
The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets

 ,
,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Core Concept of the Lipid Energy Model
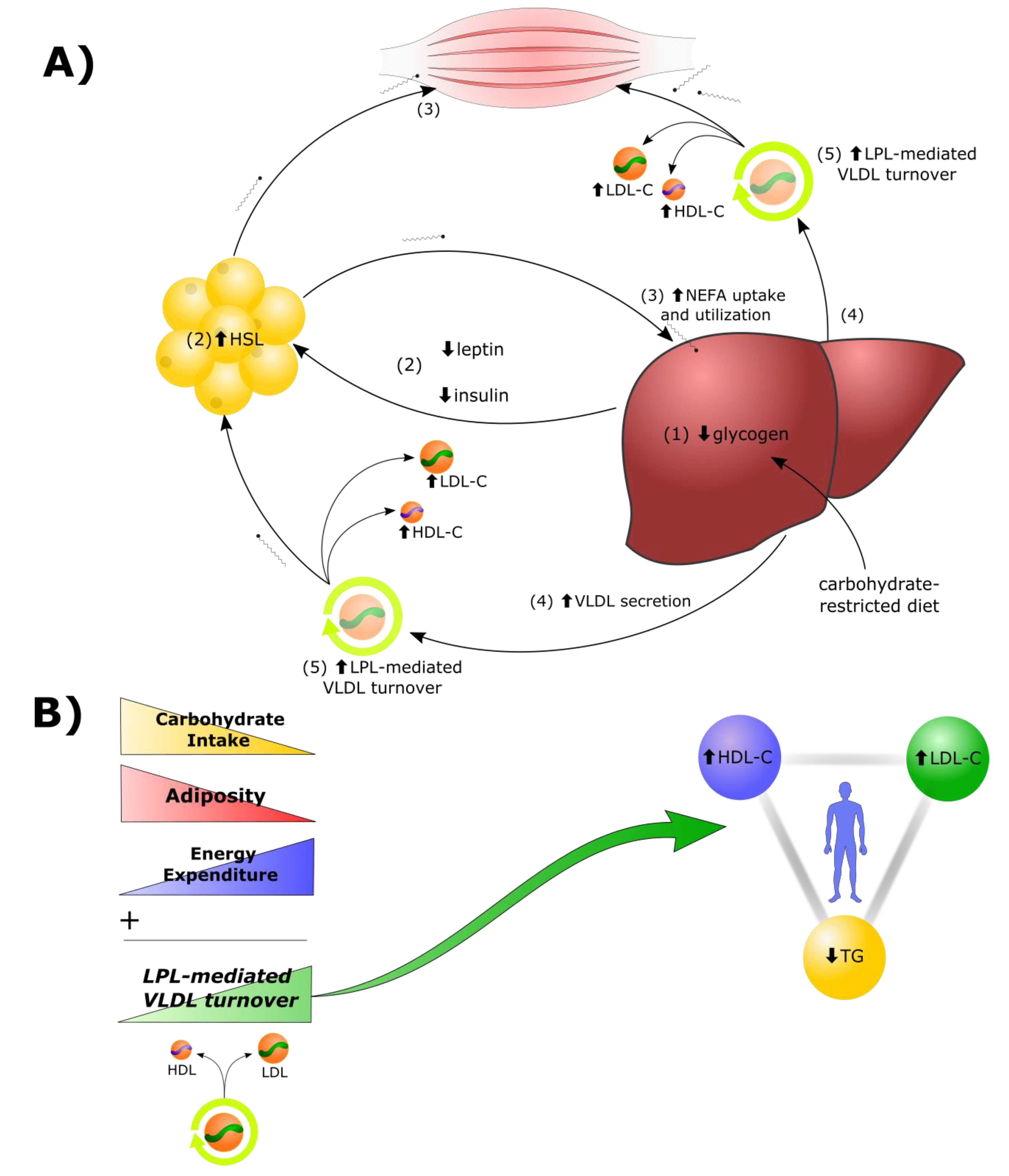
2.1. Lipid Energy Model Overview
- Reduction in dietary carbohydrates and depletion of hepatic glycogen stores results in a greater demand for fat as a metabolic fuel, to compensate for reduced glucose availability.
- Decreased insulin, leptin, and other changes to the hormonal milieu, result in increased hormone-sensitive lipase (HSL)-mediated lipolysis in adipocytes and greater secretion of non-esterified fatty acids (NEFAs) into the bloodstream.
- In addition to heightened use by tissues in the periphery, there is a greater rate of uptake of NEFAs by the liver. Under these conditions, there is a greater rate of synthesis of TGs from the increased fatty acid pool within hepatocytes.
- Increased rates of TG synthesis in the liver leads to increased rates of hepatic assembly and secretion of TG-rich VLDL.
- The increased VLDL secretion rates, in concert with greater LPL-mediated turnover of VLDL in peripheral tissues, and greater transfer of VLDL surface components (including free cholesterol) to HDL, result in higher plasma levels of LDL-C and HDL-C.
