Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Docking and Molecular Dynamics (MD) Simulations
2.3. Free Energy Perturbation (FEP) Calculations
3. Results
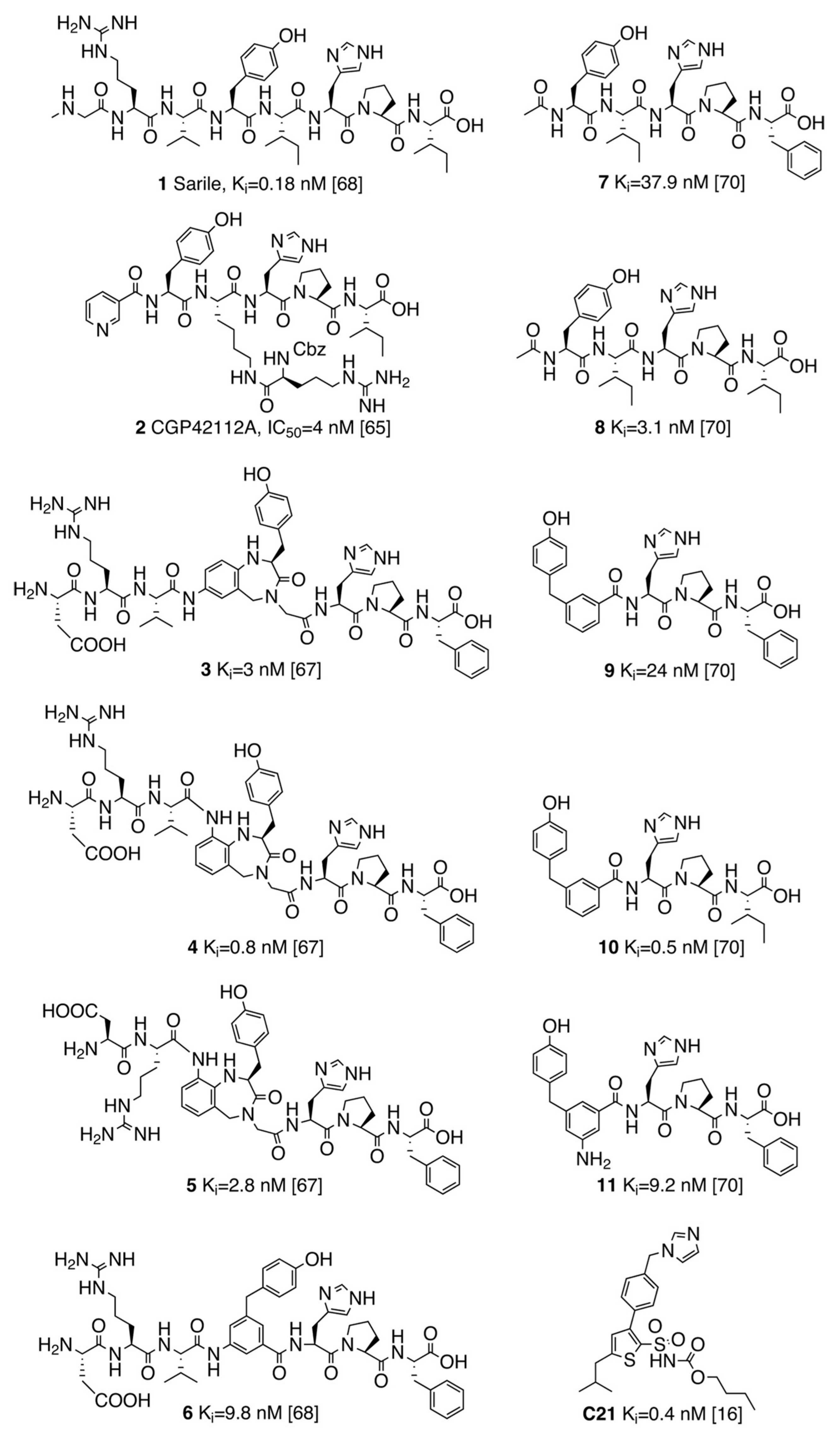
3.1. Selection of the AT2R Agonists Dataset and Initial Docking
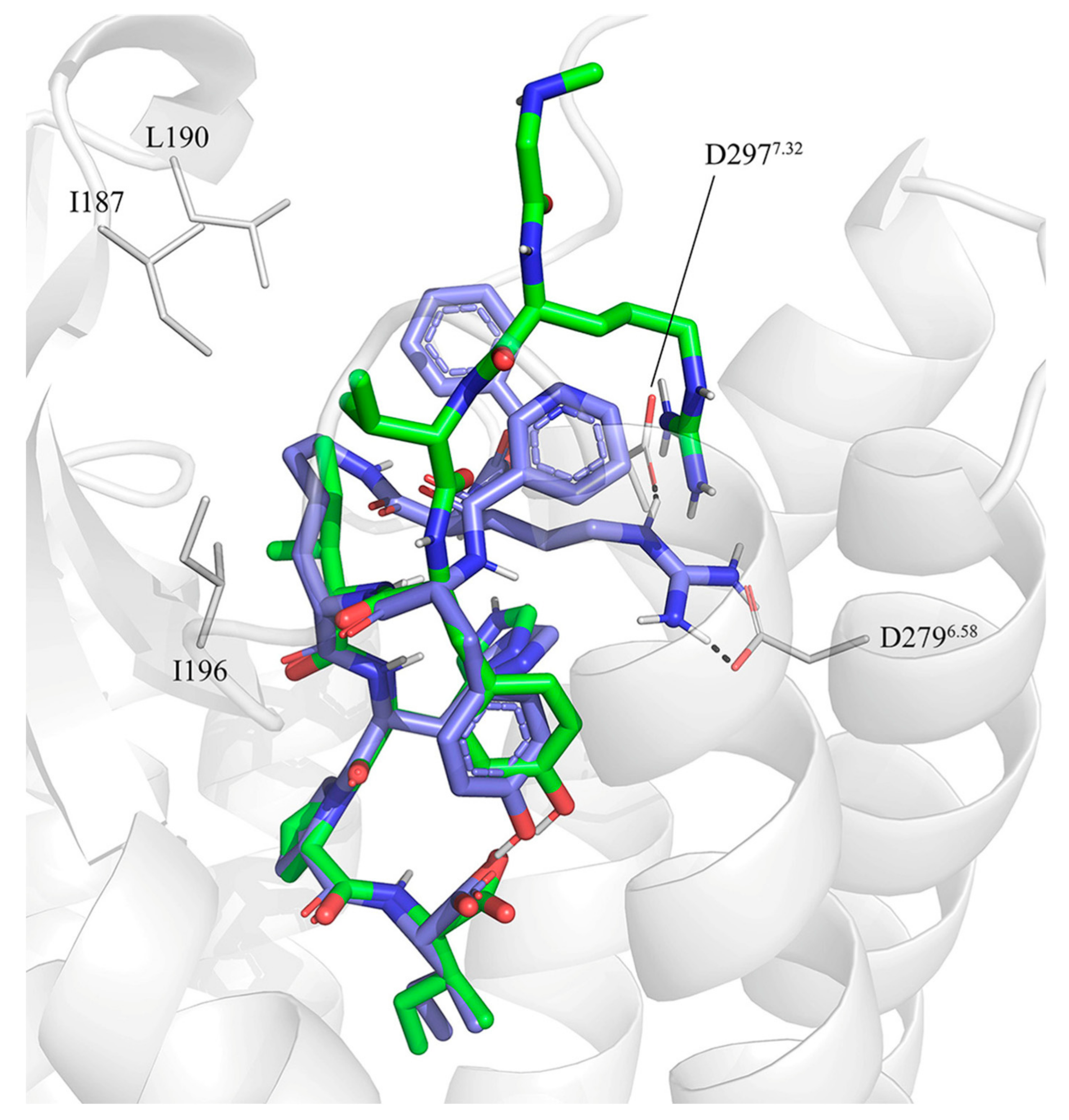
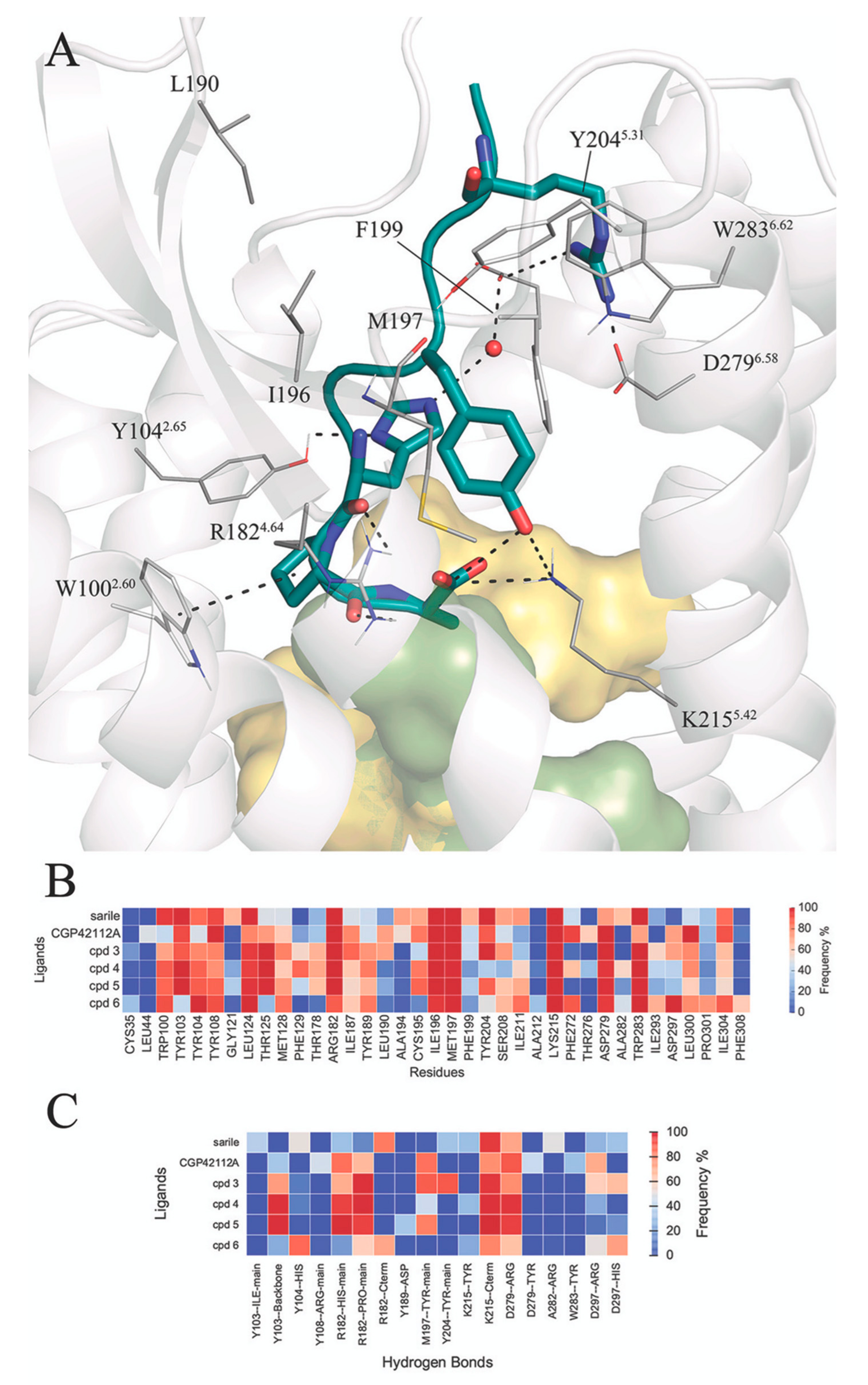
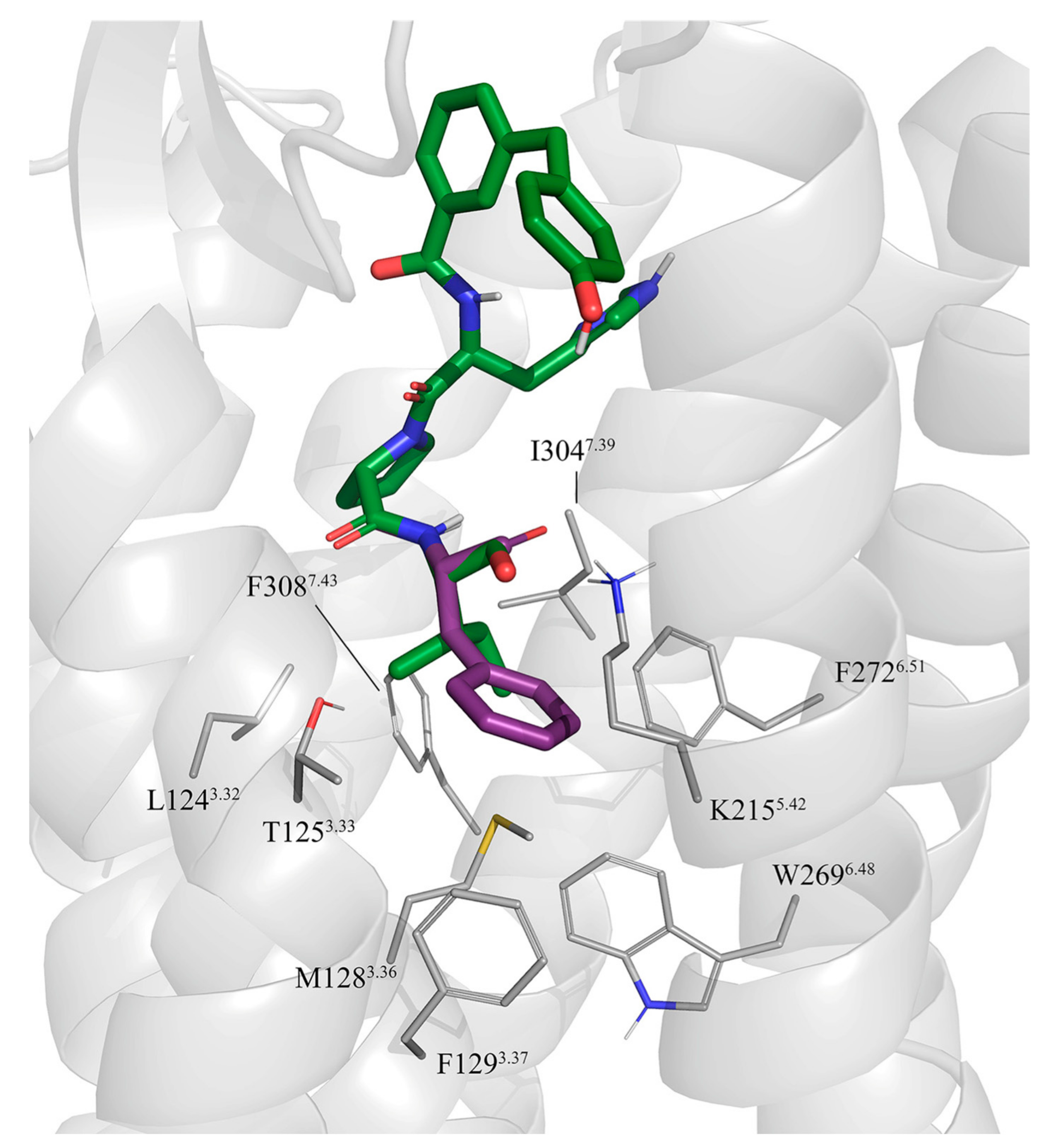
3.2. Molecular Dynamics Simulations of Octapeptides and Mimetics (Compounds 1–6)
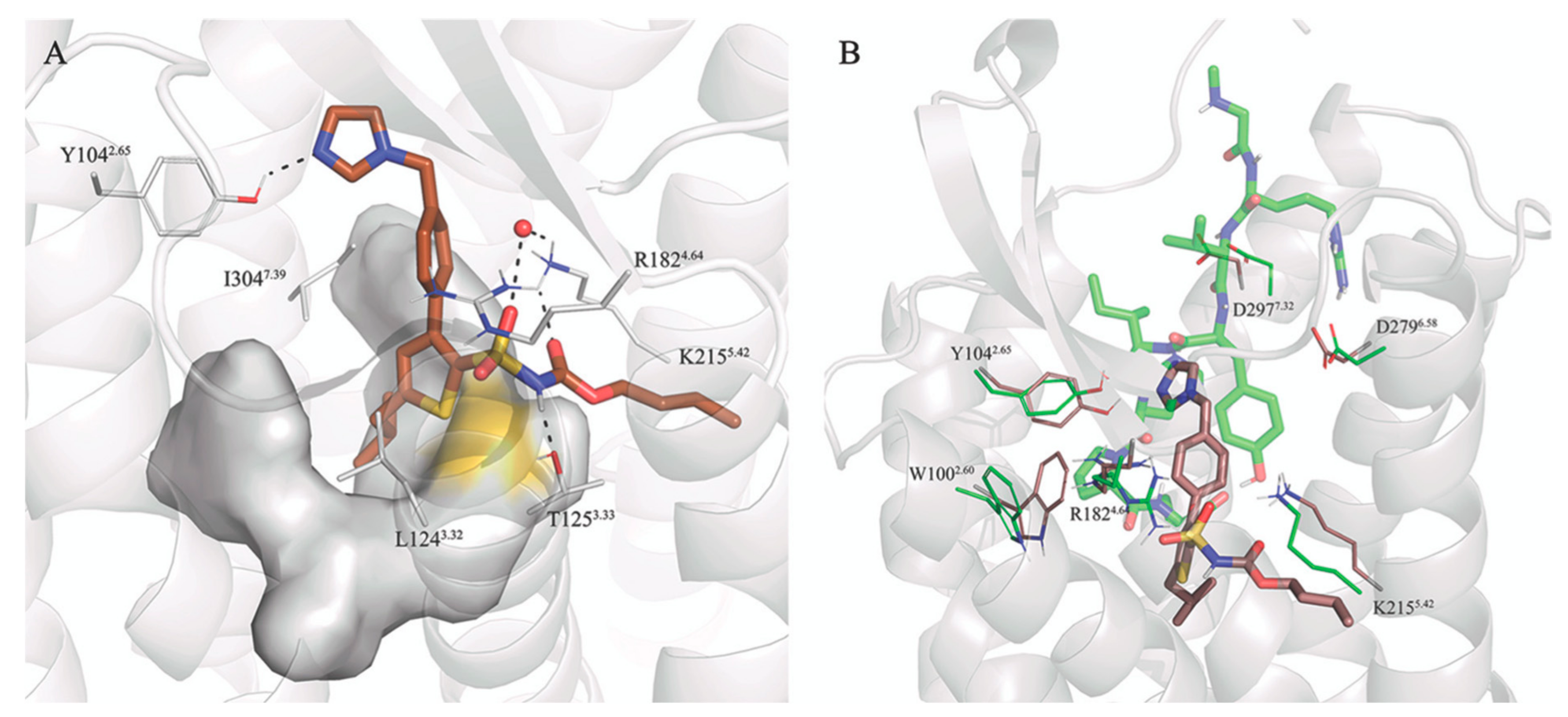
3.3. Selectivity between AT1R and AT2R
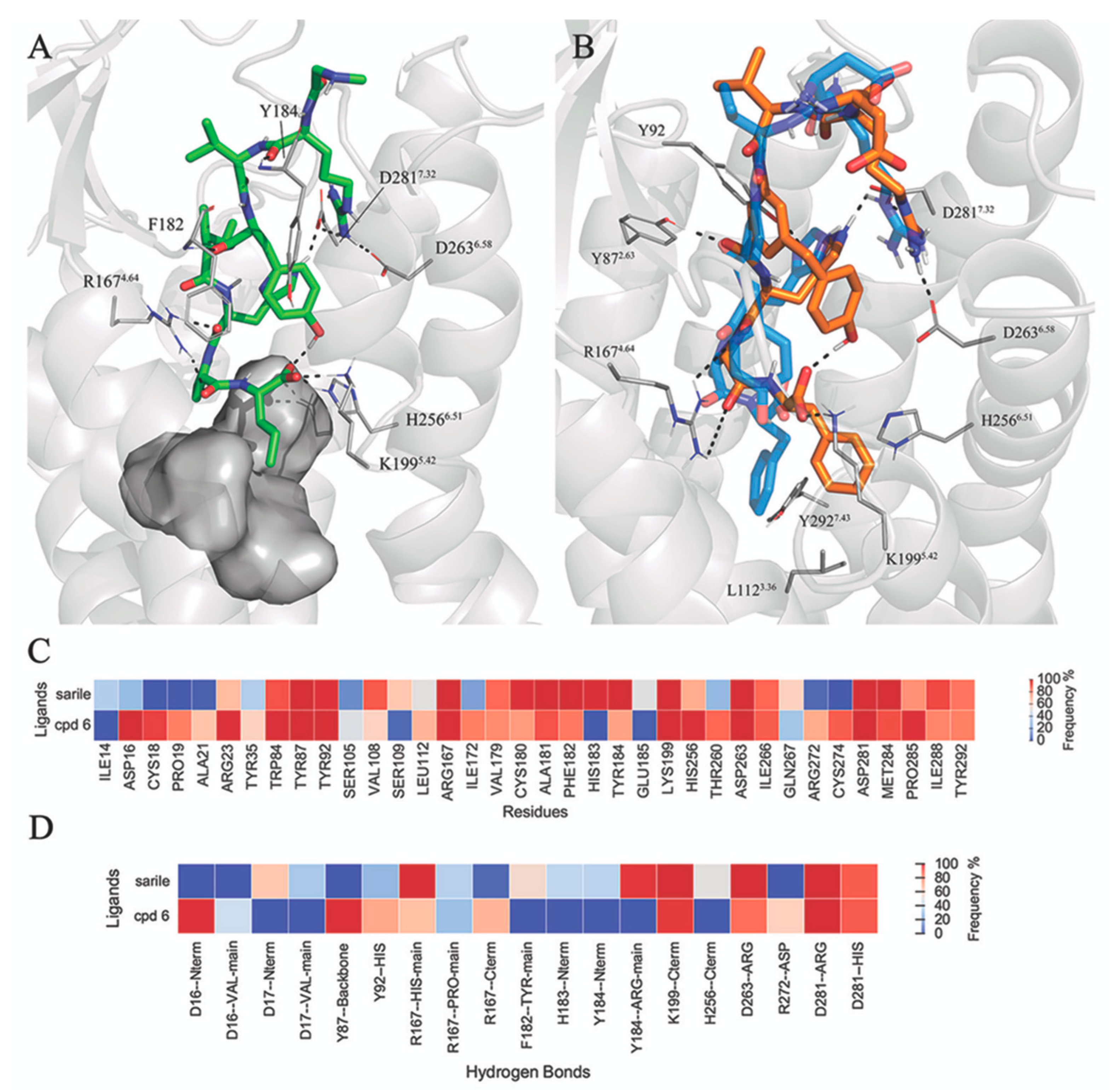
3.4. FEP Simulations of the Pentapeptides and Mimetics 7–11
3.5. Putative Binding Mode of C21
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science (80-) 1977, 196, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Ondetti, M.A.; Cushman, D.W. Inhibition of the Renin-Angiotensin System. A New Approach to the Therapy of Hypertension. J. Med. Chem. 1981, 24, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Webb, R.; Nussberger, J.; Hollenberg, N.K. Renin inhibition with aliskiren: Where are we now, and where are we going? J. Hypertens. 2006, 24, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Wexler, R.R.; Greenlee, W.J.; Irvin, J.D.; Goldberg, M.R.; Prendergast, K.; Smith, R.D.; Timmermans, P.B. Nonpeptide angiotensin II receptor antagonists: The next generation in antihypertensive therapy. J. Med. Chem. 1996, 39, 625–656. [Google Scholar] [CrossRef]
- Paulis, L.; Steckelings, U.M.; Unger, T. Key advances in antihypertensive treatment. Nat. Rev. Cardiol. 2012, 9, 276–285. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Kaschina, E.; Unger, T. The AT2 receptor—A matter of love and hate. Peptides 2005, 26, 1401–1409. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Rompe, F.; Kaschina, E.; Namsolleck, P.; Grzesiak, A.; Funke-Kaiser, H.; Bader, M.; Unger, T. The past, present and future of angiotensin II type 2 receptor stimulation. JRAAS J. Renin-Angiotensin-Aldosterone Syst. 2010, 11, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Hutchinson, H.G.; Fujinaga, M.; Hayashida, W.; Morishita, R.; Zhang, L.; Horiuchi, M.; Pratt, R.E.; Dzau, V.J. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: Gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. USA 2006, 92, 10663–10667. [Google Scholar] [CrossRef] [Green Version]
