
Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement

 , ,
, ,
Abstract
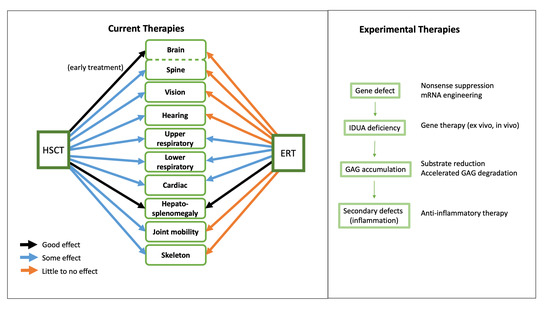
:
1. Introduction
2. Standard Therapies for MPS I
2.1. Allogeneic HSCT
2.1.1. Mortality Rates and Conditioning Regimens
2.1.2. Effectiveness of HSCT
2.2. Enzyme Replacement Therapy (ERT)
3. Impact of HSCT and ERT on Tissue-Specific Disease Manifestations
3.1. Ocular Manifestations
3.2. Respiratory System
3.3. Hearing Loss
3.4. Skeletal Manifestations
3.5. Joint Mobility
3.6. Cardiac Function
3.7. Cognitive Function
4. Experimental Therapies
4.1. Substrate Reduction
4.2. Accelerated GAG Degradation
4.3. Anti-Inflammatory Therapy
4.4. Intracerebroventricular and Intrathecal Delivery ERT
4.5. In Utero ERT Treatment
4.6. Shuttling of Enzyme Across the BBB
4.7. Molecular Therapies
4.7.1. Nonsense Suppression and mRNA Engineering
4.7.2. Ex Vivo Gene Transfer
4.7.3. In Vivo Gene Transfer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuiper, G.A.; Meijer, O.L.M.; Langereis, E.J.; Wijburg, F.A. Failure to shorten the diagnostic delay in two ultra-orphan diseases (mucopolysaccharidosis types I and III): Potential causes and implications. Orphanet J. Rare Dis. 2018, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.A.; Dickson, P.; Ellinwood, N.M.; Klein, T.L. Newborn screening for mucopolysaccharidosis I: Moving forward learning from experience. Int. J. Neonatal Screen. 2020, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44. [Google Scholar] [CrossRef] [PubMed]
- Eisengart, J.B.; Rudser, K.D.; Xue, Y.; Orchard, P.; Miller, W.; Lund, T.; Van der Ploeg, A.; Mercer, J.; Jones, S.; Mengel, K.E.; et al. Long-term outcomes of systemic therapies for Hurler syndrome: An international multicenter comparison. Genet. Med. 2018, 20, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic Cell Transplantation for Mucopolysaccharidosis Patients Is Safe and Effective: Results after Implementation of International Guidelines. Biol. Blood Marrow Transplant. 2015, 21, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Boelens, J.J.; Van Hasselt, P.M. Neurodevelopmental outcome after hematopoietic cell transplantation in inborn errors of metabolism: Current considerations and future perspectives. Neuropediatrics 2016, 47, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Lum, S.H.; Stepien, K.M.; Ghosh, A.; Broomfield, A.; Church, H.; Mercer, J.; Jones, S.; Wynn, R. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation. J. Inherit. Metab. Dis. 2017, 40, 455–460. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Nestrasil, I.; Rudser, K.; Delaney, K.; Kovac, V.; Ahmed, A.; Yund, B.; Orchard, P.J.; Eisengart, J.; Niklason, G.R.; et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015, 116, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Steward, C.G. Hematopoietic cell transplantation for inherited metabolic diseases: An overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003, 31, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Braunlin, E.A.; Rose, A.G.; Hopwood, J.J.; Candel, R.D.; Krivit, W. Coronary artery patency following long-term successful engraftment 14 years after bone marrow transplantation in the Hurler syndrome. Am. J. Cardiol. 2001, 88, 1075–1077. [Google Scholar] [CrossRef]
- Souillet, G.; Guffon, N.; Maire, I.; Pujol, M.; Taylor, P.; Sevin, F.; Bleyzac, N.; Mulier, C.; Durin, A.; Kebaili, K.; et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003, 31, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, D.; Connock, M.J.; Wraith, E.; Lavery, C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J. Rare Dis. 2008, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, N.J.; Kaizer, A.M.; Miller, W.P.; Rudser, K.D.; Orchard, P.J.; Braunlin, E.A. Mortality after hematopoietic stem cell transplantation for severe mucopolysaccharidosis type I: The 30-year University of Minnesota experience. J. Inherit. Metab. Dis. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.C.; Shapiro, E.G.; Anderson, J.; Henslee-downey, P.J.; Klemperer, M.R.; Cowan, M.J.; Saunders, E.F.; Pedro, A.; Twist, C.; Nachman, J.B.; et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood 1998, 91, 2601–2608. [Google Scholar] [CrossRef] [Green Version]
