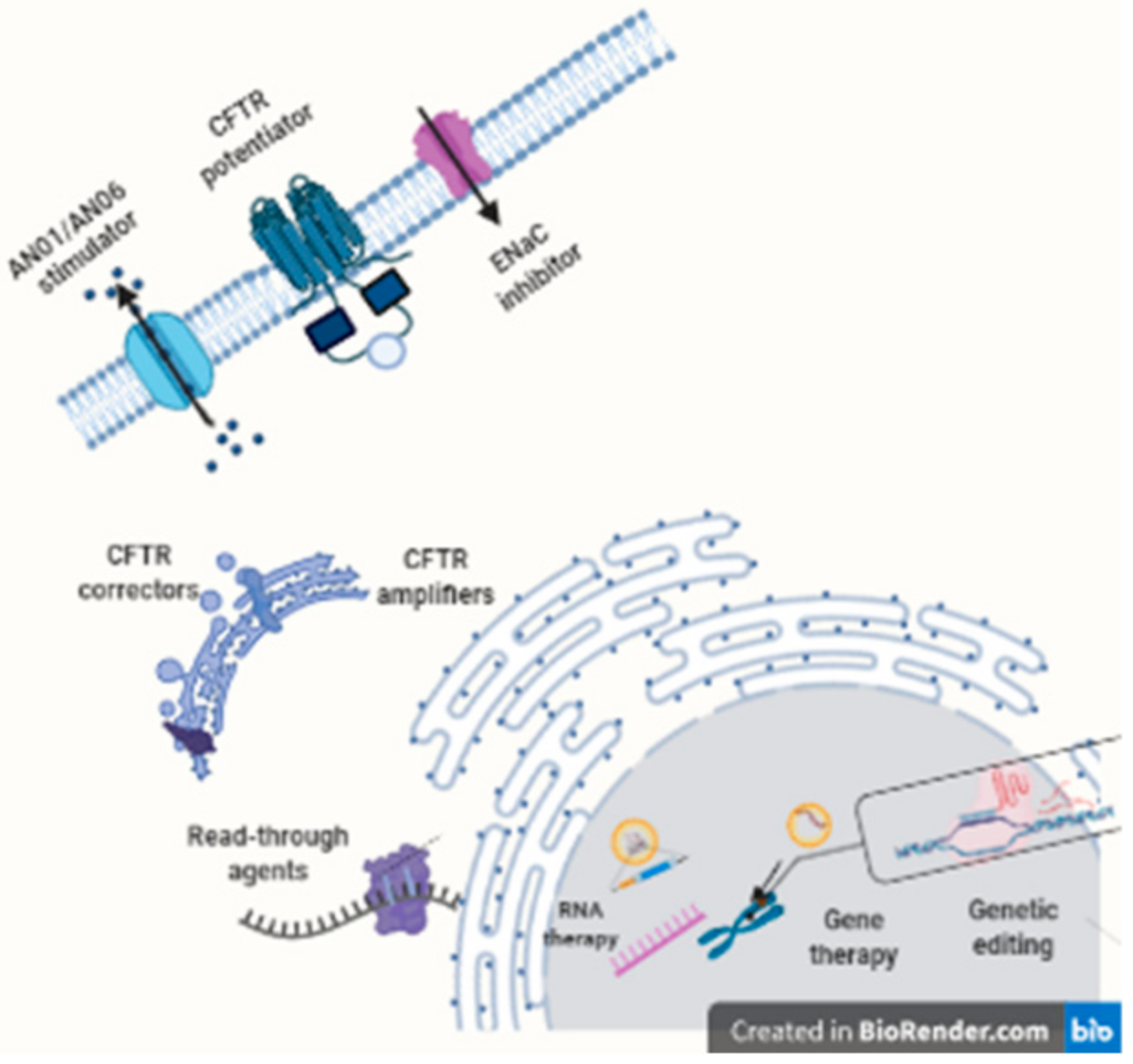
7. Modulator and Amplifiers CFTR
7.1. Ivacaftor (Kalydeco®)
7.2. Lumacaftor/Ivacaftor (Orkambi®)
7.3. Tezacaftor/Ivacaftor (Symkevi® in Europe or Symdeko® in the US)
7.4. Elexacaftor/Tezacaftor/Ivacaftor (Kaftrio® in Europe or Trikafta® in the US)
7.5. Galicaftor (ABBV-2222)
7.6. ABBV-3067
7.7. VX-121
7.8. Deutivacaftor (VX-561)
7.9. Nesolicaftor (PTI-428)
7.10. Posenacaftor (PTI-801)
7.11. Dirocaftor (PTI-808)
7.12. Cavosonstat (N91115)
7.13. Icenticaftor (QBW251)
8. Read-Through Agents
8.1. Aminoglycoside
8.2. Ataluren
8.3. ELX.02 (NB124; Eloxx Pharmaceuticals)
8.4. Others
9. Gene Therapy/Gene Editing DNA
9.1. Gene Therapy
9.1.1. Viral Vectors
9.1.2. Non-Viral Vector
9.2. Gene Editing Technologies
9.2.1. CRISPR/Cas-9
9.2.2. Zinc Finger Nucleases (ZFNs)
9.2.3. The Triplex-Forming PNA/DNA
10. RNA Therapy
10.1. mRNA
10.1.1. Antisense Oligonucleotides (ASOs)
10.1.2. MRT5005
10.2. tRNA
11. MicroRNAs Therapy
12. Cell-Based Therapy
13. Conclusions
Funding
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Gruet, M.; Troosters, T.; Verges, S. Peripheral muscle abnormalities in cystic fibrosis: Etiology, clinical implications and response to therapeutic interventions. J. Cyst. Fibros. 2017, 16, 538–552. [Google Scholar] [CrossRef] [Green Version]
- Conway, S.; Balfour-Lynn, I.M.; De Rijcke, K.; Drevinek, P.; Foweraker, J.; Havermans, T.; Heijerman, H.; Lannefors, L.; Lindblad, A.; Macek, M.; et al. European Cystic Fibrosis Society Standards of Care: Framework for the Cystic Fibrosis Centre. J. Cyst. Fibros. 2014, 13 (Suppl. 1), S3–S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell Mol Life Sci 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Cystic fibrosis: Impaired bicarbonate secretion and mucoviscidosis. Lancet 2008, 372, 415–417. [Google Scholar] [CrossRef] [Green Version]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collawn, J.F.; Matalon, S. CFTR and lung homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L917–L923. [Google Scholar] [CrossRef] [Green Version]
- Sosnay, P.R.; Salinas, D.B.; White, T.B.; Ren, C.L.; Farrell, P.M.; Raraigh, K.S.; Girodon, E.; Castellani, C. Applying Cystic Fibrosis Transmembrane Conductance Regulator Genetics and CFTR2 Data to Facilitate Diagnoses. J. Pediatr. 2017, 181S, S27–S32.e21. [Google Scholar] [CrossRef] [Green Version]
- Bombieri, C.; Seia, M.; Castellani, C. Genotypes and phenotypes in cystic fibrosis and cystic fibrosis transmembrane regulator-related disorders. Semin. Respir. Crit. Care Med. 2015, 36, 180–193. [Google Scholar] [CrossRef]
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Radtke, T.; Nevitt, S.J.; Hebestreit, H.; Kriemler, S. Physical exercise training for cystic fibrosis. Cochrane Database Syst. Rev. 2017, 11, CD002768. [Google Scholar] [CrossRef] [PubMed]
- De Carlos, E.; Pérez, M. Rehabilitación respiratoria y ejercicio físico. In Tratado de Fibrosis Quística; Salcedo Posadas, A., Gartner, S., Girón Moreno, R.M., García Novo, M.D., Eds.; Editorial Justim S.L.: Madrid, Spain, 2012; pp. 285–302. [Google Scholar]
- Button, B.M.; Wilson, C.; Dentice, R.; Cox, N.S.; Middleton, A.; Tannenbaum, E.; Bishop, J.; Cobb, R.; Burton, K.; Wood, M.; et al. Physiotherapy for cystic fibrosis in Australia and New Zealand: A clinical practice guideline. Respirology 2016, 21, 656–667. [Google Scholar] [CrossRef]
- Morrison, L.; Innes, S. Oscillating devices for airway clearance in people with cystic fibrosis. Cochrane Database Syst. Rev. 2017, 5, CD006842. [Google Scholar] [CrossRef]
- Cox, N.S.; Holland, A.E. Current perspectives of physical activity in cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 13–22. [Google Scholar] [CrossRef]
- Dwyer, T.J.; Elkins, M.R.; Bye, P.T. The role of exercise in maintaining health in cystic fibrosis. Curr. Opin. Pulm. Med. 2011, 17, 455–460. [Google Scholar] [CrossRef]
- Brand, P.L. Bronchodilators in cystic fibrosis. J. R. Soc. Med. 2000, 93 (Suppl. 38), 37–39. [Google Scholar]
- Mogayzel, P.J.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B.; et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef]
- Edmondson, C.; Davies, J.C. Current and future treatment options for cystic fibrosis lung disease: Latest evidence and clinical implications. Ther. Adv. Chronic Dis. 2016, 7, 170–183. [Google Scholar] [CrossRef] [Green Version]
- Halfhide, C.; Evans, H.J.; Couriel, J. WITHDRAWN: Inhaled bronchodilators for cystic fibrosis. Cochrane Database Syst. Rev. 2016, 2, CD003428. [Google Scholar] [CrossRef]
- Ross, K.R.; Chmiel, J.F.; Konstan, M.W. The role of inhaled corticosteroids in the management of cystic fibrosis. Paediatr. Drugs 2009, 11, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balfour-Lynn, I.M.; Welch, K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst. Rev. 2016, CD001915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.L.; Scott, S.F.; Fuchs, H.J.; Geddes, D.M.; Hodson, M.E. Medium term treatment of stable stage cystic fibrosis with recombinant human DNase I. Thorax 1995, 50, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, K.; Rietschel, E.; Ballmann, M.; Griese, M.; Worlitzsch, D.; Shute, J.; Chen, C.; Schink, T.; Döring, G.; van Koningsbruggen, S.; et al. Effect of treatment with dornase alpha on airway inflammation in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 719–725. [Google Scholar] [CrossRef]
- Amin, R.; Ratjen, F. Revisión de los tratamientos que mejoran el aclaramiento mucociliar. In Tratado de Fibrosis Quística; Salcedo Posadas, A., Gartner, S., Girón Moreno, R.M., García Novo, M.D., Eds.; Editorial Justim S.L.: Madrid, Spain, 2012; pp. 243–255. [Google Scholar]
- Jones, A.P.; Wallis, C. Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. 2010, CD001127. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Danielle Samulski, T.; LaFave, C.; Zeman, K.; Wu, J.; Trimble, A.; Ceppe, A.; Bennett, W.D.; Davis, S.D. A four week trial of hypertonic saline in children with mild cystic fibrosis lung disease: Effect on mucociliary clearance and clinical outcomes. J. Cyst. Fibros. 2020, 19, 942–948. [Google Scholar] [CrossRef]
- Elkins, M.R.; Robinson, M.; Rose, B.R.; Harbour, C.; Moriarty, C.P.; Marks, G.B.; Belousova, E.G.; Xuan, W.; Bye, P.T.; Group, National Hypertonic Saline in Cystic Fibrosis. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 2006, 354, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Wark, P.; McDonald, V.M. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst. Rev. 2018, 9, CD001506. [Google Scholar] [CrossRef]