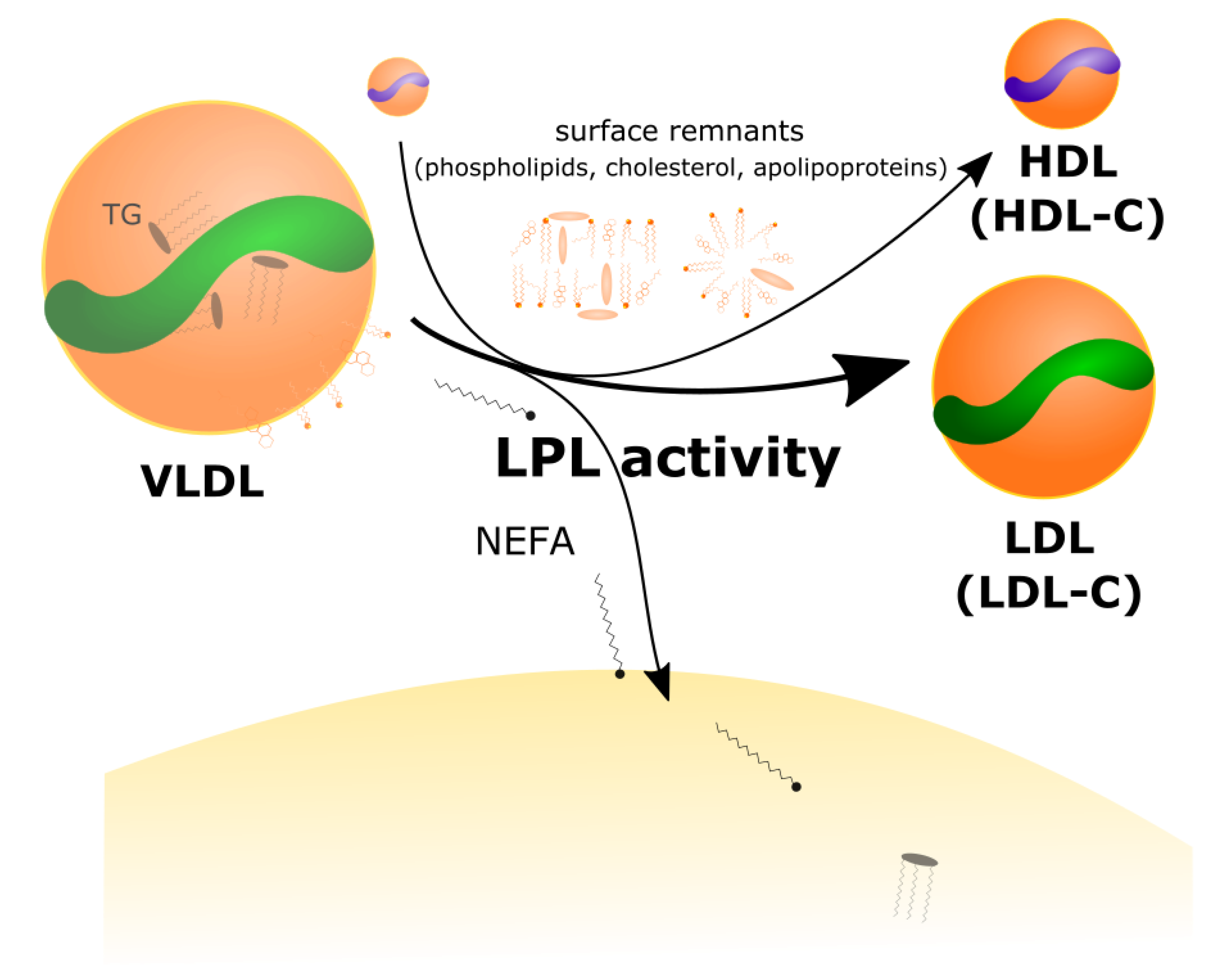
2.2. LPL Mediates the Exchange between TGRL Donors and HDL Acceptors
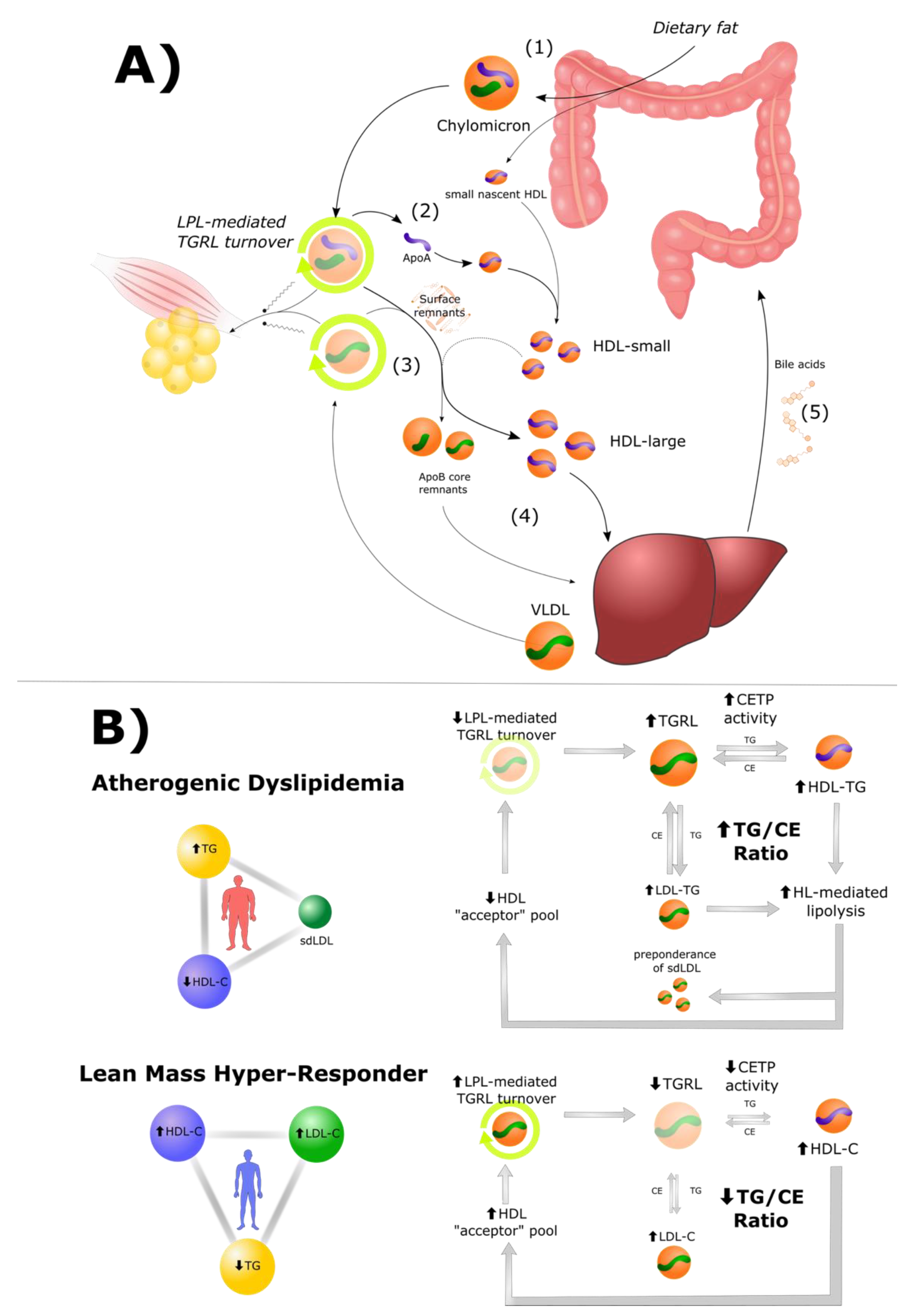
3. The Role of HDL Particles in Lipid Energy Metabolism
3.1. High HDL Is Both a Cause and Consequence of Efficient TGRL Metabolism
3.2. Cholesteryl Ester Transfer Protein (CETP) in Atherogenic Dyslipidemia and LMHR
4. Adiposity and Lean Mass Influence Lipid Energy Dynamics
4.1. Obesity Impairs the Shift from Glucose to Fat Metabolism
4.2. Lean Body Mass and Adiposity Impact VLDL Secretion, Clearance, and Turnover
4.3. Is There a “Threshold Effect” of Carbohydrate Intake on LDL-C Levels?
5. Lipoprotein Lipase Regulators
5.1. ApoC-III and Other Apolipoproteins
5.2. ANGPTL Proteins
5.3. FGF21
6. Testable Predictions of the Lipid Energy Model
- (i)
- Tracer study: Kinetic studies using stable isotope-labeled tracer methods could be employed to track (1) hepatic NEFA uptake, (2) hepatic VLDL-TG/VLDL-P secretion (3) VLDL peripheral turnover, and (4) free cholesterol transfer from TGRL to HDL.
- The LEM predicts increased rates of hepatic NEFA uptake, VLDL-TG/VLDL-P secretion and turnover, and transfer of free cholesterol from TGRL to HDL, in lean metabolically healthy persons on CRD, as compared to metabolically unhealthy subjects or those with obesity.
- (ii)
- “Gym hypothesis:” If the LMHR phenotype is a generalizable metabolic phenomenon, it should be reproducible. This possibility could be assessed with a dietary crossover study including a group of lean, athletic persons, and overweight non-diabetic controls consuming mixed diets. Both groups would undergo a controlled four-week weight-maintenance ketogenic diet and a controlled mixed diet to see if the LMHR phenotype could be induced and reversed.
- The LEM predicts implementation of a CRD in lean metabolically healthy persons with high aerobic energy demands would reproducibly induce a LMHR or LMHR-like lipid profile. The LEM also predicts increases in exercise load would increase LDL-C and HDL-C in present (or induced) LMHR when controlling for weight and carbohydrate intake.
- (iii)
