
Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus aureus: An In Silico Drug Discovery Study

 ,
,  , , , ,
, , , ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Validation of In Silico Protocol
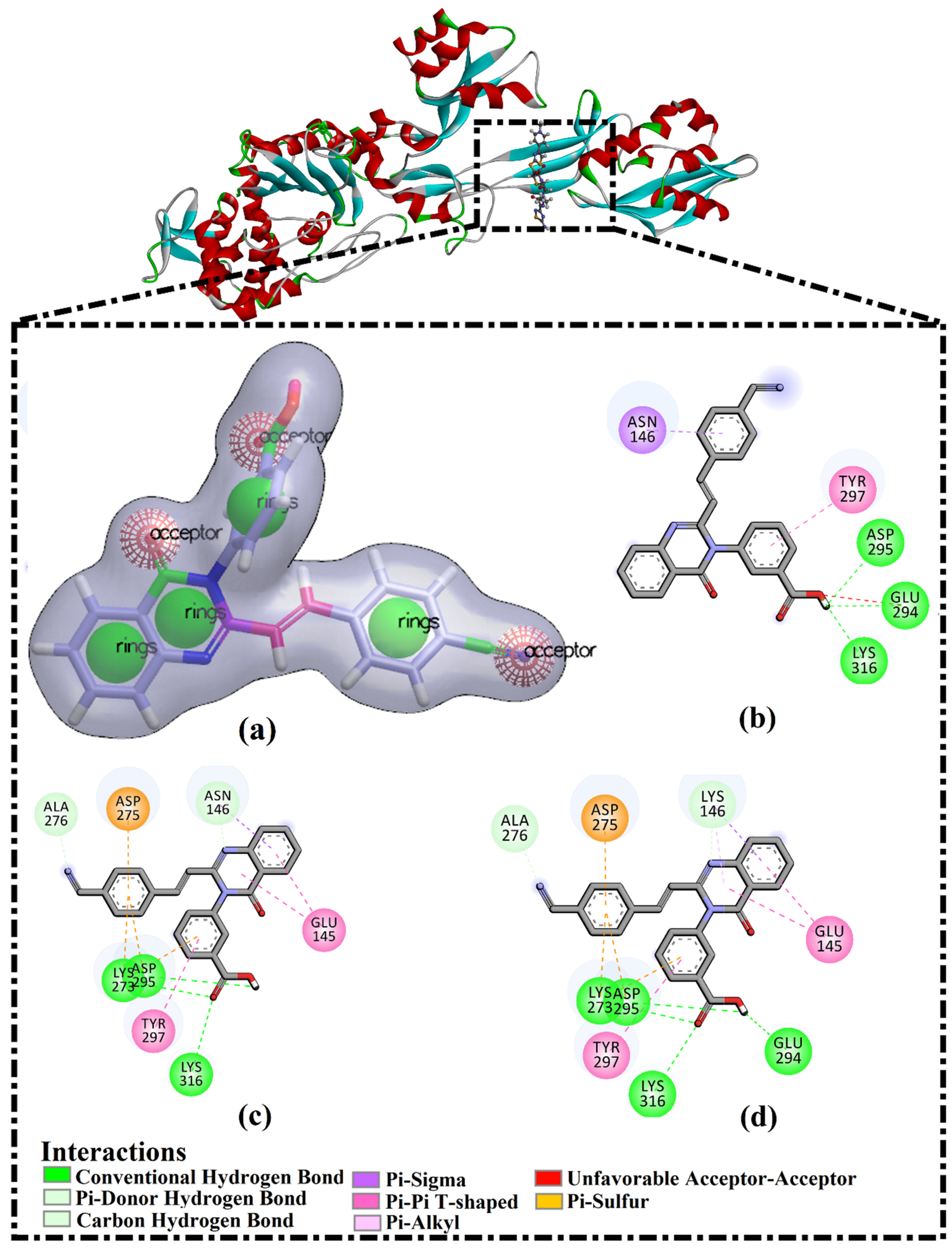
2.2. QNZ Complexed with Wild and Mutated PBP2a
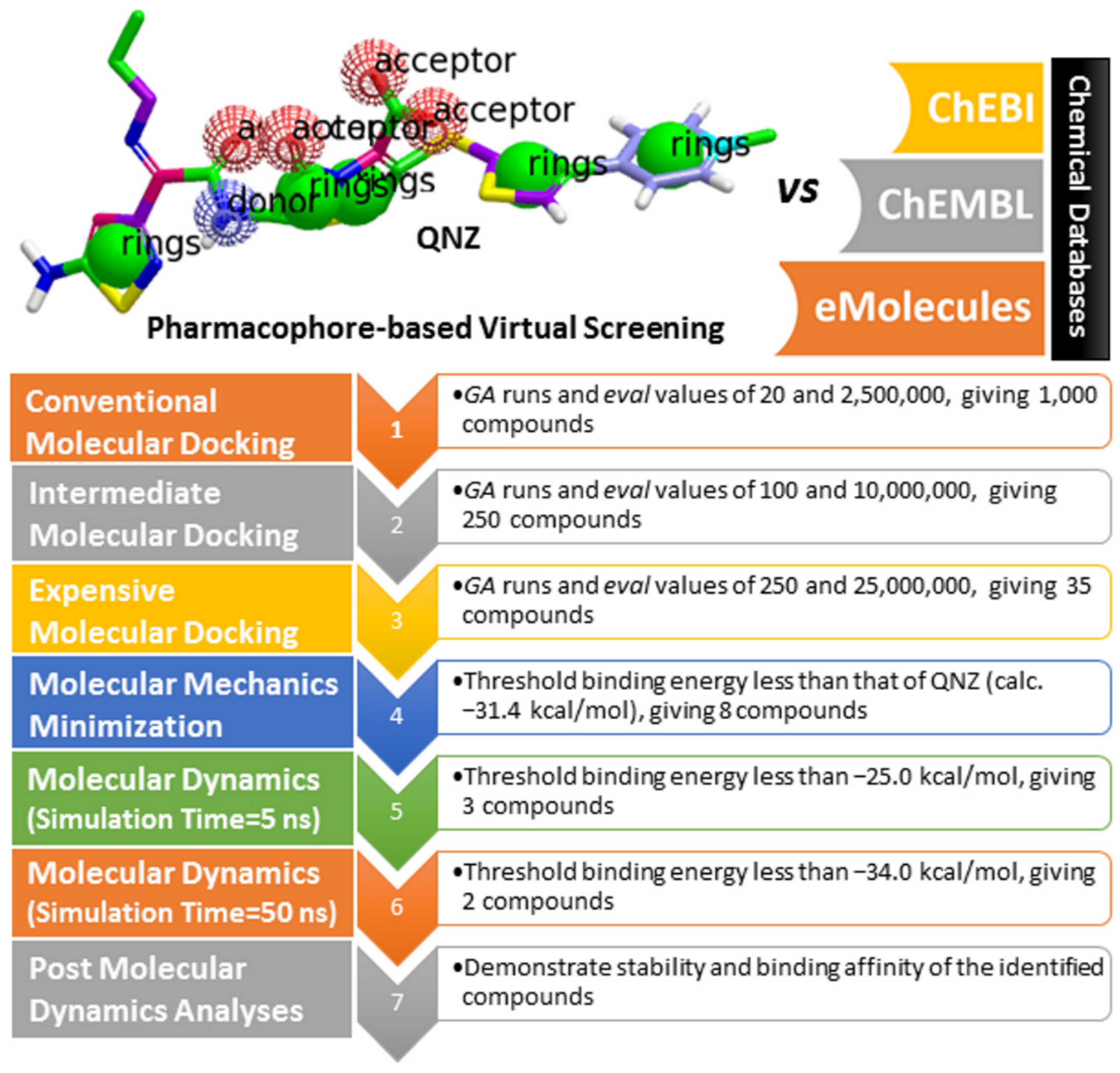
2.3. Pharmacophore-Based Virtual Screening
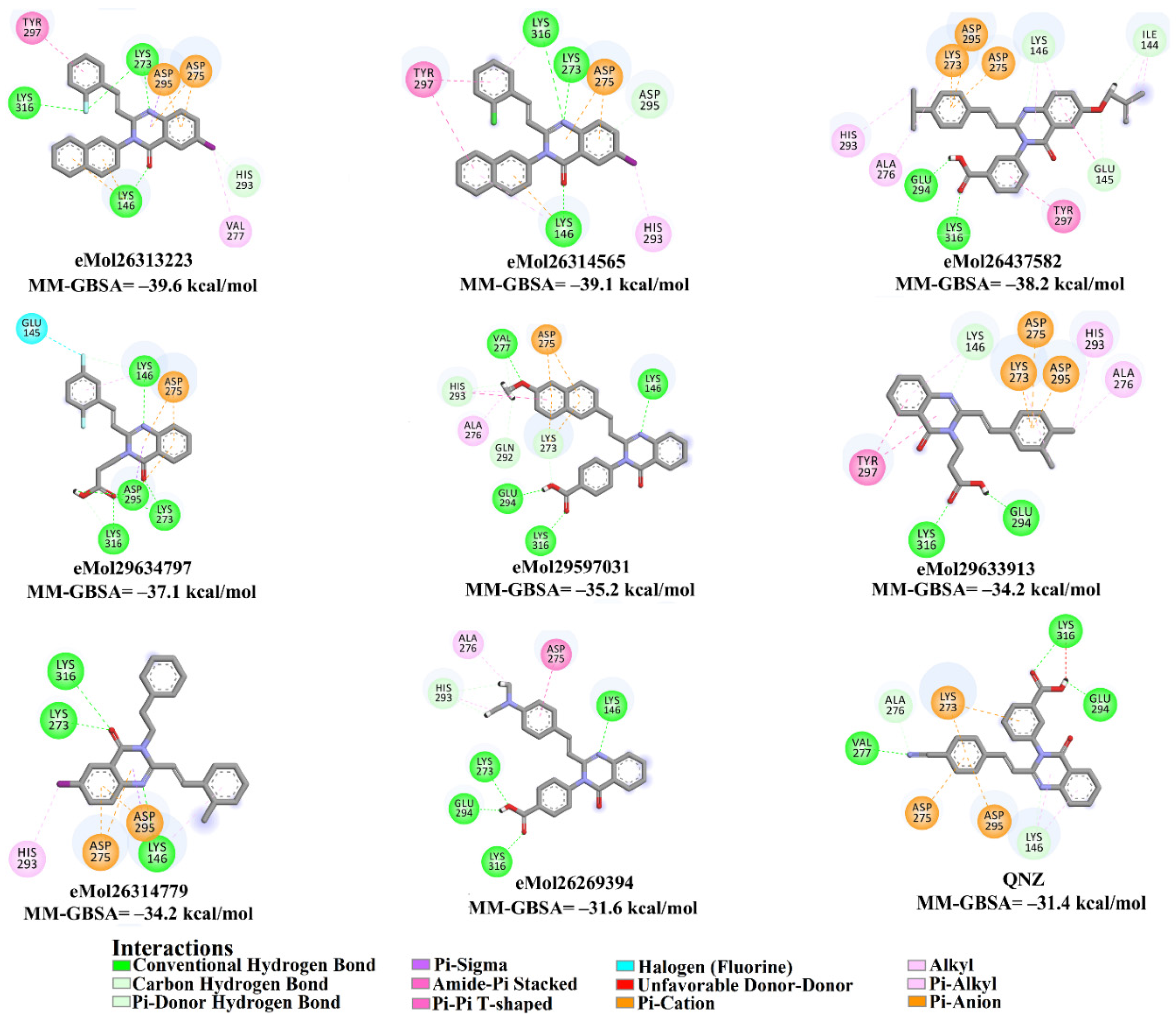
2.4. Database Filteration
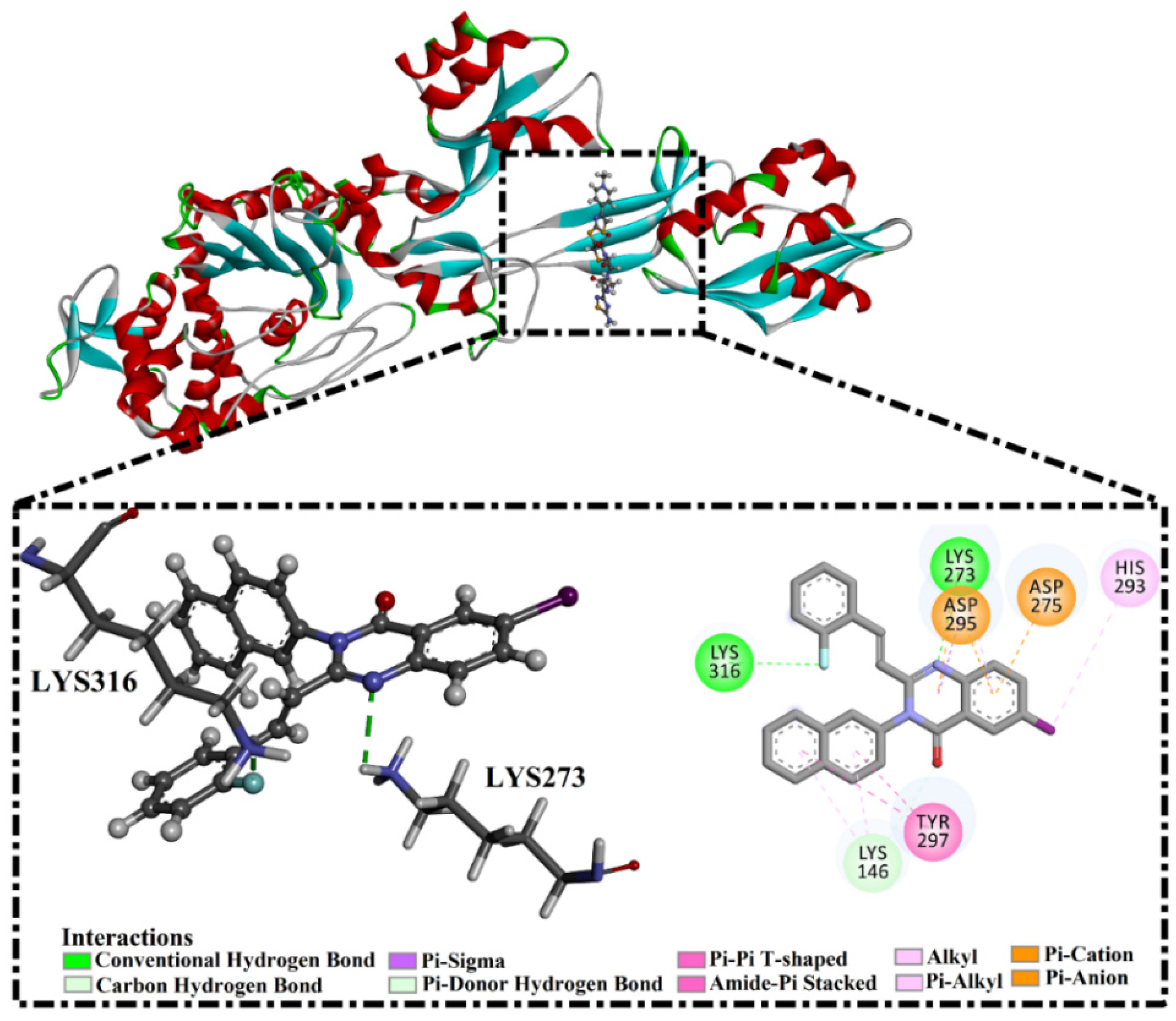
2.5. Inhibitor-PBP2a Complex Minimization
2.6. Molecular Dynamics (MD) Simulations
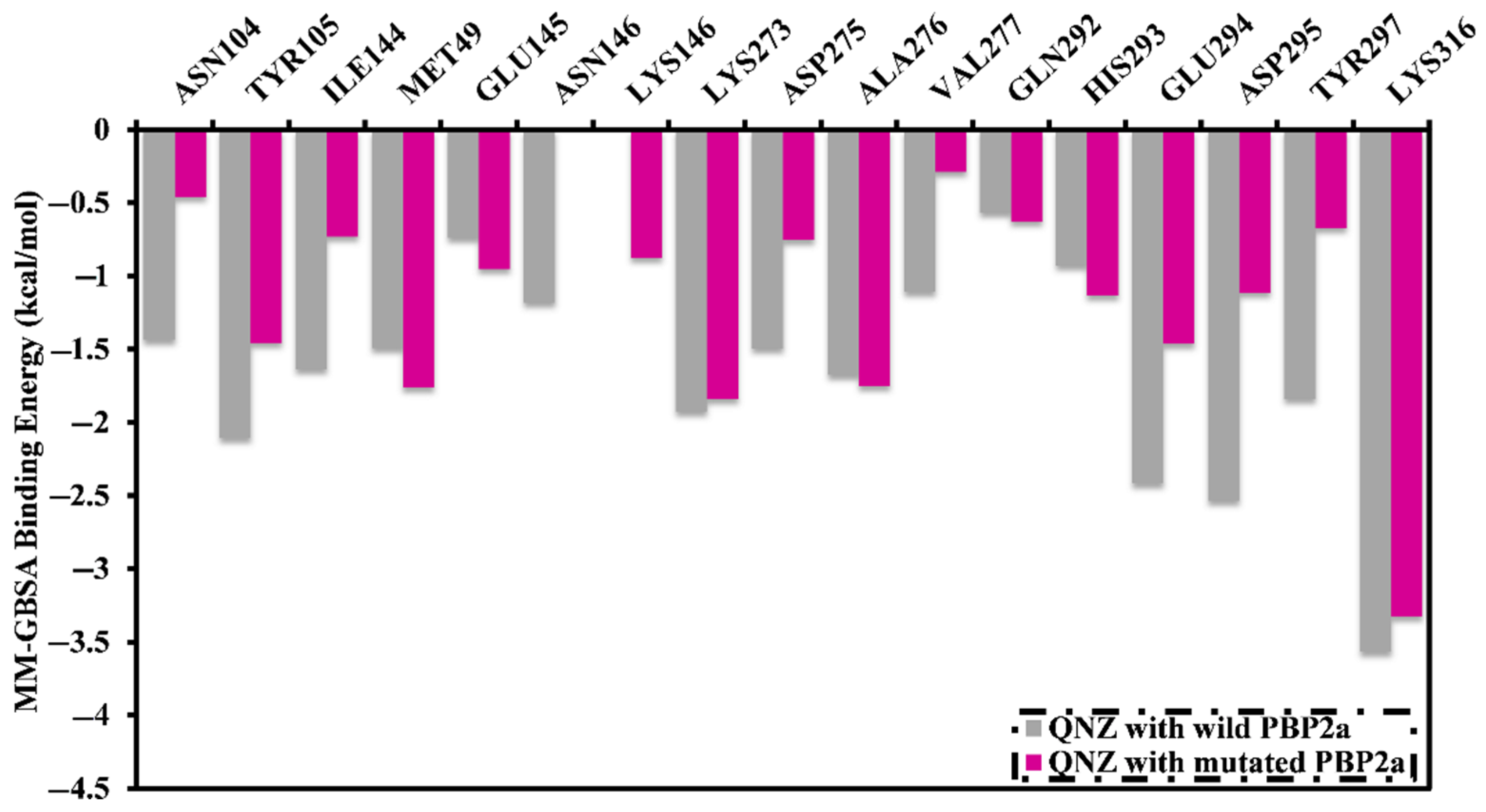
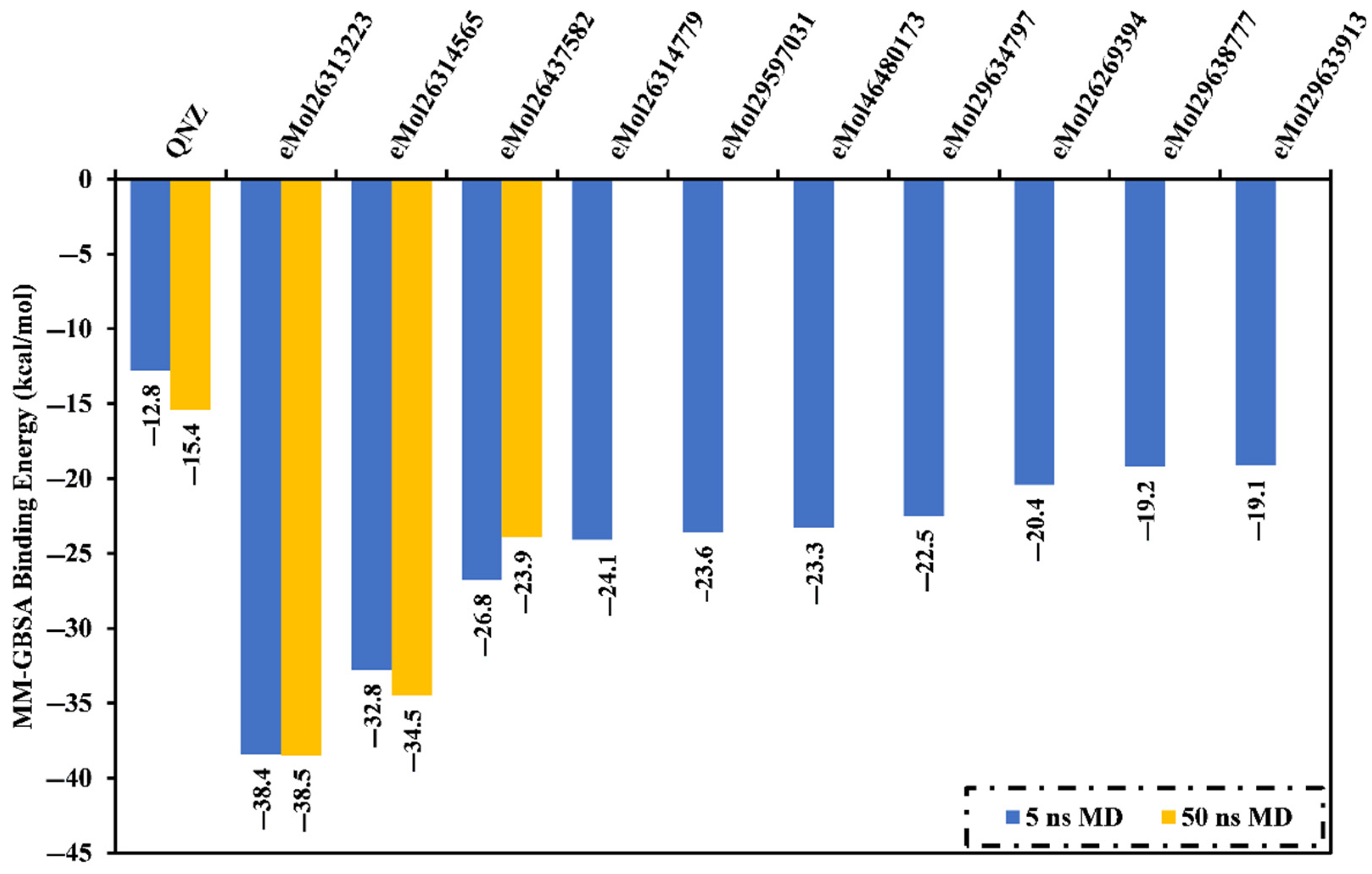
2.7. Post-Dynamics Analyses
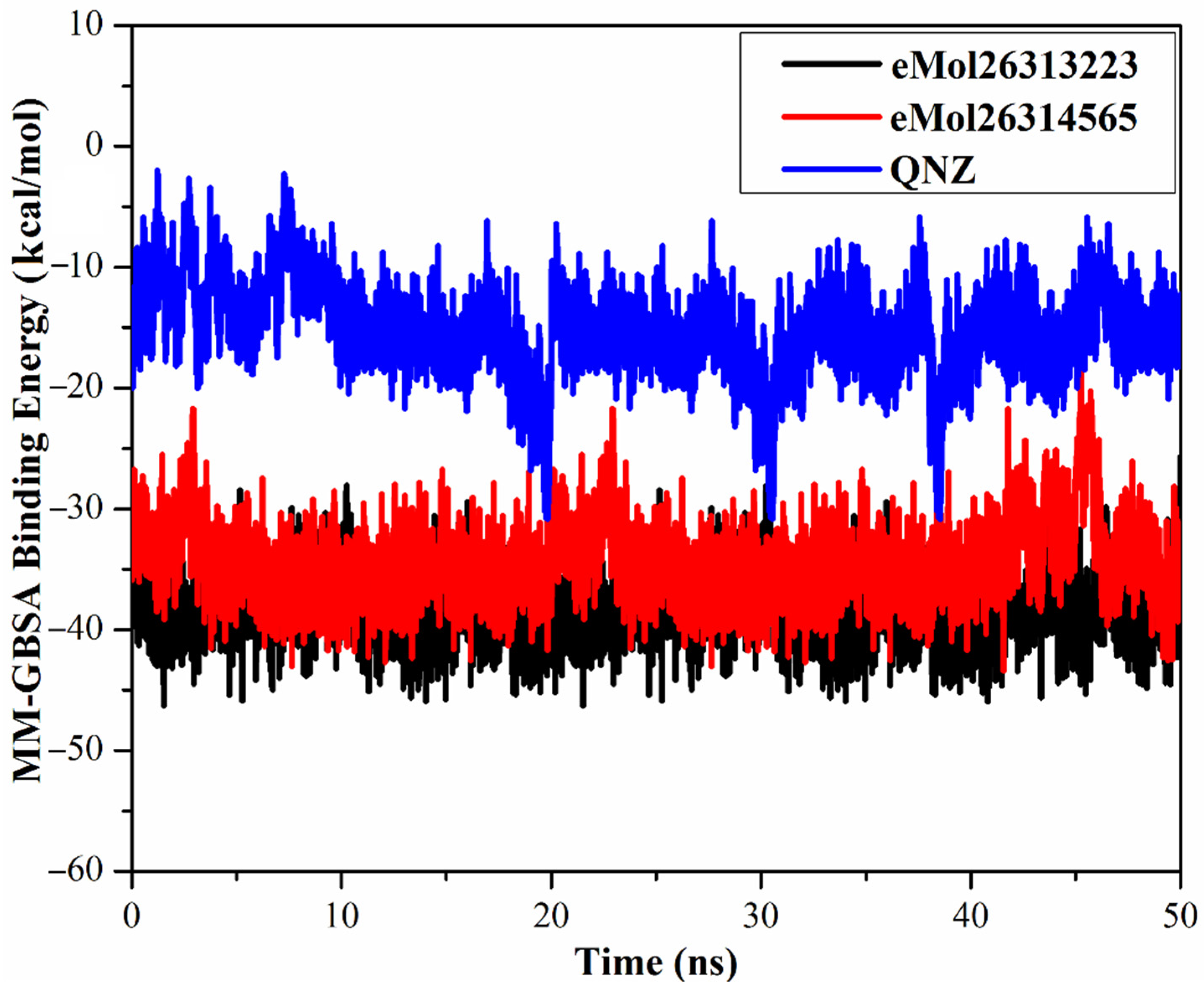
2.7.1. Binding Energy per Frame
2.7.2. Hydrogen Bond Analysis
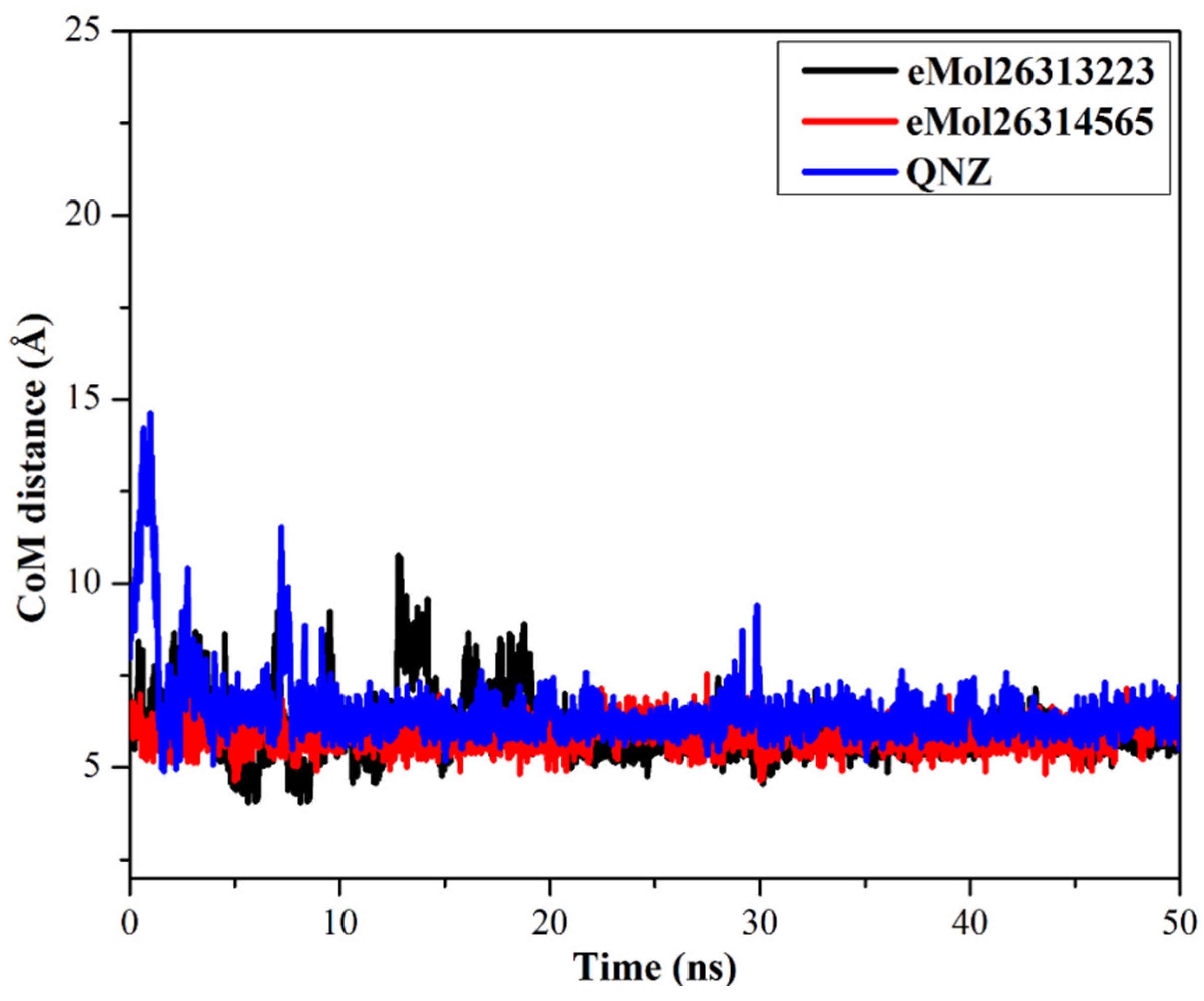
2.7.3. Center-of-Mass Distance
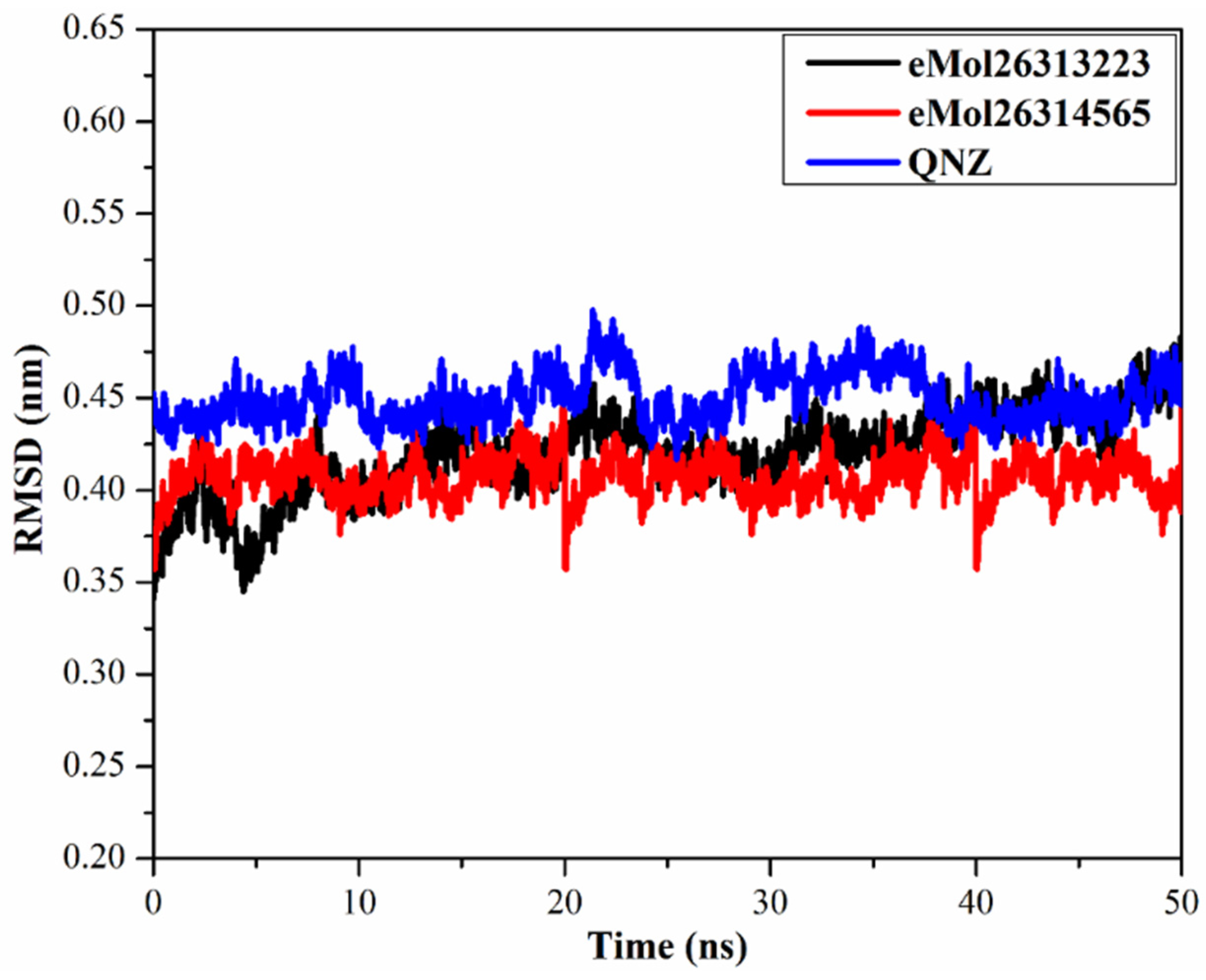
2.7.4. Root-Mean-Square Deviation
2.8. In Silico ADMET Analysis
3. Computational Methodology
3.1. PBP2a Preparation
3.2. Resolved PBP2a Allosteric Inhibitors
3.3. Pharmacophore-Based Virtual Screening
3.4. Molecular Docking
3.5. Inhibitor-PBP2a Complex Minimization
3.6. Molecular Dynamics Simulations
3.7. MM-GBSA Binding Energy
3.8. In Silico ADMET Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kadariya, J.; Smith, T.C.; Thapaliya, D. Staphylococcus aureus and staphylococcal food-borne disease: An ongoing challenge in public health. BioMed Res. Int. 2014, 2014, 827965. [Google Scholar] [CrossRef] [Green Version]