- Elkins, M.; Dentice, R. Timing of hypertonic saline inhalation for cystic fibrosis. Cochrane Database Syst. Rev. 2020, 2, CD008816. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Davis, S.D.; Stanojevic, S.; Kronmal, R.A.; Hinckley Stukovsky, K.D.; Jorgensen, N.; Rosenfeld, M.; Group, S.S. Inhaled hypertonic saline in preschool children with cystic fibrosis (SHIP): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2019, 7, 802–809. [Google Scholar] [CrossRef]
- De Boeck, K.; Haarman, E.; Hull, J.; Lands, L.C.; Moeller, A.; Munck, A.; Riethmüller, J.; Tiddens, H.; Volpi, S.; Leadbetter, J.; et al. Inhaled dry powder mannitol in children with cystic fibrosis: A randomised efficacy and safety trial. J. Cyst. Fibros. 2017, 16, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Nevitt, S.J.; Thornton, J.; Murray, C.S.; Dwyer, T. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst. Rev. 2020, 5, CD008649. [Google Scholar] [CrossRef]
- Flume, P.A.; Amelina, E.; Daines, C.L.; Charlton, B.; Leadbetter, J.; Guasconi, A.; Aitken, M.L. Efficacy and safety of inhaled dry-powder mannitol in adults with cystic fibrosis: An international, randomized controlled study. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Shei, R.J.; Peabody, J.E.; Kaza, N.; Rowe, S.M. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol. 2018, 43, 152–165. [Google Scholar] [CrossRef]
- Nickolaus, P.; Jung, B.; Sabater, J.; Constant, S.; Gupta, A. Preclinical evaluation of the epithelial sodium channel inhibitor BI 1265162 for treatment of cystic fibrosis. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- Goss, C.H.; Jain, R.; Seibold, W.; Picard, A.C.; Hsu, M.C.; Gupta, A.; Fajac, I. An innovative phase II trial to establish proof of efficacy and optimal dose of a new inhaled epithelial sodium channel inhibitor BI 1265162 in adults and adolescents with cystic fibrosis: BALANCE-CF. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Balázs, A.; Mall, M.A. Role of the SLC26A9 Chloride Channel as Disease Modifier and Potential Therapeutic Target in Cystic Fibrosis. Front. Pharmacol. 2018, 9, 1112. [Google Scholar] [CrossRef]
- Danahay, H.; Gosling, M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int. J. Mol. Sci. 2020, 21, 2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 946–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermund, A.; Recktenwald, C.V.; Skjåk-Braek, G.; Meiss, L.N.; Onsøyen, E.; Rye, P.D.; Dessen, A.; Myrset, A.H.; Hansson, G.C. OligoG CF-5/20 normalizes cystic fibrosis mucus by chelating calcium. Clin. Exp. Pharmacol. Physiol. 2017, 44, 639–647. [Google Scholar] [CrossRef]
- Narayanaswamy, V.P.; Giatpaiboon, S.A.; Uhrig, J.; Orwin, P.; Wiesmann, W.; Baker, S.M.; Townsend, S.M. In Vitro activity of novel glycopolymer against clinical isolates of multidrug-resistant Staphylococcus aureus. PLoS ONE 2018, 13, e0191522. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Petty, C.M.; Hughes, G.W.; Bowers, H.L.; Watson, J.D.; Rosen, B.H.; Townsend, S.M.; Santos, C.; Ridley, C.E.; Chu, K.K.; Birket, S.E.; et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkerman-Nijland, A.M.; Akkerman, O.W.; Grasmeijer, F.; Hagedoorn, P.; Frijlink, H.W.; Rottier, B.L.; Koppelman, G.H.; Touw, D.J. The pharmacokinetics of antibiotics in cystic fibrosis. Expert Opin. Drug Metab. Toxicol. 2021, 17, 53–68. [Google Scholar] [CrossRef]
- O’Toole, G.A. Cystic Fibrosis Airway Microbiome: Overturning the Old, Opening the Way for the New. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [Green Version]
- Ballmann, M.; Rabsch, P.; von der Hardt, H. Long-term follow up of changes in FEV1 and treatment intensity during Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Thorax 1998, 53, 732–737. [Google Scholar] [CrossRef] [Green Version]
- Emerson, J.; Rosenfeld, M.; McNamara, S.; Ramsey, B.; Gibson, R.L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 2002, 34, 91–100. [Google Scholar] [CrossRef]
- Schaedel, C.; de Monestrol, I.; Hjelte, L.; Johannesson, M.; Kornfält, R.; Lindblad, A.; Strandvik, B.; Wahlgren, L.; Holmberg, L. Predictors of deterioration of lung function in cystic fibrosis. Pediatr. Pulmonol. 2002, 33, 483–491. [Google Scholar] [CrossRef]
- Akkerman-Nijland, A.M.; Yousofi, M.; Rottier, B.L.; Van der Vaart, H.; Burgerhof, J.G.M.; Frijlink, H.W.; Touw, D.J.; Koppelman, G.H.; Akkerman, O.W. Eradication of. Ther. Adv. Respir. Dis. 2020, 14, 1753466620905279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høiby, N. Diffuse panbronchiolitis and cystic fibrosis: East meets West. Thorax 1994, 49, 531–532. [Google Scholar] [CrossRef] [Green Version]
- Labro, M.T. Immunological effects of macrolides. Curr. Opin. Infect. Dis. 1998, 11, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Tomono, K.; Sawai, T.; Hirakata, Y.; Kadota, J.; Koga, H.; Tashiro, T.; Kohno, S. Effect of clarithromycin on lymphocytes in chronic respiratory Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 1997, 155, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Blasi, F.; Esposito, S. Azithromycin use in patients with cystic fibrosis. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Kerem, E.; Ferkol, T.; Elizur, A. Airway inflammation in cystic fibrosis: Molecular mechanisms and clinical implications. Thorax 2013, 68, 1157–1162. [Google Scholar] [CrossRef] [Green Version]
- Razvi, S.; Quittell, L.; Sewall, A.; Quinton, H.; Marshall, B.; Saiman, L. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 2009, 136, 1554–1560. [Google Scholar] [CrossRef]
- Zlosnik, J.E.; Costa, P.S.; Brant, R.; Mori, P.Y.; Hird, T.J.; Fraenkel, M.C.; Wilcox, P.G.; Davidson, A.G.; Speert, D.P. Mucoid and nonmucoid Burkholderia cepacia complex bacteria in cystic fibrosis infections. Am. J. Respir. Crit. Care Med. 2011, 183, 67–72. [Google Scholar] [CrossRef]
- Foweraker, J. Recent advances in the microbiology of respiratory tract infection in cystic fibrosis. Br. Med. Bull. 2009, 89, 93–110. [Google Scholar] [CrossRef]
- Saiman, L.; Schechter, M.S. Evaluating Long-Term Benefits of Chronic Azithromycin. Furthering Our Quest for Precision Medicine. Am. J. Respir. Crit. Care Med. 2020, 201, 398–400. [Google Scholar] [CrossRef]
- Shinkai, M.; Foster, G.H.; Rubin, B.K. Macrolide antibiotics modulate ERK phosphorylation and IL-8 and GM-CSF production by human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L75–L85. [Google Scholar] [CrossRef] [Green Version]
- Bystrzycka, W.; Manda-Handzlik, A.; Sieczkowska, S.; Moskalik, A.; Demkow, U.; Ciepiela, O. Azithromycin and Chloramphenicol Diminish Neutrophil Extracellular Traps (NETs) Release. Int. J. Mol. Sci. 2017, 18, 2666. [Google Scholar] [CrossRef] [Green Version]