- Gallinat, S.; Yu, M.; Dorst, A.; Unger, T.; Herdegen, T. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor mRNA in adult rat dorsal root ganglia and sciatic nerves. Mol. Brain Res. 1998, 57, 111–122. [Google Scholar] [CrossRef]
- Altarche-Xifró, W.; Curato, C.; Kaschina, E.; Grzesiak, A.; Slavic, S.; Dong, J.; Kappert, K.; Steckelings, M.; Imboden, H.; Unger, T.; et al. Cardiac c-kit+AT2+ cell population is increased in response to ischemic injury and supports cardiomyocyte performance. Stem Cells 2009, 27, 2488–2497. [Google Scholar] [CrossRef]
- Li, J.; Culman, J.; Hörtnagl, H.; Zhao, Y.; Gerova, N.; Timm, M.; Blume, A.; Zimmermann, M.; Seidel, K.; Dirnagl, U.; et al. Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J. 2005, 19, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Sumners, C.; De Kloet, A.D.; Krause, E.G.; Unger, T.; Steckelings, U.M. Angiotensin type 2 receptors: Blood pressure regulation and end organ damage. Curr. Opin. Pharmacol. 2015, 21, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulis, L.; Foulquier, S.; Namsolleck, P.; Recarti, C.; Steckelings, U.M.; Unger, T. Combined Angiotensin Receptor Modulation in the Management of Cardio-Metabolic Disorders. Drugs 2016, 76, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larhed, M.; Hallberg, M.; Hallberg, A. Nonpeptide AT2 Receptor Agonists. In Medicinal Chemistry Reviews; Desai, M.C., Ed.; Medicinal Chemistry Division of the American Chemical Society: Washington, DC, USA, 2016; Volume 51, pp. 69–79. [Google Scholar]
- Juillerat-jeanneret, L. The Other Angiotensin II Receptor : AT2R as a Therapeutic Target. J. Med. Chem. 2020, 63, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wallinder, C.; Plouffe, B.; Beaudry, H.; Mahalingam, A.K.; Wu, X.; Johansson, B.; Holm, M.; Botoros, M.; Karlén, A.; et al. Design, synthesis, and biological evaluation, of the first selective nonpeptide AT2 receptor agonist. J. Med. Chem. 2004, 47, 5995–6008. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, M.; Sumners, C.; Steckelings, U.M.; Hallberg, A. Small-molecule AT2 receptor agonists. Med. Res. Rev. 2018, 38, 602–624. [Google Scholar] [CrossRef]