- Ansari, S.; Nepal, H.P.; Gautam, R.; Rayamajhi, N.; Shrestha, S.; Upadhyay, G.; Acharya, A.; Chapagain, M.L. Threat of drug resistant Staphylococcus aureus to health in Nepal. BMC Infect. Dis. 2014, 14, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Chambers, H.F. Methicillin resistance in staphylococci: Molecular and biochemical basis and clinical implications. Clin. Microbiol. Rev. 1997, 10, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Enright, M.C.; Robinson, D.A.; Randle, G.; Feil, E.J.; Grundmann, H.; Spratt, B.G. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. USA 2002, 99, 7687–7692. [Google Scholar] [CrossRef] [Green Version]
- Jevons, M.P. “Celbenin”-resistant staphylococci. Br. Med. J. 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Defres, S.; Marwick, C.; Nathwani, D. MRSA as a cause of lung infection including airway infection, community-acquired pneumonia and hospital-acquired pneumonia. Eur. Respir. J. 2009, 34, 1470–1476. [Google Scholar] [CrossRef]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis. 2011, 52, e18–e55. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.; Smith, D.L.; Laxminarayan, R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 2007, 13, 1840–1846. [Google Scholar] [CrossRef]
- Lyon, B.R.; Skurray, R. Antimicrobial resistance of Staphylococcus aureus: Genetic basis. Microbiol. Rev. 1987, 51, 88–134. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, F.F.; McGehee, R.F., Jr.; Finland, M. Methicillin-resistant Staphylococcus aureus at Boston City Hospital. Bacteriologic and epidemiologic observations. N. Engl. J. Med. 1968, 279, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; et al. Complete genomes of two clinical Staphylococcus aureus strains: Evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 2004, 101, 9786–9791. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.; Basit, A.; Karim, A.M.; Lee, J.H.; Jeon, J.H.; Rehman, S.U.; Lee, S.H. Mutation-based antibiotic resistance mechanism in methicillin-resistant Staphylococcus aureus clinical isolates. Pharmaceuticals 2021, 14, 420. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Strynadka, N.C. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar]
- Pinho, M.G.; de Lencastre, H.; Tomasz, A. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. USA 2001, 98, 10886–10891. [Google Scholar] [CrossRef] [Green Version]
- Mahasenan, K.V.; Molina, R.; Bouley, R.; Batuecas, M.T.; Fisher, J.F.; Hermoso, J.A.; Chang, M.; Mobashery, S. Conformational dynamics in penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus, allosteric communication network and enablement of catalysis. J. Am. Chem. Soc. 2017, 139, 2102–2110. [Google Scholar] [CrossRef] [Green Version]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [Green Version]
- Fuda, C.; Hesek, D.; Lee, M.; Morio, K.; Nowak, T.; Mobashery, S. Activation for catalysis of penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus by bacterial cell wall. J. Am. Chem. Soc. 2005, 127, 2056–2057. [Google Scholar] [CrossRef]
- Fishovitz, J.; Rojas-Altuve, A.; Otero, L.H.; Dawley, M.; Carrasco-Lopez, C.; Chang, M.; Hermoso, J.A.; Mobashery, S. Disruption of allosteric response as an unprecedented mechanism of resistance to antibiotics. J. Am. Chem. Soc. 2014, 136, 9814–9817. [Google Scholar] [CrossRef] [Green Version]
- Bouley, R.; Kumarasiri, M.; Peng, Z.; Otero, L.H.; Song, W.; Suckow, M.A.; Schroeder, V.A.; Wolter, W.R.; Lastochkin, E.; Antunes, N.T.; et al. Discovery of antibiotic (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one. J. Am. Chem. Soc. 2015, 137, 1738–1741. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, S.I.; Chaudhari, H.K. Design, synthesis, in-silico studies and biological screening of quinazolinone analogues as potential antibacterial agents against MRSA. Bioorg. Med. Chem. 2019, 27, 2676–2688. [Google Scholar] [CrossRef]