- Saiman, L.; Marshall, B.C.; Mayer-Hamblett, N.; Burns, J.L.; Quittner, A.L.; Cibene, D.A.; Coquillette, S.; Fieberg, A.Y.; Accurso, F.J.; Campbell, P.W.; et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003, 290, 1749–1756. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Saiman, L.; Mayer-Hamblett, N.; Lands, L.C.; Kloster, M.; Thompson, V.; Emmett, P.; Marshall, B.; Accurso, F.; Sagel, S.; et al. Effect of azithromycin on systemic markers of inflammation in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa. Chest 2012, 142, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Southern, K.W.; Barker, P.M.; Solis-Moya, A.; Patel, L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst. Rev. 2012, 11, CD002203. [Google Scholar] [CrossRef] [PubMed]
- Equi, A.; Balfour-Lynn, I.M.; Bush, A.; Rosenthal, M. Long term azithromycin in children with cystic fibrosis: A randomised, placebo-controlled crossover trial. Lancet 2002, 360, 978–984. [Google Scholar] [CrossRef]
- Clement, A.; Tamalet, A.; Leroux, E.; Ravilly, S.; Fauroux, B.; Jais, J.P. Long term effects of azithromycin in patients with cystic fibrosis: A double blind, placebo controlled trial. Thorax 2006, 61, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiman, L.; Anstead, M.; Mayer-Hamblett, N.; Lands, L.C.; Kloster, M.; Hocevar-Trnka, J.; Goss, C.H.; Rose, L.M.; Burns, J.L.; Marshall, B.C.; et al. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2010, 303, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.; Seeney, S.; Bell, S.; Bowler, S.; Masel, P.; McCormack, J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: A randomised trial. Thorax 2002, 57, 212–216. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.; Bell, S.; Senini, S.; Walmsley, K.; Patel, K.; Wainwright, C.; Serisier, D.; Harris, M.; Bowler, S. Daily versus weekly azithromycin in cystic fibrosis patients. Eur. Respir. J. 2007, 30, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Tramper-Stranders, G.A.; Wolfs, T.F.; Fleer, A.; Kimpen, J.L.; van der Ent, C.K. Maintenance azithromycin treatment in pediatric patients with cystic fibrosis: Long-term outcomes related to macrolide resistance and pulmonary function. Pediatr. Infect. Dis. J. 2007, 26, 8–12. [Google Scholar] [CrossRef]
- Willekens, J.; Eyns, H.; Malfroot, A. How long should we maintain long-term azithromycin treatment in cystic fibrosis patients? Pediatr. Pulmonol. 2015, 50, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Odem-Davis, K.; Cogen, J.D.; Goss, C.H.; Ren, C.L.; Skalland, M.; Somayaji, R.; Heltshe, S.L. Pulmonary Outcomes Associated with Long-Term Azithromycin Therapy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Doull, I. Cystic fibrosis 2019: Year in review. Paediatr. Respir. Rev. 2020, 35, 95–98. [Google Scholar] [CrossRef]
- Nichols, D.P.; Happoldt, C.L.; Bratcher, P.E.; Caceres, S.M.; Chmiel, J.F.; Malcolm, K.C.; Saavedra, M.T.; Saiman, L.; Taylor-Cousar, J.L.; Nick, J.A. Impact of azithromycin on the clinical and antimicrobial effectiveness of tobramycin in the treatment of cystic fibrosis. J. Cyst. Fibros. 2017, 16, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Nick, J.A.; Moskowitz, S.M.; Chmiel, J.F.; Forssén, A.V.; Kim, S.H.; Saavedra, M.T.; Saiman, L.; Taylor-Cousar, J.L.; Nichols, D.P. Azithromycin may antagonize inhaled tobramycin when targeting Pseudomonas aeruginosa in cystic fibrosis. Ann. Am. Thorac. Soc. 2014, 11, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Hamblett, N.; Retsch-Bogart, G.; Kloster, M.; Accurso, F.; Rosenfeld, M.; Albers, G.; Black, P.; Brown, P.; Cairns, A.; Davis, S.D.; et al. Azithromycin for Early Pseudomonas Infection in Cystic Fibrosis. The OPTIMIZE Randomized Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 1177–1187. [Google Scholar] [CrossRef]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of high-dose ibuprofen in patients with cystic fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef]
- Lands, L.C.; Milner, R.; Cantin, A.M.; Manson, D.; Corey, M. High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J. Pediatr. 2007, 151, 249–254. [Google Scholar] [CrossRef]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 9, CD001505. [Google Scholar] [CrossRef]
- Elborn, J.S.; Ahuja, S.; Springman, E.; Mershon, J.; Grosswald, R.; Rowe, S.M. EMPIRE-CF: A phase II randomized placebo-controlled trial of once-daily, oral acebilustat in adult patients with cystic fibrosis—Study design and patient demographics. Contemp. Clin. Trials 2018, 72, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Chmiel, J.F.; Flume, P.; Downey, D.G.; Dozor, A.J.; Colombo, C.; Mazurek, H.; Sapiejka, E.; Rachel, M.; Constantine, S.; Conley, B.; et al. Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; De Sanctis, J.B.; Shah, J.; Dumut, D.C.; Hajduch, M.; Petrof, B.J.; Radzioch, D. Age-Dependent Progression in Lung Pathophysiology can be Prevented by Restoring Fatty Acid and Ceramide Imbalance in Cystic Fibrosis. Lung 2020, 198, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Stanford, G.E.; Dave, K.; Simmonds, N.J. Pulmonary Exacerbations in Adults with Cystic Fibrosis: A Grown-up Issue in a Changing Cystic Fibrosis Landscape. Chest 2020. [Google Scholar] [CrossRef]
- Monsó, E. Look at the wood and not at the tree: The Microbiome in Chronic Obstructive Lung Disease and Cystic Fibrosis. Arch. Bronconeumol. 2020, 56, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Bell, S.C. Prevention of chronic infection with Pseudomonas aeruginosa infection in cystic fibrosis. Curr. Opin. Pulm. Med. 2019, 25, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; McLearn-Montz, A.J.; Milavetz, F.; Gates, L.K.; Fox, C.; Murry, L.T.; Sabus, A.; Porterfield, H.S.; Fischer, A.J. Pathogen acquisition in patients with cystic fibrosis receiving ivacaftor or lumacaftor/ivacaftor. Pediatr. Pulmonol. 2019, 54, 1200–1208. [Google Scholar] [CrossRef]
- Rabin, H.R.; Butler, S.M.; Wohl, M.E.; Geller, D.E.; Colin, A.A.; Schidlow, D.V.; Johnson, C.A.; Konstan, M.W.; Regelmann, W.E.; Fibrosis, E.S.o.C. Pulmonary exacerbations in cystic fibrosis. Pediatr. Pulmonol. 2004, 37, 400–406. [Google Scholar] [CrossRef]
- Sanders, D.B.; Bittner, R.C.; Rosenfeld, M.; Hoffman, L.R.; Redding, G.J.; Goss, C.H. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2010, 182, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.L.; Thayer, S.; Watkins, A.; Wagener, J.S.; Hodgkins, P.S.; Schechter, M.S. Frequency and costs of pulmonary exacerbations in patients with cystic fibrosis in the United States. Curr. Med. Res. Opin. 2017, 33, 667–674. [Google Scholar] [CrossRef] [Green Version]
- UK Cystic Fibrosis Trust. UK Cystic Fibrosis Registry Annual Data Report 2019. Available online: https://www.cysticfibrosis.org.uk/the-work-we-do/ukcf-registry/reporting-and-resources (accessed on 16 August 2020).