- Carbohydrate titration crossover: To establish the nature of the relationship between carbohydrate intake as percent of total energy intake and LDL-C levels, it would be ideal to perform a crossover study including weight-stable LMHR in which calories from fat are traded for those from carbohydrates in increments of ~5% caloric intake.
- The LEM predicts a threshold or bi-phasic relationship between carbohydrate intake and LDL-C, in which VLDL secretion, and LDL-C levels (as well as lipolytic transfer of free cholesterol from TGRL to HDL) decrease in correspondence with elevated hepatic glycogen stores and plasma insulin and leptin.
- (iv)
- Genetic heterogeneity: Even if the LMHR phenotype is reproducible, there will still likely be a meaningful degree of heterogeneity in LDL-C response to CRD among subjects with similar levels of adiposity. It would be informative to perform a cross-sectional analysis of lean persons consuming a CRD with comprehensive genetic testing of genes relevant to the model, including those coding for the LDL-receptor, ApoB, PSCK9, CETP, LPL and its regulatory proteins (GPIHBP1, ANGPTL3, 4, and 8, ApoCI-III, ApoAV, FGF21), and more.
- The LEM predicts inverse correlations between adiposity and LDL-C and HDL-C changes on CRD that would be relatively generalizable, but that the strength of the associations would be modified by gene-environment interactions that could meaningfully advance the LEM and integrate the model with present understanding of ASCVD pathology.
- (v)
- LPL regulator analysis: It would be informative to measure levels of specific LPL regulators to gain insight into the possible mechanisms behind NEFA and TGRL trafficking.
- The LEM predicts low levels of apo-CIII, ANGPTL8, and 3–8 complex. ANGPTL4 levels may be depleted or elevated in LMHR, with implications on whether NEFA are directed from TGRL directly to oxidative tissue or first trafficked through adipocytes. FGF21 may be elevated, although this prediction is complicated by the possible differing roles of FGF21 in rodent models and humans, and by the suggestion of FGF21 resistance in humans with obesity. Nevertheless, interrogating the possibility role of FGF21 signaling in LMHR may prove an informative line of investigation and one that provides broader clarification on FGF21 in human metabolism.