- Bouley, R.; Ding, D.; Peng, Z.; Bastian, M.; Lastochkin, E.; Song, W.; Suckow, M.A.; Schroeder, V.A.; Wolter, W.R.; Mobashery, S.; et al. Structure-activity relationship for the 4(3H)-quinazolinone antibacterials. J. Med. Chem. 2016, 59, 5011–5021. [Google Scholar] [CrossRef] [PubMed]
- Gatadi, S.; Gour, J.; Shukla, M.; Kaul, G.; Das, S.; Dasgupta, A.; Malasala, S.; Borra, R.S.; Madhavi, Y.V.; Chopra, S.; et al. Synthesis of 1,2,3-triazole linked 4(3H)-Quinazolinones as potent antibacterial agents against multidrug-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2018, 157, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.; Tulkens, P.M.; Van Bambeke, F. Contrasting effects of acidic pH on the extracellular and intracellular activities of the anti-gram-positive fluoroquinolones moxifloxacin and delafloxacin against Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.A.A. AMBER empirical potential describes the geometry and energy of noncovalent halogen interactions better than advanced semiempirical quantum mechanical method PM6-DH2X. J. Phys. Chem. B 2012, 116, 3659–3669. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.; de Matos, P.; Dekker, A.; Ennis, M.; Harsha, B.; Kale, N.; Muthukrishnan, V.; Owen, G.; Turner, S.; Williams, M.; et al. The ChEBI reference database and ontology for biologically relevant chemistry: Enhancements for 2013. Nucleic Acids Res. 2013, 41, D456–D463. [Google Scholar] [CrossRef]
- ROCS 3.2.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2017.