- Hewer, S.C.L.; Smyth, A.R.; Brown, M.; Jones, A.P.; Hickey, H.; Kenna, D.; Ashby, D.; Thompson, A.; Williamson, P.R.; TORPEDO-CF study group. Intravenous versus oral antibiotics for eradication of Pseudomonas aeruginosa in cystic fibrosis (TORPEDO-CF): A randomised controlled trial. Lancet Respir. Med. 2020, 8, 975–986. [Google Scholar] [CrossRef]
- Ayats Vidal, R.; Bosque García, M.; García González, M.; de la Cruz, Ó.A. Bronchial Infection due to Pseudomonas Aeruginosa in Patients with Cystic Fibrosis Diagnosed in Neonatal Screening. Arch. Bronconeumol. 2020, 56, 532–534. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Jensen, P.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Høiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.; Yang, L.; Pamp, S.J.; Tolker-Nielsen, T. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol. Med. Microbiol. 2010, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, L.; Waters, V. Factors influencing the acquisition and eradication of early Pseudomonas aeruginosa infection in cystic fibrosis. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Kronmal, R.A.; Gibson, R.L.; Rosenfeld, M.; Retsch-Bogart, G.; Treggiari, M.M.; Burns, J.L.; Khan, U.; Ramsey, B.W.; Investigators, E. Initial Pseudomonas aeruginosa treatment failure is associated with exacerbations in cystic fibrosis. Pediatr. Pulmonol. 2012, 47, 125–134. [Google Scholar] [CrossRef] [Green Version]
- McColley, S.A.; Schechter, M.S.; Morgan, W.J.; Pasta, D.J.; Craib, M.L.; Konstan, M.W. Risk factors for mortality before age 18 years in cystic fibrosis. Pediatr. Pulmonol. 2017, 52, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Kloster, M.; Rosenfeld, M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Emerson, J.; Thompson, V.; Ramsey, B.W. Impact of Sustained Eradication of New Pseudomonas aeruginosa Infection on Long-term Outcomes in Cystic Fibrosis. Clin. Infect. Dis. 2015, 61, 707–715. [Google Scholar] [CrossRef]
- Langton Hewer, S.C.; Smyth, A.R. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst. Rev. 2017, 4, CD004197. [Google Scholar] [CrossRef]
- UK Cystic Fibrosis Trust. Antibiotic Treatment for Cystic Fibrosis—Report of the UK Cystic Fibrosis Trust Antibiotic Working Group; UK Cystic Fibrosis Trust: London, UK, 2009. [Google Scholar]
- Mogayzel, P.J.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann. Am. Thorac. Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef]
- Döring, G.; Flume, P.; Heijerman, H.; Elborn, J.S.; Group, C.S. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, A.C.; Horton, E.; Stanojevic, S.; Taylor, L.; Waters, V.; Ratjen, F. Effectiveness of a stepwise Pseudomonas aeruginosa eradication protocol in children with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Waters, V.; Mathew, J.L.; Taylor, L.; Ratjen, F. Effectiveness of inhaled tobramycin in eradicating Pseudomonas aeruginosa in children with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantón, R.; Máiz, L.; Escribano, A.; Olveira, C.; Oliver, A.; Asensio, O.; Gartner, S.; Roma, E.; Quintana-Gallego, E.; Salcedo, A.; et al. Spanish consensus on the prevention and treatment of Pseudomonas aeruginosa bronchial infections in cystic fibrosis patients. Arch. Bronconeumol. 2015, 51, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.S.; Rasouliyan, L.; VanDevanter, D.R.; Pasta, D.J.; Regelmann, W.E.; Morgan, W.J.; Konstan, M.W.; Fibrosis, I.a.C.o.t.E.S.o.C. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatr. Pulmonol. 2013, 48, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orenstein, D.M.; Pattishall, E.N.; Nixon, P.A.; Ross, E.A.; Kaplan, R.M. Quality of well-being before and after antibiotic treatment of pulmonary exacerbation in patients with cystic fibrosis. Chest 1990, 98, 1081–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, J.; McAlister, O.; Elborn, S. Pulmonary function, inflammation, exercise capacity and quality of life in cystic fibrosis. Eur. Respir. J. 2001, 17, 712–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britto, M.T.; Kotagal, U.R.; Hornung, R.W.; Atherton, H.D.; Tsevat, J.; Wilmott, R.W. Impact of recent pulmonary exacerbations on quality of life in patients with cystic fibrosis. Chest 2002, 121, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; Morgan, W.J.; Butler, S.M.; Pasta, D.J.; Craib, M.L.; Silva, S.J.; Stokes, D.C.; Wohl, M.E.; Wagener, J.S.; Regelmann, W.E.; et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J. Pediatr. 2007, 151, 134–139. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Wagener, J.S.; Pasta, D.J.; Elkin, E.; Jacobs, J.R.; Morgan, W.J.; Konstan, M.W. Pulmonary outcome prediction (POP) tools for cystic fibrosis patients. Pediatr. Pulmonol. 2010, 45, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Hamblett, N.; Rosenfeld, M.; Emerson, J.; Goss, C.H.; Aitken, M.L. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am. J. Respir. Crit. Care Med. 2002, 166, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Ellaffi, M.; Vinsonneau, C.; Coste, J.; Hubert, D.; Burgel, P.R.; Dhainaut, J.F.; Dusser, D. One-year outcome after severe pulmonary exacerbation in adults with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Goss, C.H. Acute Pulmonary Exacerbations in Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 792–803. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Rosenfeld, M.; Treggiari, M.M.; Konstan, M.W.; Retsch-Bogart, G.; Morgan, W.; Wagener, J.; Gibson, R.L.; Khan, U.; Emerson, J.; et al. Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis. Pediatr. Pulmonol. 2013, 48, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Vandevanter, D.R.; Yegin, A.; Morgan, W.J.; Millar, S.J.; Pasta, D.J.; Konstan, M.W. Design and powering of cystic fibrosis clinical trials using pulmonary exacerbation as an efficacy endpoint. J. Cyst. Fibros. 2011, 10, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, A. Update on treatment of pulmonary exacerbations in cystic fibrosis. Curr. Opin. Pulm. Med. 2006, 12, 440–444. [Google Scholar] [CrossRef]
- Flume, P.A.; Mogayzel, P.J.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Committee C.P.G.f.P.T. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Butler, S.M.; Konstan, M.W.; Morgan, W.; Wohl, M.E. Factors influencing outcomes in cystic fibrosis: A center-based analysis. Chest 2003, 123, 20–27. [Google Scholar] [CrossRef]
- Briggs, E.C.; Nguyen, T.; Wall, M.A.; MacDonald, K.D. Oral antimicrobial use in outpatient cystic fibrosis pulmonary exacerbation management: A single-center experience. Clin. Respir. J. 2012, 6, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanojevic, S.; McDonald, A.; Waters, V.; MacDonald, S.; Horton, E.; Tullis, E.; Ratjen, F. Effect of pulmonary exacerbations treated with oral antibiotics on clinical outcomes in cystic fibrosis. Thorax 2017, 72, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.; Rowbotham, N.J.; Charbek, E. Inhaled antibiotics for pulmonary exacerbations in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 10, CD008319. [Google Scholar] [CrossRef] [PubMed]