- (vi)
- Evaluation of risk: While not directly related to the LEM itself, an important forthcoming line of inquiry regards an assessment of the degree to which the LMHR profile is associated with increased progression of atherosclerosis. The conservative clinical perspective is to assume that the high levels of apoB-containing lipoproteins present in LMHR are atherogenic and require treatment with lifestyle modification and/or pharmacotherapy, without exception. However, to properly provide evidence-based recommendations for this unique subgroup of patients we must systematically study them. A prospective study assessing atherosclerotic progression in 100 LMHR by coronary computed tomographic angiography, recently launched from the Lindquist institute, will provide data which will be informative regarding the clinical care of LMHR. However, given the absence of comparator coronary computed tomographic angiography data, and the fact that clinically relevant ASCVD can take years to decades to develop, there is much work to be done to assess the absolute clinical risk of the LMHR profile.
- Those that could evaluate key elements of the LEM: Demonstrating increased hepatic VLDL secretion and LPL-mediated peripheral VLDL turnover in LMHR (tracer study) and demonstrating that the LMHR phenotype is relatively reproducible (gym hypothesis).
- Those that would help to refine the LEM: By better characterizing the dose-response relationship between carbohydrate intake and LDL-C change (carbohydrate titration crossover), describing the putative contribution of genotype (genetic heterogeneity), and defining the endocrinological mechanisms by which LPL activity is regulated in lean persons on CRD (LPL regulator analysis).
- Those that would evaluate the atherosclerotic risk associated with the LMHR phenotype (evaluation of risk, ongoing).
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASCVD | atherosclerotic cardiovascular disease |
| ANGPTL | angiopoietin-like proteins |
| BMI | body mass index |
| CE | cholesteryl ester |
| CETP | cholesteryl ester transfer protein |
| CRD | carbohydrate-restricted diet |
| FGF21 | fibroblast growth factor 21 |
| HDL-C | HDL-cholesterol |
| HSL | hormone-sensitive lipase |
| IDL | intermediate-density lipoprotein |
| LBM | lean body mass |
| LDL-C | LDL-cholesterol |
| LEM | Lipid Energy Model |
| LMHR | lean mass hyper-responders |
| LPL | lipoprotein lipase |
| NAFLD | non-alcoholic fatty liver disease |
| NEFA | non-esterified fatty acids |
| PPAR | peroxisome proliferator activated receptor |
| RRT | Reverse Remnant-Cholesterol Transport |
| sdLDL | small dense LDL |
| TG | triglycerides |
| TGRL | triglyceride-rich lipoprotein |
| VLDL | very low-density lipoprotein |
References
- Ludwig, D.S. The Ketogenic Diet: Evidence for Optimism but High-Quality Research Needed. J. Nutr. 2020, 150, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Creighton, B.C.; Hyde, P.N.; Maresh, C.M.; Kraemer, W.J.; Phinney, S.D.; Volek, J.S. Paradox of hypercholesterolaemia in highly trained, keto-adapted athletes. BMJ Open Sport Exerc. Med. 2018, 4, e000429. [Google Scholar] [CrossRef] [Green Version]
- Buren, J.; Ericsson, M.; Damasceno, N.R.T.; Sjodin, A. A Ketogenic Low-Carbohydrate High-Fat Diet Increases LDL Cholesterol in Healthy, Young, Normal-Weight Women: A Randomized Controlled Feeding Trial. Nutrients 2021, 13, 814. [Google Scholar] [CrossRef]
- Retterstol, K.; Svendsen, M.; Narverud, I.; Holven, K.B. Effect of low carbohydrate high fat diet on LDL cholesterol and gene expression in normal-weight, young adults: A randomized controlled study. Atherosclerosis 2018, 279, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valsdottir, T.D.; Henriksen, C.; Odden, N.; Nellemann, B.; Jeppesen, P.B.; Hisdal, J.; Westerberg, A.C.; Jensen, J. Effect of a Low-Carbohydrate High-Fat Diet and a Single Bout of Exercise on Glucose Tolerance, Lipid Profile and Endothelial Function in Normal Weight Young Healthy Females. Front. Physiol. 2019, 10, 1499. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Ibrahim, N.; Bredefeld, C.; Foo, S.; Lim, V.; Gutman, D.; Huggins, L.A.; Hegele, R.A. Ketogenic diets, not for everyone. J. Clin. Lipidol. 2021, 15, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Soto-Mota, A.; Feldman, D.; Parpos, S.; Budoff, M. Case report: Hypercholesterolemia “Lean Mass Hyper-Responder” phenotype presents in the context of a low saturated fat carbohydrate-restricted diet. Front. Endocrinol. 2022, 13. [Google Scholar] [CrossRef]
- Sharman, M.J.; Gomez, A.L.; Kraemer, W.J.; Volek, J.S. Very low-carbohydrate and low-fat diets affect fasting lipids and postprandial lipemia differently in overweight men. J. Nutr. 2004, 134, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, P.; Iqbal, N.; Stern, L.; Williams, M.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Gracely, E.J.; Rader, D.J.; Samaha, F.F. A randomized study comparing the effects of a low-carbohydrate diet and a conventional diet on lipoprotein subfractions and C-reactive protein levels in patients with severe obesity. Am. J. Med. 2004, 117, 398–405. [Google Scholar] [CrossRef]