- Braunlin, E.A.; Stauffer, N.R.; Peters, C.H.; Bass, J.L.; Berry, J.M.; Hopwood, J.J.; Krivit, W. Usefulness of bone marrow transplantation in the Hurler syndrome. Am. J. Cardiol. 2003, 92, 882–886. [Google Scholar] [CrossRef]
- Kunin-Batson, A.S.; Shapiro, E.G.; Rudser, K.D.; Lavery, C.A.; Bjoraker, K.J.; Jones, S.A.; Wynn, R.F.; Vellodi, A.; Tolar, J.; Orchard, P.J. Long-term cognitive and functional outcomes in children with mucopolysaccharidosis (MPS)-IH (Hurler syndrome) treated with hematopoietic cell transplantation. JIMD Rep. 2016, 29, 95–102. [Google Scholar]
- Boelens, J.J.; Wynn, R.F.; O’Meara, A.; Veys, P.; Bertrand, Y.; Souillet, G.; Wraith, J.E.; Fischer, A.; Cavazzana-Calvo, M.; Sykora, K.W.; et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: A risk factor analysis for graft failure. Bone Marrow Transplant. 2007, 40, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.; Théoret, Y.; Rezgui, M.A.; Peters, C.; Mezziani, S.; Desjean, C.; Vachon, M.F.; Champagne, M.A.; Duval, M.; Krajinovic, M.; et al. Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematopoietic stem cell transplantation. Ther. Drug Monit. 2014, 36, 93–99. [Google Scholar] [CrossRef]
- Giorgiani, G.; Bozzola, M.; Locatelli, F.; Picco, P.; Zecca, M.; Cisternino, M.; Dallorso, S.; Bonetti, F.; Dini, G.; Borrone, C. Role of busulfan and total body irradiation on growth of prepubertal children receiving bone marrow transplantation and results of treatment with recombinant human growth hormone. Blood 1995, 86, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Allewelt, H.; El-Khorazaty, J.; Mendizabal, A.; Taskindoust, M.; Martin, P.L.; Prasad, V.; Page, K.; Sanders, J.; Kurtzberg, J. Late effects after umbilical cord blood transplantation in very young children after busulfan-based, myeloablative conditioning. Biol. Blood Marrow Transpl. 2016, 22, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Vatanen, A.; Wilhelmsson, M.; Borgström, B.; Gustafsson, B.; Taskinen, M.; Saarinen-Pihkala, U.M.; Winiarski, J.; Jahnukainen, K. Ovarian function after allogeneic hematopoietic stem cell transplantation in childhood and adolescence. Eur. J. Endocrinol. 2014, 170, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tichelli, A.; Rovó, A. Fertility issues following hematopoietic stem cell transplantation. Expert Rev. Hematol. 2013, 6, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Borgmann-Staudt, A.; Rendtorff, R.; Reinmuth, S.; Hohmann, C.; Keil, T.; Schuster, F.R.; Holter, W.; Ehlert, K.; Keslova, P.; Lawitschka, A.; et al. Fertility after allogeneic haematopoietic stem cell transplantation in childhood and adolescence. Bone Marrow Transplant. 2012, 47, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Dalle, J.H.; Lucchini, G.; Balduzzi, A.; Ifversen, M.; Jahnukainen, K.; MacKlon, K.T.; Ahler, A.; Jarisch, A.; Ansari, M.; Beohou, E.; et al. State-of-the-art fertility preservation in children and adolescents undergoing haematopoietic stem cell transplantation: A report on the expert meeting of the Paediatric Diseases Working Party (PDWP) of the European Society for Blood and Marrow Transplantation (EBMT) in Baden, Austria, 29–30 September 2015. Bone Marrow Transplant. 2017, 52, 1029–1035. [Google Scholar] [CrossRef] [Green Version]