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Rush, T.S., 3rd; Grant, J.A.; Mosyak, L.; Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 95–113. [Google Scholar] [CrossRef]
- Aanismaa, P.; Seelig, A. P-glycoprotein kinetics measured in plasma membrane vesicles and living cells. Biochemistry 2007, 46, 3394–3404. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and Toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Sams, C.; Cocker, J.; Lennard, M.S. Biotransformation of chlorpyrifos and diazinon by human liver microsomes and recombinant human cytochrome P450s (CYP). Xenobiotica 2004, 34, 861–873. [Google Scholar] [CrossRef]
- Motohashi, H.; Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013, 15, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Frisch, J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- SZYBKI, 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- OMEGA 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- QUACPAC, 1.7.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC international chemical identifier. J. Cheminform. 2015, 7, 23–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Morales, J.L.; Nocedal, J. Automatic preconditioning by limited memory quasi-Newton updating. SIAM J. Optim. 2000, 10, 1079–1096. [Google Scholar] [CrossRef] [Green Version]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Amiri, S.; Sansom, M.S.; Biggin, P.C. Molecular dynamics studies of AChBP with nicotine and carbamylcholine: The role of water in the binding pocket. Protein Eng. Des. Sel. 2007, 20, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle—An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y.J.M. ADMET-score—A comprehensive scoring function for evaluation of chemical drug-likeness. MedChemComm 2019, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Tian, S.; Li, Y.; Li, Q.; Xu, X.; Wang, J.; Hou, T. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J. Cheminform. 2012, 4, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBP2a | MM-GBSA//MM a (kcal/mol) | Calculated MM-GBSA Binding Energy (kcal/mol) b | ||||||
|---|---|---|---|---|---|---|---|---|
| ∆Evdw | ∆Eele | ∆EGB | ∆ESUR | ∆Ggas | ∆GSolv | ∆Gbinding | ||
| Wild | −30.5 | −25.3 | −9.4 | 22.5 | −3.2 | −34.6 | 19.3 | −15.3 |
| Mutated | −31.4 | −24.7 | −15.7 | 28.3 | −3.3 | −40.4 | 25.0 | −15.4 |
| Compound Name/Code | Chemical Structure | Docking Score (kcal/mol) | Compound Name/Code | Chemical Structure | Docking Score (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Conv. b | Inter. c | Exp. d | Conv. b | Inter. c | Exp. d | ||||
| QNZ |  | −6.9 | −8.0 | −8.3 | eMol29597031 |  | −9.2 | −9.3 | −9.3 |
| eMol26313223 |  | −8.3 | −8.4 | −10.0 | eMol26269394 |  | −8.5 | −8.7 | −9.1 |
| eMol26437582 |  | −8.2 | −8.9 | −9.9 | eMol27202760 |  | −8.0 | −8.1 | −9.1 |
| eMol26314565 |  | −8.2 | −8.2 | −9.6 | eMol26242018 |  | −8.0 | −8.8 | −9.0 |
| eMol26313117 |  | −8.9 | −9.4 | −9.5 | eMol29633913 |  | −7.8 | −8.6 | −8.9 |
| eMol26293960 |  | −8.1 | −9.2 | −9.4 | CHEMBL1215080 |  | −8.4 | −8.4 | −8.9 |
| eMol3021959 |  | −7.1 | −8.9 | −9.4 | eMol30017880 |  | −7.4 | −8.3 | −8.7 |
| eMol27252412 |  | −8.1 | −8.2 | −8.7 | eMol29634797 |  | −7.6 | −8.1 | −8.6 |
| eMol300094331 |  | −8.2 | −8.3 | −8.7 | eMol27202252 |  | −8.0 | −8.1 | −8.6 |
| eMol26264570 |  | −8.1 | −8.4 | −8.7 | eMol26262168 |  | −7.0 | −8.0 | −8.5 |
| eMol26319231 |  | −8.2 | −8.6 | −8.7 | CHEMBL1215082 |  | −7.4 | −8.0 | −8.5 |