- Hays, J.H.; DiSabatino, A.; Gorman, R.T.; Vincent, S.; Stillabower, M.E. Effect of a high saturated fat and no-starch diet on serum lipid subfractions in patients with documented atherosclerotic cardiovascular disease. Mayo Clin. Proc. 2003, 78, 1331–1336. [Google Scholar] [CrossRef] [Green Version]
- Hyde, P.N.; Sapper, T.N.; Crabtree, C.D.; LaFountain, R.A.; Bowling, M.L.; Buga, A.; Fell, B.; McSwiney, F.T.; Dickerson, R.M.; Miller, V.J.; et al. Dietary carbohydrate restriction improves metabolic syndrome independent of weight loss. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.; Guo, M.; Zhang, P.; Sun, G.; Chen, B. The effects of low-carbohydrate diets on cardiovascular risk factors: A meta-analysis. PLoS ONE 2020, 15, e0225348. [Google Scholar] [CrossRef]
- Dashti, H.M.; Al-Zaid, N.S.; Mathew, T.C.; Al-Mousawi, M.; Talib, H.; Asfar, S.K.; Behbahani, A.I. Long term effects of ketogenic diet in obese subjects with high cholesterol level. Mol. Cell Biochem. 2006, 286, 1–9. [Google Scholar] [CrossRef]
- Samaha, F.F.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, T.; Williams, M.; Gracely, E.J.; Stern, L. A low-carbohydrate as compared with a low-fat diet in severe obesity. N. Engl. J. Med. 2003, 348, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.W.; Greco, K.F.; Ma, C.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia-a randomized controlled feeding trial. Am. J. Clin. Nutr. 2021, 115, 154–162. [Google Scholar] [CrossRef]
- Abbasi, J. Interest in the Ketogenic Diet Grows for Weight Loss and Type 2 Diabetes. JAMA 2018, 319, 215–217. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Feldman, D.; Soto-Mota, A.; Kalayjian, T.; Ludwig, D.S. Elevated LDL-cholesterol with a carbohydrate-restricted diet: Evidence for a ‘lean mass hyper-responder’ phenotype. Curr. Dev. Nutr. 2021, 6, nzab144. [Google Scholar] [CrossRef]
- Magill, P.; Rao, S.N.; Miller, N.E.; Nicoll, A.; Brunzell, J.; St Hilaire, J.; Lewis, B. Relationships between the metabolism of high-density and very-low-density lipoproteins in man: Studies of apolipoprotein kinetics and adipose tissue lipoprotein lipase activity. Eur. J. Clin. Investig. 1982, 12, 113–120. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Nikkila, E.A.; Kuusi, T.; Tulikoura, I. Changes of high density lipoprotein subfraction concentration and composition by intralipid in vivo and by lipolysis of intralipid in vitro. Arteriosclerosis 1983, 3, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Patsch, J.R.; Gotto, A.M., Jr.; Olivercrona, T.; Eisenberg, S. Formation of high density lipoprotein2-like particles during lipolysis of very low density lipoproteins in vitro. Proc. Natl. Acad. Sci. USA 1978, 75, 4519–4523. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Blum, C.B.; Forester, G.P.; Nelson, C.A. Changes in the distribution and composition of plasma high density lipoproteins after ingestion of fat. J. Biol. Chem. 1982, 257, 198–207. [Google Scholar] [CrossRef]
- Groot, P.H.; Scheek, L.M. Effects of fat ingestion on high density lipoprotein profiles in human sera. J. Lipid Res. 1984, 25, 684–692. [Google Scholar] [CrossRef]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A. HDL and Reverse Remnant-Cholesterol Transport (RRT): Relevance to Cardiovascular Disease. Trends Mol. Med. 2020, 26, 1086–1100. [Google Scholar] [CrossRef]
- Randolph, G.J.; Miller, N.E. Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Investig. 2014, 124, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Green, P.H.; Glickman, R.M. Intestinal lipoprotein metabolism. J. Lipid Res. 1981, 22, 1153–1173. [Google Scholar] [CrossRef]
- Han, Y.-H.; Onufer, E.J.; Huang, L.-H.; Sprung, R.W.; Davidson, W.S.; Czepielewski, R.S.; Wohltmann, M.; Sorci-Thomas, M.G.; Warner, B.W.; Randolph, G.J. Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science 2021, 373, eabe6729. [Google Scholar] [CrossRef]
- Imaizumi, K.; Havel, R.J.; Fainaru, M.; Vigne, J.L. Origin and transport of the A-I and arginine-rich apolipoproteins in mesenteric lymph of rats. J. Lipid Res. 1978, 19, 1038–1046. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Lyssenko, N.N.; Nickel, M.; Nguyen, D.; Chetty, P.S.; Weibel, G.; Phillips, M.C. Mechanisms responsible for the compositional heterogeneity of nascent high density lipoprotein. J. Biol. Chem. 2013, 288, 23150–23160. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, E.J.; Wetzel, M.G.; Bengtsson, G.; Scow, R.O.; Brewer, H.B., Jr.; Olivecrona, T. Transfer of human lymph chylomicron constituents to other lipoprotein density fractions during in vitro lipolysis. J. Lipid Res. 1982, 23, 1259–1273. [Google Scholar] [CrossRef]
- Gillard, B.K.; Rosales, C.; Xu, B.; Gotto, A.M., Jr.; Pownall, H.J. Rethinking reverse cholesterol transport and dysfunctional high-density lipoproteins. J. Clin. Lipidol. 2018, 12, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Hopkins, G.J.; Calvert, G.D. Transfers and exchanges of esterified cholesterol between plasma lipoproteins. Biochem. J. 1982, 208, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. Plasma cholesteryl ester transfer protein. J. Lipid Res. 1993, 34, 1255–1274. [Google Scholar] [CrossRef]