- West, N.E.; Beckett, V.V.; Jain, R.; Sanders, D.B.; Nick, J.A.; Heltshe, S.L.; Dasenbrook, E.C.; VanDevanter, D.R.; Solomon, G.M.; Goss, C.H.; et al. Standardized Treatment of Pulmonary Exacerbations (STOP) study: Physician treatment practices and outcomes for individuals with cystic fibrosis with pulmonary Exacerbations. J. Cyst. Fibros. 2017, 16, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Bilton, D.; Canny, G.; Conway, S.; Dumcius, S.; Hjelte, L.; Proesmans, M.; Tümmler, B.; Vavrova, V.; De Boeck, K. Pulmonary exacerbation: Towards a definition for use in clinical trials. Report from the EuroCareCF Working Group on outcome parameters in clinical trials. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S79–S81. [Google Scholar] [CrossRef] [Green Version]
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Schwarzenberg, S.J.; et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J. Cyst. Fibros. 2014, 13 (Suppl. 1), S23–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, M.N.; Prayle, A.P.; Flume, P. Intravenous antibiotics for pulmonary exacerbations in people with cystic fibrosis. Cochrane Database Syst. Rev. 2015, CD009730. [Google Scholar] [CrossRef]
- Waters, V.J.; Kidd, T.J.; Canton, R.; Ekkelenkamp, M.B.; Johansen, H.K.; LiPuma, J.J.; Bell, S.C.; Elborn, J.S.; Flume, P.A.; VanDevanter, D.R.; et al. Reconciling Antimicrobial Susceptibility Testing and Clinical Response in Antimicrobial Treatment of Chronic Cystic Fibrosis Lung Infections. Clin. Infect. Dis. 2019, 69, 1812–1816. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.; Burgel, P.R.; Taccetti, G.; Holmes, A.; Ratjen, F.; Byrnes, C.A.; Waters, V.J.; Bell, S.C.; VanDevanter, D.R.; Stuart Elborn, J.; et al. Antimicrobial resistance in cystic fibrosis: A Delphi approach to defining best practices. J. Cyst. Fibros. 2020, 19, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Einarsson, G.; Flanagan, E.; Lee, A.; Elborn, J.S.; Tunney, M.; Plant, B.J. WS03.1 Longitudinal airway microbiota profiling in cystic fibrosis patients enrolled in the CFMATTERS clinical trial. J. Cyst. Fibros. 2017, 16, S4. [Google Scholar] [CrossRef]
- Plant, B. Final Report Summary—Cystic Fibrosis Microbiomedetermined Antibiotic Therapy Trial in Exacerbations: Results Stratified. Available online: https://cordis.europa.eu/docs/results/603/603038/final1-cfmattersfinal-report-draft-v3-mp-31082017.pdf (accessed on 11 November 2020).
- Kraynack, N.C.; Gothard, M.D.; Falletta, L.M.; McBride, J.T. Approach to treating cystic fibrosis pulmonary exacerbations varies widely across US CF care centers. Pediatr. Pulmonol. 2011, 46, 870–881. [Google Scholar] [CrossRef]
- Conway, S.P.; Pond, M.N.; Watson, A.; Etherington, C.; Robey, H.L.; Goldman, M.H. Intravenous colistin sulphomethate in acute respiratory exacerbations in adult patients with cystic fibrosis. Thorax 1997, 52, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Tümmler, B. Emerging therapies against infections with. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Ng, C.; Nadig, T.; Smyth, A.R.; Flume, P. Treatment of pulmonary exacerbations in cystic fibrosis. Curr. Opin. Pulm. Med. 2020, 26, 679–684. [Google Scholar] [CrossRef] [PubMed]
- VanDevanter, D.R.; Flume, P.A.; Morris, N.; Konstan, M.W. Probability of IV antibiotic retreatment within thirty days is associated with duration and location of IV antibiotic treatment for pulmonary exacerbation in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Abbott, L.; Plummer, A.; Hoo, Z.H.; Wildman, M. Duration of intravenous antibiotic therapy in people with cystic fibrosis. Cochrane Database Syst. Rev. 2019, 9, CD006682. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry. 2018 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MA, USA, 2019. [Google Scholar]
- Collaco, J.M.; Green, D.M.; Cutting, G.R.; Naughton, K.M.; Mogayzel, P.J. Location and duration of treatment of cystic fibrosis respiratory exacerbations do not affect outcomes. Am. J. Respir. Crit. Care Med. 2010, 182, 1137–1143. [Google Scholar] [CrossRef]
- Heltshe, S.L.; West, N.E.; VanDevanter, D.R.; Sanders, D.B.; Beckett, V.V.; Flume, P.A.; Goss, C.H.; Group, S.S. Study design considerations for the Standardized Treatment of Pulmonary Exacerbations 2 (STOP2): A trial to compare intravenous antibiotic treatment durations in CF. Contemp. Clin. Trials 2018, 64, 35–40. [Google Scholar] [CrossRef]
- Plummer, A.; Wildman, M. Duration of intravenous antibiotic therapy in people with cystic fibrosis. Cochrane Database Syst. Rev. 2013, CD006682. [Google Scholar] [CrossRef]
- Schelstraete, P.; Haerynck, F.; Van daele, S.; Deseyne, S.; De Baets, F. Eradication therapy for Pseudomonas aeruginosa colonization episodes in cystic fibrosis patients not chronically colonized by P. aeruginosa. J. Cyst. Fibros. 2013, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Flume, P.A.; O’Sullivan, B.P.; Robinson, K.A.; Goss, C.H.; Mogayzel, P.J.; Willey-Courand, D.B.; Bujan, J.; Finder, J.; Lester, M.; Quittell, L.; et al. Cystic fibrosis pulmonary guidelines: Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2007, 176, 957–969. [Google Scholar] [CrossRef]
- Ryan, G.; Singh, M.; Dwan, K. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2011, CD001021. [Google Scholar] [CrossRef]
- Heijerman, H.; Westerman, E.; Conway, S.; Touw, D.; Döring, G.; Gerd Döring for the Consensus Working Group. Inhaled medication and inhalation devices for lung disease in patients with cystic fibrosis: A European consensus. J. Cyst. Fibros. 2009, 8, 295–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girón Moreno, R.M.; Salcedo Posadas, A.; Mar Gómez-Punter, R. Inhaled antibiotic therapy in cystic fibrosis. Arch. Bronconeumol. 2011, 47 (Suppl. 6), 14–18. [Google Scholar] [CrossRef]
- Smith, S.; Rowbotham, N.J.; Regan, K.H. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 3, CD001021. [Google Scholar] [CrossRef]
- Robinson, T.E.; Leung, A.N.; Chen, X.; Moss, R.B.; Emond, M.J. Cystic fibrosis HRCT scores correlate strongly with Pseudomonas infection. Pediatr. Pulmonol. 2009, 44, 1107–1117. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Pepe, M.S.; Quan, J.M.; Otto, K.L.; Montgomery, A.B.; Williams-Warren, J.; Vasiljev, K.M.; Borowitz, D.; Bowman, C.M.; Marshall, B.C.; et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N. Engl. J. Med. 1999, 340, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assael, B.M.; Pressler, T.; Bilton, D.; Fayon, M.; Fischer, R.; Chiron, R.; LaRosa, M.; Knoop, C.; McElvaney, N.; Lewis, S.A.; et al. Inhaled aztreonam lysine vs. inhaled tobramycin in cystic fibrosis: A comparative efficacy trial. J. Cyst. Fibros. 2013, 12, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Lo, D.; VanDevanter, D.R.; Flume, P.; Smyth, A. Aerosolized antibiotic therapy for chronic cystic fibrosis airway infections: Continuous or intermittent? Respir. Med. 2011, 105 (Suppl. 2), S9–S17. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.; Pedersen, S.S.; Garne, S.; Heilmann, C.; Høiby, N.; Koch, C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J. Antimicrob. Chemother. 1987, 19, 831–838. [Google Scholar] [CrossRef]