- Bartelink, I.H.; van Reij, E.M.L.; Gerhardt, C.E.; van Maarseveen, E.M.; de Wildt, A.; Versluys, B.; Lindemans, C.A.; Bierings, M.B.; Boelens, J.J. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: Maintaining efficacy with less toxicity. Biol. Blood Marrow Transpl. 2014, 20, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Ben-Barouch, S.; Cohen, O.; Vidal, L.; Avivi, I.; Ram, R. Busulfan fludarabine vs busulfan cyclophosphamide as a preparative regimen before allogeneic hematopoietic cell transplantation: Systematic review and meta-analysis. Bone Marrow Transplant. 2016, 51, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Gyurkocza, B.; Sandmaier, B.M. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 2014, 124, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.D.; Filipovich, A.H.; Davies, S.M.; Mehta, P.; Bleesing, J.; Jodele, S.; Hayashi, R.; Barnes, Y.; Shenoy, S. Allogeneic hematopoietic cell transplantation (HCT) in Hurler’s syndrome using a reduced intensity preparative regimen. Bone Marrow Transplant. 2008, 41, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Langereis, E.J.; van Vlies, N.; Church, H.J.; Geskus, R.B.; Hollak, C.E.M.; Jones, S.A.; Kulik, W.; van Lenthe, H.; Mercer, J.; Schreider, L.; et al. Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long-term enzyme replacement therapy. Mol. Genet. Metab. 2015, 114, 129–137. [Google Scholar] [CrossRef]
- Wynn, R.; Wraith, E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.; Bigger, B. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.R.; Langereis, E.J.; Saif, M.A.; Mercer, J.; Church, H.J.; Tylee, K.L.; Wynn, R.F.; Wijburg, F.A.; Jones, S.A.; Bruce, I.A.; et al. Sleep disordered breathing in mucopolysaccharidosis I: A multivariate analysis of patient, therapeutic and metabolic correlators modifying long term clinical outcome. Orphanet J. Rare Dis. 2015, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; Defor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013, 121, 3981–3987. [Google Scholar] [CrossRef] [PubMed]
- Staba, S.L.; Escolar, M.L.; Poe, M.; Kim, Y.; Martin, P.L.; Szabolcs, P.; Allison-Thacker, J.; Wood, S.; Wenger, D.A.; Rubinstein, P.; et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N. Engl. J. Med. 2004, 350, 1960–1969. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, J.; Hugh-Jones, K.; Barrett, A.; Byrom, N.; James, D.C.O.; Lucas, C.F. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Kuiper, G.A.; van Hasselt, P.M.; Boelens, J.J.; Wijburg, F.A.; Langereis, E.J. Incomplete biomarker response in mucopolysaccharidosis type I after successful hematopoietic cell transplantation. Mol. Genet. Metab. 2017, 122, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Church, H.; Tylee, K.; Cooper, A.; Thornley, M.; Mercer, J.; Wraith, E.; Carr, T.; O’Meara, A.; Wynn, R.F. Biochemical monitoring after haemopoietic stem cell transplant for Hurler syndrome (MPSIH): Implications for functional outcome after transplant in metabolic disease. Bone Marrow Transplant. 2007, 39, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, A.; Webber, C.; Pidoux, I.; Choi, H.; Rosenberg, L. Localization of a Dermatan Sulfate Proteoglycan (DS-PGII) in Cartilage and the Presence of an Immunologically Related Species in Other Tissues. J. Histochem. Cytochem. 1986, 34, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande-Allen, K.J.; Clabro, A.; Gupta, V.; Wight, T.N.; Hascall, V.C.; Vesely, I. Glycosaminoglycans and proteoglycans in normal mitral valve leaflets and chordae: Association with regions of tensile and compressive loading. Glycobiology 2004, 14, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Ellinwood, N.M.; Colle, M.A.; Weil, M.A.; Casal, M.L.; Vite, C.H.; Wiemelt, S.; Hasson, C.W.; O’Malley, T.M.; He, X.; Prociuk, U.; et al. Bone marrow transplantation for feline mucopolysaccharidosis I. Mol. Genet. Metab. 2007, 91, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Pievani, A.; Azario, I.; Antolini, L.; Shimada, T.; Patel, P.; Remoli, C.; Rambaldi, B.; Valsecchi, M.; Riminucci, M.; Biondi, A.; et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 2014, 124. [Google Scholar] [CrossRef]