- Chapman, M.J.; Le Goff, W.; Guerin, M.; Kontush, A. Cholesteryl ester transfer protein: At the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur. Heart J. 2010, 31, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C.C.; VandenBroek, J.M.; Cooper, P.S. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid Res. 2004, 45, 1594–1607. [Google Scholar] [CrossRef] [Green Version]
- Mann, C.J.; Yen, F.T.; Grant, A.M.; Bihain, B.E. Mechanism of plasma cholesteryl ester transfer in hypertriglyceridemia. J. Clin. Investig. 1991, 88, 2059–2066. [Google Scholar] [CrossRef] [Green Version]
- Boren, J.; Watts, G.F.; Adiels, M.; Soderlund, S.; Chan, D.C.; Hakkarainen, A.; Lundbom, N.; Matikainen, N.; Kahri, J.; Verges, B.; et al. Kinetic and Related Determinants of Plasma Triglyceride Concentration in Abdominal Obesity: Multicenter Tracer Kinetic Study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2218–2224. [Google Scholar] [CrossRef] [Green Version]
- Boren, J.; Packard, C.J.; Taskinen, M.R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Chapman, M.J. Therapeutic elevation of HDL-cholesterol to prevent atherosclerosis and coronary heart disease. Pharmacol. Ther. 2006, 111, 893–908. [Google Scholar] [CrossRef]
- Guerin, M.; Lassel, T.S.; Le Goff, W.; Farnier, M.; Chapman, M.J. Action of atorvastatin in combined hyperlipidemia: Preferential reduction of cholesteryl ester transfer from HDL to VLDL1 particles. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Guerin, M.; Egger, P.; Soudant, C.; Le Goff, W.; van Tol, A.; Dupuis, R.; Chapman, M.J. Cholesteryl ester flux from HDL to VLDL-1 is preferentially enhanced in type IIB hyperlipidemia in the postprandial state. J. Lipid Res. 2002, 43, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, W.; Guerin, M.; Chapman, M.J. Pharmacological modulation of cholesteryl ester transfer protein, a new therapeutic target in atherogenic dyslipidemia. Pharmacol. Ther. 2004, 101, 17–38. [Google Scholar] [CrossRef]
- McPherson, R.; Mann, C.J.; Tall, A.R.; Hogue, M.; Martin, L.; Milne, R.W.; Marcel, Y.L. Plasma concentrations of cholesteryl ester transfer protein in hyperlipoproteinemia. Relation to cholesteryl ester transfer protein activity and other lipoprotein variables. Arterioscler. Thromb. 1991, 11, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J. Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996, 37, 693–707. [Google Scholar] [CrossRef]
- Rashid, S.; Watanabe, T.; Sakaue, T.; Lewis, G.F. Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: The combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin. Biochem. 2003, 36, 421–429. [Google Scholar] [CrossRef]
- Deeb, S.S.; Zambon, A.; Carr, M.C.; Ayyobi, A.F.; Brunzell, J.D. Hepatic lipase and dyslipidemia: Interactions among genetic variants, obesity, gender, and diet. J. Lipid Res. 2003, 44, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Charles, M.A.; Kane, J.P. New molecular insights into CETP structure and function: A review. J. Lipid Res. 2012, 53, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Guendouzi, K.; Collet, X.; Perret, B.; Chap, H.; Barbaras, R. Remnant high density lipoprotein2 particles produced by hepatic lipase display high-affinity binding and increased endocytosis into a human hepatoma cell line (HEPG2). Biochemistry 1998, 37, 14974–14980. [Google Scholar] [CrossRef]
- Newnham, H.H.; Barter, P.J. Synergistic effects of lipid transfers and hepatic lipase in the formation of very small high-density lipoproteins during incubation of human plasma. Biochim. Biophys. Acta 1990, 1044, 57–64. [Google Scholar] [CrossRef]
- Rye, K.A.; Clay, M.A.; Barter, P.J. Remodelling of high density lipoproteins by plasma factors. Atherosclerosis 1999, 145, 227–238. [Google Scholar] [CrossRef]
- Xiao, C.; Watanabe, T.; Zhang, Y.; Trigatti, B.; Szeto, L.; Connelly, P.W.; Marcovina, S.; Vaisar, T.; Heinecke, J.W.; Lewis, G.F. Enhanced cellular uptake of remnant high-density lipoprotein particles: A mechanism for high-density lipoprotein lowering in insulin resistance and hypertriglyceridemia. Circ. Res. 2008, 103, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagor, W.R.; Brown, R.J.; Toh, S.A.; Millar, J.S.; Fuki, I.V.; de la Llera-Moya, M.; Yuen, T.; Rothblat, G.; Billheimer, J.T.; Rader, D.J. Overexpression of apolipoprotein F reduces HDL cholesterol levels in vivo. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.Y.; Chen, S.F.; Lai, M.D.; Chang, T.T.; Chen, T.L.; Li, P.Y.; Shieh, D.B.; Young, K.C. Comparative proteomic profiling of plasma very-low-density and low-density lipoproteins. Clin. Chim. Acta 2010, 411, 336–344. [Google Scholar] [CrossRef]
- Deckelbaum, R.J.; Eisenberg, S.; Oschry, Y.; Butbul, E.; Sharon, I.; Olivecrona, T. Reversible modification of human plasma low density lipoproteins toward triglyceride-rich precursors. A mechanism for losing excess cholesterol esters. J. Biol. Chem. 1982, 257, 6509–6517. [Google Scholar] [CrossRef]
- Deckelbaum, R.J.; Ramakrishnan, R.; Eisenberg, S.; Olivecrona, T.; Bengtsson-Olivecrona, G. Triacylglycerol and phospholipid hydrolysis in human plasma lipoproteins: Role of lipoprotein and hepatic lipase. Biochemistry 1992, 31, 8544–8551. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Le, N.A.; Paterniti, J.R., Jr.; Ginsberg, H.N.; Lindgren, F.T.; Brown, W.V. Lipoprotein metabolism during acute inhibition of hepatic triglyceride lipase in the cynomolgus monkey. J. Clin. Investig. 1982, 70, 1184–1192. [Google Scholar] [CrossRef]