- Rathinasabapathy, A.; Horowitz, A.; Horton, K.; Kumar, A.; Gladson, S.; Unger, T.; Martinez, D.; Bedse, G.; West, J.; Raizada, M.K.; et al. The selective angiotensin II type 2 receptor agonist, compound 21, attenuates the progression of lung fibrosis and pulmonary hypertension in an experimental model of bleomycin-induced lung injury. Front. Physiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Murugaiah, A.M.S.; Wallinder, C.; Mahalingam, A.K.; Wu, X.; Wan, Y.; Plouffe, B.; Botros, M.; Karlén, A.; Hallberg, M.; Gallo-Payet, N.; et al. Selective angiotensin II AT2 receptor agonists devoid of the imidazole ring system. Bioorg. Med. Chem. 2007, 15, 7166–7183. [Google Scholar] [CrossRef]
- Wallinder, C.; Botros, M.; Rosenström, U.; Guimond, M.O.; Beaudry, H.; Nyberg, F.; Gallo-Payet, N.; Hallberg, A.; Alterman, M. Selective angiotensin II AT2 receptor agonists: Benzamide structure-activity relationships. Bioorg. Med. Chem. 2008, 16, 6841–6849. [Google Scholar] [CrossRef]
- Wu, X.; Wan, Y.; Mahalingam, A.K.; Murugaiah, A.M.S.; Plouffe, B.; Botros, M.; Karlén, A.; Hallberg, M.; Gallo-Payet, N.; Alterman, M. Selective angiotensin II AT2 receptor agonists: Arylbenzylimidazole structure-activity relationships. J. Med. Chem. 2006, 49, 7160–7168. [Google Scholar] [CrossRef]
- Sallander, J.; Wallinder, C.; Hallberg, A.; Åqvist, J.; Gutiérrez-De-Terán, H. Structural determinants of subtype selectivity and functional activity of angiotensin II receptors. Bioorganic Med. Chem. Lett. 2016, 26, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, M.; Sävmarker, A.; Hallberg, A. Angiotensin peptides as AT2 receptor agonists. Curr. Protein Pept. Sci. 2017, 18, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, M. Neuropeptides: Metabolism to Bioactive Fragments and the Pharmacology of their Receptors. Med. Res. Rev. 2015, 36, 464–519. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, F.; Hallberg, M. Peptide Conversion—A Potential Pathway Modulating G-Protein Signaling. Curr. Drug Targets 2006, 8, 147–154. [Google Scholar] [CrossRef]