| eMol299980544 |  | −8.2 | −8.2 | −8.6 | eMol26242042 |  | −7.6 | −8.5 | −8.5 |
| eMol27406062 |  | −7.8 | −8.3 | −8.6 | eMol300154219 |  | −7.3 | −8.3 | −8.5 |
| eMol29565259 |  | −7.8 | −8.2 | −8.6 | eMol27091498 |  | −8.0 | −8.1 | −8.4 |
| eMol27202248 |  | −8.4 | −8.3 | −8.6 | eMol26242026 |  | −7.8 | −8.2 | −8.4 |
| CHEMBL1215004 |  | −7.7 | −8.2 | −8.4 | eMol301527162 |  | −8.0 | −8.1 | −8.3 |
| eMol26330545 |  | −8.2 | −8.2 | −8.4 | eMol27202948 |  | −7.4 | −8.0 | −8.3 |
| eMol26314779 |  | −8.1 | −8.2 | −8.3 | eMol26314779 |  | −8.1 | −8.2 | −8.3 |
| Compound Name/Code | Estimated MM-GBSA Binding Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ∆Evdw | ∆Eele | ∆EGB | ∆ESUR | ∆Ggas | ∆Gsolv | ∆Gbinding | |
| QNZ | −24.7 | −15.7 | 28.3 | −3.3 | −40.4 | 25.0 | −15.4 |
| eMol26313223 | −50.8 | −26.9 | 44.9 | −5.6 | −73.3 | 39.3 | −38.4 |
| eMol26314565 | −47.1 | −19.0 | 36.8 | −5.2 | −64.3 | 31.6 | −34.5 |
| Compound Name/Code | Acceptor | Donor | Distance (Å) a | Angle (Degree) a | Occupied (%) b |
|---|---|---|---|---|---|
| QNZ | GLU294@O | QNZ@O-H | 2.8 | 160 | 50.9 |
| QNZ@O | LYS316@N-H | 2.6 | 151 | 79.7 | |
| eMol26313223 | eMol26313223@O | LYS146@N-H | 2.9 | 141 | 60.6 |
| eMol26313223@N | LYS273@N-H | 2.9 | 148 | 90.0 | |
| eMol26313223@F | LYS316@N-H | 2.7 | 162 | 94.2 | |
| eMol26314565 | eMol26314565@O | LYS146@N-H | 2.9 | 141 | 52.0 |
| eMol26314565@N | LYS273@N-H | 2.9 | 139 | 54.8 | |
| eMol26314565@N | LYS316@N-H | 3.0 | 143 | 85.7 |
| ADME Parameters | QNZ | eMol26313223 | eMol26314565 |
|---|---|---|---|
| Absorption | |||
| Human Intestinal Absorption (% Absorbed) | + | + | + |
| + 98.3% | + 99.4% | + 99.4% | |
| P-glycoprotein Inhibitor | − | + | + |
| P-glycoprotein Substrate | − | − | − |
| Distribution | |||