- Hodson, M.E.; Gallagher, C.G.; Govan, J.R. A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur. Respir. J. 2002, 20, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.D.; Anbar, R.D.; Lester, L.A.; Nasr, S.Z.; Nickerson, B.; VanDevanter, D.R.; Colin, A.A. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatr. Pulmonol. 2004, 38, 314–320. [Google Scholar] [CrossRef]
- Sawicki, G.S.; Signorovitch, J.E.; Zhang, J.; Latremouille-Viau, D.; von Wartburg, M.; Wu, E.Q.; Shi, L. Reduced mortality in cystic fibrosis patients treated with tobramycin inhalation solution. Pediatr. Pulmonol. 2012, 47, 44–52. [Google Scholar] [CrossRef]
- McCoy, K.S.; Quittner, A.L.; Oermann, C.M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Montgomery, A.B. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, A.; Haliburn, C.; Döring, G.; Goldman, M.H.; Group, F.S. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: A randomised study. Thorax 2013, 68, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstan, M.W.; Geller, D.E.; Minić, P.; Brockhaus, F.; Zhang, J.; Angyalosi, G. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: The EVOLVE trial. Pediatr. Pulmonol. 2011, 46, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstan, M.W.; Flume, P.A.; Kappler, M.; Chiron, R.; Higgins, M.; Brockhaus, F.; Zhang, J.; Angyalosi, G.; He, E.; Geller, D.E. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J. Cyst. Fibros. 2011, 10, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassetti, M.; Vena, A.; Russo, A.; Peghin, M. Inhaled Liposomal Antimicrobial Delivery in Lung Infections. Drugs 2020, 80, 1309–1318. [Google Scholar] [CrossRef]
- Elhissi, A. Liposomes for Pulmonary Drug Delivery: The Role of Formulation and Inhalation Device Design. Curr. Pharm. Des. 2017, 23, 362–372. [Google Scholar] [CrossRef]
- Kapnadak, S.G.; Dimango, E.; Hadjiliadis, D.; Hempstead, S.E.; Tallarico, E.; Pilewski, J.M.; Faro, A.; Albright, J.; Benden, C.; Blair, S.; et al. Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease. J. Cyst. Fibros. 2020, 19, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.; Flume, P.A. Pulmonary Complications of Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Prados, C.; Máiz, L.; Antelo, C.; Baranda, F.; Blázquez, J.; Borro, J.M.; Gartner, S.; Garzón, G.; Girón, R.; de Gracia, J.; et al. Cystic fibrosis: Consensus on the treatment of pneumothorax and massive hemoptysis and on the indications for lung transplantation. Arch. Bronconeumol. 2000, 36, 411–416. [Google Scholar] [CrossRef]
- Quintana-Gallego, E.; Delgado-Pecellín, I.; Calero Acuña, C. CFTR protein repair therapy in cystic fibrosis. Arch. Bronconeumol. 2014, 50, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M.; VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: A double-blind, randomised controlled trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.J.; Krainer, G.; Ng, D.R.S.; Schenkel, M.; Shishido, H.; Yoon, J.S.; Haggie, P.M.; Schlierf, M.; Sheppard, D.N.; Skach, W.R. Towards next generation therapies for cystic fibrosis: Folding, function and pharmacology of CFTR. J. Cyst. Fibros. 2020, 19 (Suppl. 1), S25–S32. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- McKone, E.F.; Borowitz, D.; Drevinek, P.; Griese, M.; Konstan, M.W.; Wainwright, C.; Ratjen, F.; Sermet-Gaudelus, I.; Plant, B.; Munck, A.; et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: A phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2014, 2, 902–910. [Google Scholar] [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Tian, S.; Schneider, J.; Cunningham, S.; Davies, J.C.; et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): A phase 3 single-arm study. Lancet Respir. Med. 2018, 6, 545–553. [Google Scholar] [CrossRef]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Jaques, R.; Shakeel, A.; Hoyle, C. Novel therapeutic approaches for the management of cystic fibrosis. Multidiscip. Respir. Med. 2020, 15, 690. [Google Scholar] [CrossRef]
- Elborn, J.S.; Ramsey, B.W.; Boyle, M.P.; Konstan, M.W.; Huang, X.; Marigowda, G.; Waltz, D.; Wainwright, C.E.; VX-809 TRAFFIC and TRANSPORT study groups. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: A pooled analysis. Lancet Respir. Med. 2016, 4, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Diab-Cáceres, L.; Girón-Moreno, R.M.; Pastor-Sanz, M.T.; Quintana-Gallego, E.; Delgado-Pecellín, I.; Blanco-Aparicio, M.; Maiz, L.; García-Clemente, M.M.; Luna-Paredes, C.; Mondéjar-López, P.; et al. Compassionate Use of Lumacaftor/Ivacaftor in Cystic Fibrosis: Spanish Experience. Arch. Bronconeumol. 2018, 54, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Munck, A.; Durieu, I.; Chiron, R.; Mely, L.; Prevotat, A.; Murris-Espin, M.; Porzio, M.; Abely, M.; Reix, P.; et al. Real-Life Safety and Effectiveness of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 188–197. [Google Scholar] [CrossRef]
- McNamara, J.J.; McColley, S.A.; Marigowda, G.; Liu, F.; Tian, S.; Owen, C.A.; Stiles, D.; Li, C.; Waltz, D.; Wang, L.T.; et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2-5 years with cystic fibrosis homozygous for F508del-CFTR: An open-label phase 3 study. Lancet Respir. Med. 2019, 7, 325–335. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Nichols, D.P.; Donaldson, S.H.; Frederick, C.A.; Freedman, S.D.; Gelfond, D.; Hoffman, L.R.; Kelly, A.; Narkewicz, M.R.; Pittman, J.E.; Ratjen, F.; et al. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A.M.; Vortherms, T.; et al. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid (ABBV/GLPG-2222), a Potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for the Treatment of Cystic Fibrosis. J. Med. Chem. 2018, 61, 1436–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, S.H.; Solomon, G.M.; Zeitlin, P.L.; Flume, P.A.; Casey, A.; McCoy, K.; Zemanick, E.T.; Mandagere, A.; Troha, J.M.; Shoemaker, S.A.; et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros. 2017, 16, 371–379. [Google Scholar] [CrossRef]
- Kazani, S.; Rowlands, D.J.; Bottoli, I.; Milojevic, J.; Alcantara, J.; Jones, I.; Kulmatycki, K.; Machineni, S.; Mostovy, L.; Nicholls, I.; et al. Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251). J. Cyst. Fibros. 2021, 20, 250–256. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Keeling, K.M.; Rowe, S.M. Pharmacological approaches for targeting cystic fibrosis nonsense mutations. Eur. J. Med. Chem. 2020, 200, 112436. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sermet-Gaudelus, I.; Renouil, M.; Fajac, A.; Bidou, L.; Parbaille, B.; Pierrot, S.; Davy, N.; Bismuth, E.; Reinert, P.; Lenoir, G.; et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: A pilot study. BMC Med. 2007, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; VanDevanter, D.R.; Rowe, S.M.; Wilschanski, M.; Kerem, E.; Sermet-Gaudelus, I.; DiMango, E.; Melotti, P.; McIntosh, J.; De Boeck, K.; et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J. Cyst. Fibros. 2020, 19, 595–601. [Google Scholar] [CrossRef]
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; van Duzer, J.; Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 984–994. [Google Scholar] [CrossRef]