- Fransson, R.; Botros, M.; Nyberg, F.; Lindeberg, G.; Sandström, A.; Hallberg, M. Small peptides mimicking substance P (1-7) and encompassing a C-terminal amide functionality. Neuropeptides 2008, 42, 31–37. [Google Scholar] [CrossRef]
- Hallberg, M.; Sandstrom, A. From the Anti-Nociceptive Substance P Metabolite Substance P (1-7) to Small Peptidomimetics. Curr. Protein Pept. Sci. 2018, 19, 1038–1048. [Google Scholar] [CrossRef]
- Braszko, J.J.; Kupryszewski, G.; Witczuk, B.; Wiśniewski, K. Angiotensin ii-(3-8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience 1988, 27, 777–783. [Google Scholar] [CrossRef]
- Lee, J.; Albiston, A.L.; Allen, A.M.; Mendelsohn, F.A.O.; Ping, S.E.; Barrett, G.L.; Murphy, M.; Morris, M.J.; Mcdowall, S.G.; Chai, S.Y. Effect of I.C.V. injection of AT4 receptor ligands, NLE 1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience 2004, 124, 341–349. [Google Scholar] [CrossRef]
- Diwakarla, S.; Nylander, E.; Grönbladh, A.; Vanga, S.R.; Khan, Y.S.; Gutierrez-de-Teran, H.; Ng, L.; Pham, V.; Sävmarker, J.; Lundbäck, T.; et al. Binding to and Inhibition of Insulin-Regulated Aminopeptidase by Macrocyclic Disulfides Enhances Spine Density. Mol. Pharmacol. 2016, 89, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Hallberg, M. Targeting the insulin-regulated aminopeptidase/AT4 receptor for cognitive disorders. Drug News Perspect. 2009, 22, 133–139. [Google Scholar] [CrossRef]
- Andersson, H.; Hallberg, M. Discovery of inhibitors of insulin-regulated aminopeptidase as cognitive enhancers. Int. J. Hypertens. 2012, 2012, 789671. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Pérodin, J.; Plante, H.; Řihakova, L.; Bernier, S.G.; Maletínská, L.; Guillemette, G.; Escher, E. N- and C-terminal structure-activity study of angiotensin II on the angiotensin AT2 receptor. Eur. J. Pharmacol. 1998, 343, 323–331. [Google Scholar] [CrossRef]