- Krauss, R.M.; Burke, D.J. Identification of multiple subclasses of plasma low density lipoproteins in normal humans. J. Lipid Res. 1982, 23, 97–104. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.T.; Vranizan, K.M.; Krauss, R.M. Correlations of plasma lipoproteins with LDL subfractions by particle size in men and women. J. Lipid Res. 1992, 33, 765–774. [Google Scholar] [CrossRef]
- Goschke, H. Mechanism of glucose intolerance during fasting: Differences between lean and obese subjects. Metabolism 1977, 26, 1147–1153. [Google Scholar] [CrossRef]
- Bak, A.M.; Vendelbo, M.H.; Christensen, B.; Viggers, R.; Bibby, B.M.; Rungby, J.; Jorgensen, J.O.L.; Moller, N.; Jessen, N. Prolonged fasting-induced metabolic signatures in human skeletal muscle of lean and obese men. PLoS ONE 2018, 13, e0200817. [Google Scholar] [CrossRef] [Green Version]
- Wijngaarden, M.A.; van der Zon, G.C.; van Dijk, K.W.; Pijl, H.; Guigas, B. Effects of prolonged fasting on AMPK signaling, gene expression, and mitochondrial respiratory chain content in skeletal muscle from lean and obese individuals. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1012–E1021. [Google Scholar] [CrossRef] [Green Version]
- Sondergaard, E.; Nellemann, B.; Sorensen, L.P.; Christensen, B.; Gormsen, L.C.; Nielsen, S. Lean body mass, not FFA, predicts VLDL-TG secretion rate in healthy men. Obesity 2015, 23, 1379–1385. [Google Scholar] [CrossRef]
- Bartelt, A.; John, C.; Schaltenberg, N.; Berbee, J.F.P.; Worthmann, A.; Cherradi, M.L.; Schlein, C.; Piepenburg, J.; Boon, M.R.; Rinninger, F.; et al. Thermogenic adipocytes promote HDL turnover and reverse cholesterol transport. Nat. Commun. 2017, 8, 15010. [Google Scholar] [CrossRef]
- Perry, R.J.; Wang, Y.; Cline, G.W.; Rabin-Court, A.; Song, J.D.; Dufour, S.; Zhang, X.M.; Petersen, K.F.; Shulman, G.I. Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 2018, 172, 234–248.e217. [Google Scholar] [CrossRef]
- Steinhauser, M.L.; Olenchock, B.A.; O’Keefe, J.; Lun, M.; Pierce, K.A.; Lee, H.; Pantano, L.; Klibanski, A.; Shulman, G.I.; Clish, C.B.; et al. The circulating metabolome of human starvation. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Goossens, N.; Jornayvaz, F.R. Translational Aspects of Diet and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 1077. [Google Scholar] [CrossRef] [Green Version]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.M.; Hakkarainen, A.; Lehtimaki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Jarvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, O.; Bassi, A.; Stranieri, C.; Trabetti, E.; Martinelli, N.; Pizzolo, F.; Girelli, D.; Friso, S.; Pignatti, P.F.; Corrocher, R. Apolipoprotein C-III, metabolic syndrome, and risk of coronary artery disease. J. Lipid Res. 2003, 44, 2374–2381. [Google Scholar] [CrossRef] [Green Version]
- Juntti-Berggren, L.; Berggren, P.O. Apolipoprotein CIII is a new player in diabetes. Curr. Opin Lipidol. 2017, 28, 27–31. [Google Scholar] [CrossRef]
- Paola Gutierrez Castro, K.; Patricia Gonzalez, A.; Caccavello, R.; Garay-Sevilla, M.E.; Gugliucci, A. Lean adolescents with insulin resistance display higher angiopoietin like protein 3, ApoC-III and chylomicron remnant dyslipidemia. Clin. Chim. Acta 2022, 526, 43–48. [Google Scholar] [CrossRef]
- Luo, M.; Su, X.; Yi, Y.; Yang, Y.; Peng, D. Apolipoprotein CIII may mediate the impacts of angiopoietin-like protein 8 on triglyceride metabolism. Lipids Health Dis. 2018, 17, 160. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.J.; Blanche, P.J.; Rawlings, R.S.; Fernstrom, H.S.; Krauss, R.M. Increased plasma concentrations of lipoprotein(a) during a low-fat, high-carbohydrate diet are associated with increased plasma concentrations of apolipoprotein C-III bound to apolipoprotein B-containing lipoproteins. Am. J. Clin. Nutr. 2007, 85, 1527–1532. [Google Scholar] [CrossRef]
- Furtado, J.D.; Campos, H.; Appel, L.J.; Miller, E.R.; Laranjo, N.; Carey, V.J.; Sacks, F.M. Effect of protein, unsaturated fat, and carbohydrate intakes on plasma apolipoprotein B and VLDL and LDL containing apolipoprotein C-III: Results from the OmniHeart Trial. Am. J. Clin. Nutr. 2008, 87, 1623–1630. [Google Scholar] [CrossRef]
- Mendoza, S.; Trenchevska, O.; King, S.M.; Nelson, R.W.; Nedelkov, D.; Krauss, R.M.; Yassine, H.N. Changes in low-density lipoprotein size phenotypes associate with changes in apolipoprotein C-III glycoforms after dietary interventions. J. Clin. Lipidol. 2017, 11, 224–233.e222. [Google Scholar] [CrossRef] [Green Version]
- Huff, M.W.; Nestel, P.J. Metabolism of apolipoproteins CII, CIII1, CIII2 and VLDL-B in human subjects consuming high carbohydrate diets. Metabolism 1982, 31, 493–498. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Rasanen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietilainen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e555. [Google Scholar] [CrossRef] [Green Version]