- Kerem, E. ELX-02: An investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opin Investig Drugs 2020, 29, 1347–1354. [Google Scholar] [CrossRef]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Mutyam, V.; Du, M.; Xue, X.; Keeling, K.M.; White, E.L.; Bostwick, J.R.; Rasmussen, L.; Liu, B.; Mazur, M.; Hong, J.S.; et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. Am. J. Respir. Crit. Care Med. 2016, 194, 1092–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, B.R.; Pickles, R.J.; Ye, H.; Yankaskas, J.R.; Vick, R.N.; Engelhardt, J.F.; Wilson, J.M.; Johnson, L.G.; Boucher, R.C. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and humans. Nature 1994, 371, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C.; Knowles, M.R.; Johnson, L.G.; Olsen, J.C.; Pickles, R.; Wilson, J.M.; Engelhardt, J.; Yang, Y.; Grossman, M. Gene therapy for cystic fibrosis using E1-deleted adenovirus: A phase I trial in the nasal cavity. The University of North Carolina at Chapel Hill. Hum. Gene Ther. 1994, 5, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Choi, H.; Burda, J.; Chen, S.J.; Wilson, J.M. Recombinant adenovirus deleted of all viral genes for gene therapy of cystic fibrosis. Virology 1996, 217, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, J.F.; Simon, R.H.; Yang, Y.; Zepeda, M.; Weber-Pendleton, S.; Doranz, B.; Grossman, M.; Wilson, J.M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: Biological efficacy study. Hum. Gene Ther. 1993, 4, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, F.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: A randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef]
- Griesenbach, U.; Pytel, K.M.; Alton, E.W. Cystic Fibrosis Gene Therapy in the UK and Elsewhere. Hum. Gene Ther. 2015, 26, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Repeated Nebulisation of Non-Viral Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene Therapy in Patients with Cystic Fibrosis; NIHR Journals Library: Southampton, UK, 2016. [Google Scholar]
- Marangi, M.; Pistritto, G. Innovative Therapeutic Strategies for Cystic Fibrosis: Moving Forward to CRISPR Technique. Front. Pharmacol. 2018, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Flynn, R.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Correction of the ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Gene by Zinc-Finger Nuclease Homology-Directed Repair. BioRes. Open Access 2012, 1, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Economos, N.G.; Oyaghire, S.; Quijano, E.; Ricciardi, A.S.; Saltzman, W.M.; Glazer, P.M. Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules 2020, 25, 735. [Google Scholar] [CrossRef] [Green Version]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beumer, W.; Swildens, J.; Leal, T.; Noel, S.; Anthonijsz, H.; van der Horst, G.; Kuiperij-Boersma, H.; Potman, M.; van Putten, C.; Biasutto, P.; et al. Evaluation of eluforsen, a novel RNA oligonucleotide for restoration of CFTR function in in vitro and murine models of p.Phe508del cystic fibrosis. PLoS ONE 2019, 14, e0219182. [Google Scholar] [CrossRef]
- Drevinek, P.; Pressler, T.; Cipolli, M.; De Boeck, K.; Schwarz, C.; Bouisset, F.; Boff, M.; Henig, N.; Paquette-Lamontagne, N.; Montgomery, S.; et al. Antisense oligonucleotide eluforsen is safe and improves respiratory symptoms in F508DEL cystic fibrosis. J. Cyst. Fibros. 2020, 19, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Sermet-Gaudelus, I.; Clancy, J.P.; Nichols, D.P.; Nick, J.A.; De Boeck, K.; Solomon, G.M.; Mall, M.A.; Bolognese, J.; Bouisset, F.; den Hollander, W.; et al. Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J. Cyst. Fibros. 2019, 18, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, M.E. Emerging technologies for cystic fibrosis transmembrane conductance regulator restoration in all people with CF. Pediatr. Pulmonol. 2021, 56 (Suppl. 1), S32–S39. [Google Scholar] [CrossRef]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-β Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Miller, S.M.; Mascenik, T.M.; Lewis, C.A.; Dang, H.; Boggs, Z.H.; Tarran, R.; Randell, S.H. Assessing Human Airway Epithelial Progenitor Cells for Cystic Fibrosis Cell Therapy. Am. J. Respir. Cell Mol. Biol. 2020, 63, 374–385. [Google Scholar] [CrossRef]
- Berical, A.; Lee, R.E.; Randell, S.H.; Hawkins, F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 74. [Google Scholar] [CrossRef]
- Huang, S.X.; Green, M.D.; de Carvalho, A.T.; Mumau, M.; Chen, Y.W.; D’Souza, S.L.; Snoeck, H.W. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, S.; Salahudeen, A.A.; Sellers, Z.M.; Bravo, D.T.; Choi, S.S.; Batish, A.; Le, W.; Baik, R.; de la, O.S.; Kaushik, M.P.; et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26, 161–171.e164. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment of Obstruction |
| Manual and instrumental physiotherapy |
| Physical exercise |
| Bronchodilators |
| Mucolytics: human dornase alfa |
| Hypertonic substances: 6 or 7% saline/Mannitol |
| Treatment of Inflammation |
| Oral/inhaled corticosteroids (ICS) |
| Ibuprofen |
| Azithromycin |
| Treatment of Infection |
| Treatment of initial colonization by Pseudomonas aeruginosa |
| Treatment of other pathogens |
| Chronic maintenance treatment |
| Treatment of exacerbations |
| Treatment of Chronic Respiratory Failure |
| Oxygen therapy |
| Noninvasive mechanical ventilation (transplant bridge) |
| Pulmonary transplant |
| Goal of the Clinical Trials | ClinicalTrials.gov Identifier | Study Phase | Status | Results | Country |
|---|---|---|---|---|---|
| Continuous Azithromycin in CF patients beyond two years (AZITHRO) | NCT02803944 | Phase 4 | Completed | Not available | France |
| Effect of Azithromycin on Lung Function in 6-18 years old with CF Not Infected with P. aeruginosa | NCT00431964. | Phase 4 | Completed | Available | EEUU |
| Scandinavian CF Azithromycin Study | NCT00411736 | Phase 4 | Completed | Not available | Denmark Norway Sweden |
| Effect of Azithromycin on Fatty Acids in CF | NCT03045198 | Phase 4 | Unknown | Not available | Germany |
| Testing the effect of adding chronic oral azithromycin to inhaled tobramycin in people with CF (TEACH) | NCT02677701. | Phase 4 | Completed | Not available | USA |
| Azithromycin in patients with CF, infected with Burkholderia cepacia complex | NCT00298922 | Phase 2 | Unknown | Not available | Canada |
| Prevention of bronchiectasis in infants with CF (COMBATCF) | NCT01270074 | Phase 3 | Active | Not available | USA |
| OPTIMIZing treatment for early Pseudomonas aeruginosa infection in Cystic Fibrosis. | NCT02054156 | Phase 3 | Completed | Available | USA |
| Inhaled Antibiotics | Dose/Posology | Inhalation System |
| Colistimethate solution for inhalation | 2 million U/12 h (1 million = 80 mg) BID Continue | e-Flow® Pari LC |
| Colistimethate dry powder for inhalation | 1,662,500 U/12 h (125 mg) BID Continue | Turbospin® |
| Tobramycin solution for inhalation | 300 mg/4–5 mL/12 h BID On-off cycles 28 days | e-Flow® Pari LC plus® |
| Tobramycin dry powder for inhalation | 300 mg/12 h BID On-off cycles 28 days | T-326 inhalator® |
| Aztreonam lysine solution for inhalation | 75 mg/8 h TID On-Off cycles 28 days | e-Flow® (Altera) |
| Amikacin | 400 mg/24 h On-off cycles 28 days | e-Flow® (Lamira) |