- Rosenström, U.; Sköld, C.; Lindeberg, G.; Botros, M.; Nyberg, F.; Hallberg, A.; Karlén, A. Synthesis and AT2 receptor-binding properties of angiotensin II analogues. J. Pept. Res. 2004, 64, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M. Update on angiotensin AT2 receptors. Curr. Opin. Nephrol. Hypertens. 2017, 26, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Carey, R.M. Blood Pressure and the Renal Actions of AT2Receptors. Curr. Hypertens. Rep. 2017, 19, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Padia, S.H.; Howell, N.L.; Siragy, H.M.; Carey, R.M. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension 2006, 47, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Siragy, H.M.; Fournie-Zaluski, M.C.; Roques, B.P.; Carey, R.M. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension 2007, 49, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Fournie-Zaluski, M.C.; Roques, B.P.; Carey, R.M. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension 2008, 51, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Kemp, B.A.; Bell, J.F.; Rottkamp, D.M.; Howell, N.L.; Shao, W.; Navar, L.G.; Padia, S.H.; Carey, R.M. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension 2012, 60, 387–395. [Google Scholar] [CrossRef]
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O.; et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 1–9. [Google Scholar] [CrossRef]
- Guimond, M.O.; Hallberg, M.; Gallo-Payet, N.; Wallinder, C. Saralasin and sarile are AT2 receptor agonists. ACS Med. Chem. Lett. 2014, 5, 1129–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksson, R.; Lindman, J.; Wannberg, J.; Sallander, J.; Backlund, M.; Baraldi, D.; Widdop, R.; Hallberg, M.; Åqvist, J.; Gutierrez de Teran, H.; et al. A Series of Analogues to the AT2R Prototype Antagonist C38 Allow Fine Tuning of the Previously Reported Antagonist Binding Mode. ChemistryOpen 2019, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-de-Terán, H.; Bello, X.; Rodríguez, D. Characterization of the dynamic events of GPCRs by automated computational simulations. Biochem. Soc. Trans. 2013, 41, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides †. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647–3661. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.S.; Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992, 97, 3100–3107. [Google Scholar] [CrossRef]
- Åqvist, J. Calculation of absolute binding free energies for charged ligands and effects of long-range electrostatic interactions. J. Comput. Chem. 1996, 17, 1587–1597. [Google Scholar] [CrossRef]