- van Capelleveen, J.C.; Bernelot Moens, S.J.; Yang, X.; Kastelein, J.J.P.; Wareham, N.J.; Zwinderman, A.H.; Stroes, E.S.G.; Witztum, J.L.; Hovingh, G.K.; Khaw, K.T.; et al. Apolipoprotein C-III Levels and Incident Coronary Artery Disease Risk: The EPIC-Norfolk Prospective Population Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1206–1212. [Google Scholar] [CrossRef] [Green Version]
- Taskinen, M.R.; Packard, C.J.; Boren, J. Emerging Evidence that ApoC-III Inhibitors Provide Novel Options to Reduce the Residual CVD. Curr. Atheroscler. Rep. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, P.M.M.; Michielsen, C.; Hazebroek, E.J.; Pirayesh, A.; Olivecrona, G.; Afman, L.A.; Kersten, S. Fasting induces ANGPTL4 and reduces LPL activity in human adipose tissue. Mol. Metab. 2020, 40, 101033. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Price, N.L.; Suarez, Y.; Fernandez-Hernando, C. ANGPTL4 in Metabolic and Cardiovascular Disease. Trends Mol. Med. 2019, 25, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Cha, J.Y.; Gopal, H.; Harp, C.; Yu, X.; Repa, J.J.; Li, C. Differential regulation and properties of angiopoietin-like proteins 3 and 4. J. Lipid Res. 2005, 46, 1484–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhang, K. An updated ANGPTL3-4-8 model as a mechanism of triglyceride partitioning between fat and oxidative tissues. Prog. Lipid Res. 2021, 85, 101140. [Google Scholar] [CrossRef]
- Ye, J.; Qin, Y.; Wang, D.; Yang, L.; Yuan, G. The Relationship between Circulating ANGPTL8/Betatrophin Concentrations and Adult Obesity: A Meta-Analysis. Dis. Markers 2019, 2019, 5096860. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Kim, J.Y.; Smas, C.M. Identification of RIFL, a novel adipocyte-enriched insulin target gene with a role in lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E334–E351. [Google Scholar] [CrossRef] [Green Version]
- Cinkajzlova, A.; Mraz, M.; Lacinova, Z.; Klouckova, J.; Kavalkova, P.; Kratochvilova, H.; Trachta, P.; Krizova, J.; Haluzikova, D.; Skrha, J.; et al. Angiopoietin-like protein 3 and 4 in obesity, type 2 diabetes mellitus, and malnutrition: The effect of weight reduction and realimentation. Nutr. Diabetes 2018, 8, 21. [Google Scholar] [CrossRef]
- Dijk, W.; Kersten, S. Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol. Metab. 2014, 25, 146–155. [Google Scholar] [CrossRef]
- Holmes, D. Metabolism: Fasting induces FGF21 in humans. Nat. Rev. Endocrinol. 2016, 12, 3. [Google Scholar] [CrossRef]
- Tanimura, Y.; Aoi, W.; Takanami, Y.; Kawai, Y.; Mizushima, K.; Naito, Y.; Yoshikawa, T. Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.S.; Lim, C.; Choi, H.Y.; Lee, Y.K.; Ku, E.J.; Moon, J.H.; Park, K.S.; Jang, H.C.; Choi, S.H. Plasma fibroblast growth factor 21 levels increase with ectopic fat accumulation and its receptor levels are decreased in the visceral fat of patients with type 2 diabetes. BMJ Open Diabetes Res. Care 2019, 7, e000776. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef] [Green Version]
- Domouzoglou, E.M.; Maratos-Flier, E.E. Fibroblast growth factor 21 is a metabolic regulator that plays a role in the adaptation to ketosis. Am. J. Clin. Nutr. 2011, 93, 901S–905S. [Google Scholar] [CrossRef] [Green Version]
- Hron, B.M.; Ebbeling, C.B.; Feldman, H.A.; Ludwig, D.S. Hepatic, adipocyte, enteric and pancreatic hormones: Response to dietary macronutrient composition and relationship with metabolism. Nutr. Metab. 2017, 14, 44. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Tillman, E.J.; Rolph, T. FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front. Endocrinol. 2020, 11, 601290. [Google Scholar] [CrossRef]
- Sancar, G.; Liu, S.; Gasser, E.; Alvarez, J.G.; Moutos, C.; Kim, K.; van Zutphen, T.; Wang, Y.; Huddy, T.F.; Ross, B.; et al. FGF1 and insulin control lipolysis by convergent pathways. Cell Metab. 2022, 34, 171–183. [Google Scholar] [CrossRef]
- Wang, A.; Yan, X.; Zhang, C.; Du, C.; Long, W.; Zhan, D.; Luo, X. Characterization of fibroblast growth factor 1 in obese children and adolescents. Endocr. Connect. 2018, 7, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286.e6. [Google Scholar] [CrossRef]
- Flippo, K.H.; Trammell, S.A.J.; Gillum, M.P.; Aklan, I.; Prez, M.B.; Yavuz, Y.; Smith, N.K.; Jensen-Cody, S.O.; Zhou, B.; Claflin, K.E.; et al. FGF21 suppresses alcohol consumption through an amygdalo-striatal circuit. Cell Metab. 2022, 34, 317–328.e6. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norwitz, N.G.; Soto-Mota, A.; Kaplan, B.; Ludwig, D.S.; Budoff, M.; Kontush, A.; Feldman, D. The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets. Metabolites 2022, 12, 460. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050460
Norwitz NG, Soto-Mota A, Kaplan B, Ludwig DS, Budoff M, Kontush A, Feldman D. The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets. Metabolites. 2022; 12(5):460. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050460
Chicago/Turabian StyleNorwitz, Nicholas G., Adrian Soto-Mota, Bob Kaplan, David S. Ludwig, Matthew Budoff, Anatol Kontush, and David Feldman. 2022. "The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets" Metabolites 12, no. 5: 460. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050460