| Blood–Brain Barrier | + | + | + |
| Subcellular localization | Mitochondria | Mitochondria | Mitochondria |
| Metabolism | |||
| CYP450 2D6 Inhibition | − | − | − |
| CYP450 2D6 Substrate | − | − | − |
| CYP450 3A4 Inhibition | − | − | − |
| CYP450 3A4 Substrate | − | − | + |
| Excretion | |||
| OCT1 Inhibitor | − | − | − |
| OCT2 Inhibitor | − | − | − |
| MATE1 Inhibitor | − | − | − |
| Toxicity | |||
| Carcinogens | − | − | − |
| Acute Toxicity (Class) | II | III | III |
| Eye corrosion | − | − | − |
| Eye irritation | − | − | − |
| Human Ether-a-go-go-Related Inhibition | − | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Alzahrani, O.R.; Alshabrmi, F.M.; Khalaf, E.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; Soliman, M.E.S.; et al. Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus aureus: An In Silico Drug Discovery Study. Antibiotics 2021, 10, 934. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10080934
Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, Alzahrani OR, Alshabrmi FM, Khalaf E, Moustafa MF, Alrumaihi F, Allemailem KS, Soliman MES, et al. Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus aureus: An In Silico Drug Discovery Study. Antibiotics. 2021; 10(8):934. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10080934
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Khlood A. A. Abdeljawaad, Alaa H. M. Abdelrahman, Othman R. Alzahrani, Fahad M. Alshabrmi, Esraa Khalaf, Mahmoud F. Moustafa, Faris Alrumaihi, Khaled S. Allemailem, Mahmoud E. S. Soliman, and et al. 2021. "Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus aureus: An In Silico Drug Discovery Study" Antibiotics 10, no. 8: 934. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10080934