- Keränen, H.; Gutiérrez-de-Terán, H.; Åqvist, J. Structural and Energetic Effects of A2A Adenosine Receptor Mutations on Agonist and Antagonist Binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef]
- Boukharta, L.; Gutiérrez-de-Terán, H.; Åqvist, J.; Impey, R.; Klein, M. Computational Prediction of Alanine Scanning and Ligand Binding Energetics in G-Protein Coupled Receptors. PLoS Comput. Biol. 2014, 10, e1003585. [Google Scholar] [CrossRef] [Green Version]
- Keränen, H.; Åqvist, J.; Gutiérrez-de-Terán, H. Free energy calculations of A 2A adenosine receptor mutation effects on agonist binding. Chem. Commun. 2015, 51, 3522–3525. [Google Scholar] [CrossRef] [Green Version]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science (80-) 2020, 367, 888–892. [Google Scholar] [CrossRef]
- Regoli, D.; Rioux, F.; Park, W.K.; Choi, C. Role of the N-terminal Amino Acid for the Biological Activities of Angiotensin and Inhibitory Analogues. Can. J. Phisiology Pharmacol. 1974, 52, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Behrends, M.; Wallinder, C.; Wieckowska, A.; Guimond, M.O.; Hallberg, A.; Gallo-Payet, N.; Larhed, M. N-aryl isoleucine derivatives as angiotensin II AT2 receptor ligands. ChemistryOpen 2014, 3, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, D.K.; Heerding, J.N.; Krichavsky, M.Z.; Fluharty, S.J. Role of the amino terminus in ligand binding for the angiotensin II type 2 receptor. Mol. Brain Res. 1998, 57, 325–329. [Google Scholar] [CrossRef]
- Hines, J.; Heerding, J.N.; Fluharty, S.J.; Yee, D.K. Identification of angiotensin ii type 2 (at(2)) receptor domains mediating high-affinity cgp 42112a binding and receptor activation. J. Pharmacol. Exp. Ther. 2001, 298, 665–673. [Google Scholar]
- Whitebread, S.; Mele, M.; Kamber, B.; de Gasparo, M. Preliminary biochemical characterization of two angiotensin II receptor subtypes. Biochem. Biophys. Res. Commun. 1989, 163, 284–291. [Google Scholar] [CrossRef]