| Levofloxacin | 240 mg/12h On-off cycles 28 days | e-Flow®(Zirela) |
| Gentamicin | 80 mg/12 h BID | Pari LC |
| Title | Clinical Trial | Product | Study Phase |
|---|---|---|---|
| A Study of the Safety and Tolerability of inhaled SNSP113 in Healthy Subjects and Subjects with Stable CF | NCT03309358 | SNSP113 | Phase 1 |
| Dose Escalation Study of ALX-009 in Healthy Men and CF and Non-CF Bronchiectasis Patients | NCT02598999 | ALX-009 | Phase 1 |
| SPI-1005 for Prevention and Treatment of Tobramycin Induced Ototoxicity | NCT02819856 | SPI-1005 | Phase 2 |
| Study to evaluate inhaled AP-PA02 in adults with CF and chronic Pseudomonas aeruginosa (Armata Phase 1b/2 SAD) (Armata AP-PA02-101) | NCT04596319 | AP-PA02 | Phase 1 Phase 2 |
| SAD and MAD of Inhaled AR-501 in Health Adults and P. Aeruginosa Infected CF Subjects | NCT03669614 | AR-501 Inhaled gallium | Phase 1 Phase 2 |
| A Phase 2 IV Gallium Study for Patients with CF (IGNITE Study) | NCT02354859 | Intravenous gallium | Phase 2 |
| IV Gallium Study for Patients with CF who have NTM (ABATE Study) (ABATE) | NCT04294043 | Intravenous gallium | Phase 1 |
| Phase 2 study of inhaled nitric oxide in people with CF (Novoteris NO-CF-02E) | NCT02498535 | Inhaled Nitric Oxide (NO) | Phase 2 |
| Antibiotic | Title | Clinical Trial | Study Phase | Results |
|---|---|---|---|---|
| Amikacin liposome inhalation suspension (Arikayce) | Safety/Tolerability Study of Arikayce™ in CF Patients with Chronic Infection Due to Pseudomonas aeruginosa | NCT00558844 | Phase 1 Phase 2 |
Additionally, Liposomal Amikacin was associated with improvement in lung function and reduction in Pseudomonas aeruginosa density. No more frequency of adverse events |
| Study to Evaluate Arikayce™ in CF Patients with Chronic Pseudomonas aeruginosa infection | NCT01315678 | Phase 3 | This study found that the drug Arikayce® was comparable to the approved drug TOBI® (Tobramycin Solution for Inhalation) | |
| Aztreonam for inhalation solution (AZLI) | International Safety and Efficacy Study of Aztreonam for Inhalation Solution (AZLI) in CF Patients with P. aeruginosa (AIR-CF1). | NCT00112359 | Phase 3 | After 28-days treatment, AZLI improved mean CFQ-R (Cystic Fibrosis Questionnaire-Revised)-Respiratory scores (9.7 points, p < 0.001) compared with placebo. Adverse events for AZLI and placebo were comparable |
| Safety and Efficacy Study of Aztreonam for Inhalation Solution (AZLI) in CF Patients with P. Aeruginosa (AIR-CF2) | NCT00104520 | Phase 3 | AZLI also improved mean CFQ-R Respiratory scores (5.01 points, p = 0.02), improved FEV1 (6.3%, p = 0.001), and decreased sputum PA density (−0.66 log10 CFU/gram, p = 0.006) compared with placebo. No difference in adverse events | |
| Safety and Efficacy Study of Aztreonam for Inhalation Solution (AZLI) in CF Patients with Pseudomonas aeruginosa (PA) (AIR-CF3) | NCT00128492 | Phase 3 | Patients who received AZLI three times a day had greater improvement in FEV1 and in patient reported outcomes (CFQ-R) | |
| Inhaled levofloxacin (Quinsair TM) | Trial of Aeroquin Versus Tobramycin Inhalation Solution (TIS) in CF Patients (TIS) | NCT01270347 | Phase 3 | Study results showed that levofloxacin was not inferior to inhaled tobramycin as measured by lung function. The adverse event profile was similar for both the inhaled levofloxacin and tobramycin solution for inhalation groups; however, levofloxacin treated participants complained more frequently about the taste of the medication |
| MP-376 (Aeroquin™, Levofloxacin for Inhalation) in Patients with CF | NCT01180634 | Phase 3 | Inhaled levofloxacin was generally well-tolerated; however, the study did not demonstrate a benefit after 28 days of treatment on reducing or delaying pulmonary exacerbations | |
| Safety, Tolerability and Pharmacokinetics of MP-376 Administered for 14 Days to Stable Paediatric (CF) Patients | NCT00840333 | Phase 1 |
Closed to enrolment No results yet |
| CFTR Modulator | Country | Mutations | Age | Adverse Events | Expansion Label Studies |
|---|---|---|---|---|---|
| Ivacaftor Kalydeco® | FDA | 711 + 3A→G, F311del, I148T, R75Q, S589N, 2789 + 5G→A, F311L, I175V, R117C, S737F, 3272-26A→G, F508C, I807M, R117G, S945L, 3849 + 10kbC→T, F508C, I1027T, S977F, A120T, F1052V, I1139V, R117L, S1159F, A234D, F1074L, K1060T, R117P, S1159P, A349V, G178E, L206W, R170H, A455E, L320V, R347H, S1255P, A1067T, G194R, L967S, R347L, T338I, D110E, G314E, L997F, R352Q, T1053I, D110H, L1480P, R553Q, V232D, D192G, M152V, R668C, V562I, D579G, G576A, M952I, R792G, V754M, D924N, G970D, M952T, R933G, V1293G, D1152H, G1069R, P67L, R1070Q, W1282, D1270N, Q237E, R1070W, Y1014C, E56K, G1249R, Q237H, R1162L, Y1032C, E193K, Q359R, R1283M, E822K, H939R, Q1291R, E831X, H1375P, R74W | >12 months | Sore throat Increases in transaminase levels | |
| G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, G1349D, R117H. | 4 Months | ||||
| EMA | R117H, G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R. | 6 months | |||
| Lumacaftor/Ivacaftor Orkambi® | FDA and EMA | F508del homozygous | >2 years old | Elevated alanine or aspartate aminotransferase levels. Chest pain Dyspnea | 1–2 years old (On going) |
| Tezacaftor/Ivacaftor Symdeko® Symkevi® | FDA | F508del homozygous Have a single copy: A455E, E56K, R74W, A1067T, E193K, R117C, D110E, F1052V, R347H, D110H, F1074L, R352Q, D579G, K1060T, R1070W, D1152H, L206W, S945L, D1270N, P67L, S977F, E831X, 711 + 3A→G, 3272-26A→G, 2789 + 5G→A, 3849 + 10kbC→T | >6 years old | Headache Nasopharyngitis Elevated alanine or aspartate aminotransferase levels. | |
| EMA | F508del homozygous F508del heterozygous with one of this: P67L, R117C, L206W, R352Q, A455E, D579G, 711þ3A > G, S945L, S977F, R1070W, D1152H, 2789þ5G > A, 3272 26A > G, 3849þ10kbC > T | >12 years old | |||
| Elexacaftor/Tezacaftor/Ivacaftor Trikafta® Kaftrio® | FDA | F508del homozygous All F508del heterozygous | >12 years old | Rash Elevated alanine or aspartate aminotransferase levels. Headache Diarrhea | 2–5 years old (On going) 6–11 years old (Completed) |
| EMA | F508del homozygous F508del heterozygous with minimal function: G542X, W1282X, R553X, R1162X, 621 + 1G→T, 1717-1G→A, 1898 + 1G→A, 3659delC, 394delTT, CFTRdele2,3, N1303K, I507del, G85E, R347P, R560T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girón Moreno, R.M.; García-Clemente, M.; Diab-Cáceres, L.; Martínez-Vergara, A.; Martínez-García, M.Á.; Gómez-Punter, R.M. Treatment of Pulmonary Disease of Cystic Fibrosis: A Comprehensive Review. Antibiotics 2021, 10, 486. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10050486
Girón Moreno RM, García-Clemente M, Diab-Cáceres L, Martínez-Vergara A, Martínez-García MÁ, Gómez-Punter RM. Treatment of Pulmonary Disease of Cystic Fibrosis: A Comprehensive Review. Antibiotics. 2021; 10(5):486. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10050486
Chicago/Turabian StyleGirón Moreno, Rosa María, Marta García-Clemente, Layla Diab-Cáceres, Adrián Martínez-Vergara, Miguel Ángel Martínez-García, and Rosa Mar Gómez-Punter. 2021. "Treatment of Pulmonary Disease of Cystic Fibrosis: A Comprehensive Review" Antibiotics 10, no. 5: 486. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10050486