- Rosenström, U.; Sköld, C.; Plouffe, B.; Beaudry, H.; Lindeberg, G.; Botros, M.; Nyberg, F.; Wolf, G.; Karlén, A.; Gallo-Payet, N.; et al. New selective AT2 receptor ligands encompassing a γ-turn mimetic replacing the amino acid residues 4-5 of angiotensin II act as agonists. J. Med. Chem. 2005, 48, 4009–4024. [Google Scholar] [CrossRef]
- Georgsson, J.; Sköld, C.; Plouffe, B.; Lindeberg, G.; Botros, M.; Larhed, M.; Nyberg, F.; Gallo-Payet, N.; Gogoll, A.; Karlén, A.; et al. Angiotensin II pseudopeptides containing 1,3,5-trisubstituted benzene scaffolds with high AT2 receptor affinity. J. Med. Chem. 2005, 48, 6620–6631. [Google Scholar] [CrossRef]
- Rosenström, U.; Sköld, C.; Lindeberg, G.; Botros, M.; Nyberg, F.; Karlén, A.; Hallberg, A. A Selective AT2 Receptor Ligand with a γ-Turn-Like Mimetic Replacing the Amino Acid Residues 4-5 of Angiotensin II. J. Med. Chem. 2004, 47, 859–870. [Google Scholar] [CrossRef]
- Georgsson, J.; Rosenström, U.; Wallinder, C.; Beaudry, H.; Plouffe, B.; Lindeberg, G.; Botros, M.; Nyberg, F.; Karlén, A.; Gallo-Payet, N.; et al. Short pseudopeptides containing turn scaffolds with high AT2 receptor affinity. Bioorganic Med. Chem. 2006, 14, 5963–5972. [Google Scholar] [CrossRef]
- Georgsson, J.; Sköld, C.; Botros, M.; Lindeberg, G.; Nyberg, F.; Karlén, A.; Hallberg, A.; Larhed, M. Synthesis of a new class of druglike angiotensin II C-terminal mimics with affinity for the AT2 receptor. J. Med. Chem. 2007, 50, 1711–1715. [Google Scholar] [CrossRef]
- De Gasparo, M.; Whitebread, S.; Kamber, B.; Criscione, L.; Thomann, H.; Riniker, B.; Andreatta, R. Effect of covalent dimer conjugates of angiotensin II on receptor affinity and activity in vitro. J. Recept. Signal Transduct. 1991, 11, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Jespers, W.; Isaksen, G.V.; Andberg, T.A.H.; Vasile, S.; van Veen, A.; Åqvist, J.; Brandsdal, B.O.; Gutiérrez-de-Terán, H. QresFEP: An Automated Protocol for Free Energy Calculations of Protein Mutations in Q. J. Chem. Theory Comput. 2019, 15, 5461–5473. [Google Scholar] [CrossRef] [PubMed]
- Vasile, S.; Esguerra, M.; Jespers, W.; Oliveira, A.; Sallander, J.; Åqvist, J.; Gutiérrez-de-Terán, H. Characterization of Ligand Binding to GPCRs Through Computational Methods. In Computational Methods for GPCR Drug Discovery. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1705, ISBN 9781493974658. [Google Scholar]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, H.; Inoue, A.; Ngako Kadji, F.M.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T.; et al. The Crystal Structure of Angiotensin II Type 2 Receptor with Endogenous Peptide Hormone. Structure 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jespers, W.; Oliveira, A.; Prieto-Díaz, R.; Majellaro, M.; Åqvist, J.; Sotelo, E.; Gutiérrez-De-Terán, H. Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors. Molecules 2017, 22, 1–17. [Google Scholar] [CrossRef]
- Wallinder, C.; Sköld, C.; Botros, M.; Guimond, M.O.; Hallberg, M.; Gallo-Payet, N.; Karlén, A.; Alterman, M. Interconversion of functional activity by minor structural alterations in nonpeptide AT2 receptor ligands. ACS Med. Chem. Lett. 2015, 6, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Wallinder, C.; Sköld, C.; Sundholm, S.; Guimond, M.-O.; Yahiaoui, S.; Lindeberg, G.; Gallo-Payet, N.; Hallberg, M.; Alterman, M. High affinity rigidified AT 2 receptor ligands with indane scaffolds. MedChemComm 2019, 10, 2146–2160. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Pair | Chemical Modification | ΔΔGexp ± SEM (kcal/mol) | ΔΔGcalc ± SEM (kcal/mol) |
|---|---|---|---|
| 8 → 7 | Ile → Phe | 1.54 ± 0.06 | 0.97 ± 0.64 |
| 10 → 9 | Ile → Phe | 2.38 ± 0.07 | 0.55 ± 0.67 |
| 11 → 9 | Aniline → Phenyl | 0.59 ± 0.06 | 0.46 ± 0.36 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasile, S.; Hallberg, A.; Sallander, J.; Hallberg, M.; Åqvist, J.; Gutiérrez-de-Terán, H. Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists. Biomolecules 2020, 10, 649. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040649
Vasile S, Hallberg A, Sallander J, Hallberg M, Åqvist J, Gutiérrez-de-Terán H. Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists. Biomolecules. 2020; 10(4):649. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040649
Chicago/Turabian StyleVasile, Silvana, Anders Hallberg, Jessica Sallander, Mathias Hallberg, Johan Åqvist, and Hugo Gutiérrez-de-Terán. 2020. "Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists" Biomolecules 10, no. 4: 649. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040649