- Azario, I.; Pievani, A.; Del Priore, F.; Antolini, L.; Santi, L.; Corsi, A.; Cardinale, L.; Sawamoto, K.; Kubaski, F.; Gentner, B.; et al. Neonatal umbilical cord blood transplantation halts skeletal disease progression in the murine model of MPS-I. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogerbrugge, P.M.; Brouwer, O.F.; Bordigoni, P.; Cornu, G.; Kapaun, P.; Ortega, J.J.; O’Meara, A.; Souillet, G.; Frappaz, D.; Blanche, S.; et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. Lancet 1995, 345, 1398–1402. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 2008, 41, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, E.; Sung, J.; Manivel, J.C.; Chenggis, M.; Blazar, B.; Krivit, W. Male donor-derived cells in the brain of female sex-mismatched bone marrow transplant recipients: A y-chromosome specific in situ hybridization study. J. Neuropathol. Exp. Neurol. 1993, 52, 460–470. [Google Scholar] [CrossRef]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef]
- de Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; van der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme Replacement Therapy and/or Hematopoietic Stem Cell Transplantation at diagnosis in patients with Mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J. Rare Dis. 2011, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Krivit, W.; Sung, J.; Shapiro, E.G.; Lockman, L.A. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transpl. 1995, 4, 385–392. [Google Scholar] [CrossRef]
- Eisengart, J.; Rudser, K.; Tolar, J.; Orchared, P.; Kivisto, T.; Ziegler, R.S.; Whitley, C.; Shapiro, E. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J. Pediatrics 2013, 162, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Beck, M.; Lane, R.; van der Ploeg, A.; Shapiro, E.; Xue, Y.; Kakkis, E.D.; Guffon, N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 2007, 120, e37–e46. [Google Scholar] [CrossRef]
- Kakkis, E.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Rojas, V.M.; Martins, A.M.; Valadares, E.R.; Clarke, J.T.; Goes, J.E.; Kakkis, E.D.; Worden, M.A.; Sidman, M.; Cox, G.F. A dose-optimization trial of laronidase (Aldurazyme) in patients with mucopolysaccharidosis I. Mol. Genet. Metab. 2009, 96, 13–19. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous enzyme replacement therapy in mucopolysaccharidoses: Clinical effectiveness and limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef]
- Tylki-Szymanska, A.; Marucha, J.; Jurecka, A.; Syczewska, M.; Czartoryska, B. Efficacy of recombinant human α-L-iduronidase (laronidase) on restricted range of motion of upper extremities in mucopolysaccharidosis type I patients. J. Inherit. Metab. Dis. 2010, 33, 151–157. [Google Scholar] [CrossRef]
- Wiseman, D.H.; Mercer, J.; Tylee, K.; Malaiya, N.; Bonney, D.K.; Jones, S.A.; Wraith, J.E.; Wynn, R.F. Management of mucopolysaccharidosis type IH (Hurler’s syndrome) presenting in infancy with severe dilated cardiomyopathy: A single institution’s experience. J. Inherit. Metab. Dis. 2013, 36, 263–270. [Google Scholar] [CrossRef]
- Muenzer, J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 79, ISBN 4026501901147. [Google Scholar]
- White, K.K. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology 2011, 50, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Spina, V.; Barbuti, D.; Gaeta, A.; Palmucci, S.; Soscia, E.; Grimaldi, M.; Leone, A.; Manara, R.; Polonara, G. The role of imaging in the skeletal involvement of mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Breyer, S.; Löbel, U.; Yarar, S.; Stücker, R.; Ullrich, K.; Müller, I.; Muschol, N. Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J. Rare Dis. 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teär Fahnehjelm, K.; Olsson, M.; Chen, E.; Hengstler, J.; Naess, K.; Winiarski, J. Children with mucopolysaccharidosis risk progressive visual dysfunction despite haematopoietic stem cell transplants. Acta Paediatr. Int. J. Paediatr. 2018, 107, 1995–2003. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Richards, S.M.; Mahmood, A.; Cox, G.F. Effect of anti-laronidase antibodies on efficacy and safety of laronidase enzyme replacement therapy for MPS I: A comprehensive meta-analysis of pooled data from multiple studies. Mol. Genet. Metab. 2016, 117, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Eisengart, J.B.; Jarnes, J.; Ahmed, A.; Nestrasil, I.; Ziegler, R.; Delaney, K.; Shapiro, E.; Whitley, C. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol. Genet. Metab. Rep. 2017, 13, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, L.E.; Lund, T.C.; Braunlin, E.; Tolar, J.; Miller, B.S.; Fung, E.; Whitley, C.B.; Eisengart, J.B.; Northrop, E.; Rudser, K.; et al. Clinical trial of laronidase in Hurler syndrome after hematopoietic cell transplantation. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Kakavanos, R.; Turner, C.T.; Hopwood, J.J.; Kakkis, E.D.; Brooks, D.A. Immune tolerance after long-term enzyme-replacement therapy among patients who have mucopolysaccharidosis I. Lancet 2003, 361, 1608–1613. [Google Scholar] [CrossRef]
- Lund, T.C.; Miller, W.P.; Liao, A.Y.; Tolar, J.; Shanley, R.; Pasquali, M.; Sando, N.; Bigger, B.W.; Polgreen, L.E.; Orchard, P.J. Post-transplant laronidase augmentation for children with Hurler syndrome: Biochemical outcomes. Sci. Rep. 2019, 9, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Giugliani, R.; Vieira, T.A.; Carvalho, C.G.; Munoz-Rojas, M.V.; Semyachkina, A.N.; Voinova, V.Y.; Richards, S.; Cox, G.F.; Xue, Y. Immune tolerance induction for laronidase treatment in mucopolysaccharidosis I. Mol. Genet. Metab. Rep. 2017, 10, 61–66. [Google Scholar] [CrossRef]
- Ghosh, A.; Liao, A.; O’Leary, C.; Mercer, J.; Tylee, K.; Goenka, A.; Holley, R.; Jones, S.A.; Bigger, B.W. Strategies for the Induction of Immune Tolerance to Enzyme Replacement Therapy in Mucopolysaccharidosis Type I. Mol. Ther. Methods Clin. Dev. 2019, 13, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Kakkis, E.; Lester, T.; Yang, R.; Tanaka, C.; Anand, V.; Lemontt, J.; Peinovich, M.; Passage, M. Successful induction of immune tolerance to enzyme replacement therapy in canine mucopolysaccharidosis I. Proc. Natl. Acad. Sci. USA 2004, 101, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox-Brinkman, J.; Boelens, J.J.; Wraith, J.E.; O’Meara, A.; Veys, P.; Wijburg, F.A.; Wulffraat, N.; Wynn, R.F. Haematopoietic cell transplantation (HCT) in combination with enzyme replacement therapy (ERT) in patients with Hurler syndrome. Bone Marrow Transplant. 2006, 38, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, G.; Maximova, N.; Zennaro, F.; Gregori, M.; Tamaro, P. Hematopoietic stem cell transplantation effects on spinal cord compression in Hurler. Pediatr. Transplant. 2014, 18, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Miller, W.; Orchard, P.J.; Jones, S.A.; Mercer, J.; Church, H.J.; Tylee, K.; Lund, T.; Bigger, B.W.; Tolar, J.; et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in Mucopolysaccharidosis Type I: 10 year combined experience of 2 centres. Mol. Genet. Metab. 2016, 117, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolar, J.; Grewal, S.S.; Bjoraker, K.J.; Whitley, C.B.; Shapiro, E.G.; Charnas, L.; Orchard, P.J. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008, 41, 531–535. [Google Scholar] [CrossRef]
- Grewal, S.S.; Wynn, R.; Abdenur, J.E.; Burton, B.K.; Gharib, M.; Haase, C.; Hayashi, R.J.; Shenoy, S.; Sillence, D.; Tiller, G.E.; et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet. Med. 2005, 7, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Saif, M.A.; Bigger, B.W.; Brookes, K.E.; Mercer, J.; Tylee, K.L.; Church, H.J.; Bonney, D.K.; Jones, S.; Wraith, J.E.; Wynn, R.F. Hematopoietic stem cell transplantation improves the high incidence of neutralizing allo-antibodies observed in Hurler’s syndrome after pharmacological enzyme replacement therapy. Haematologica 2012, 97, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; Van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef] [Green Version]
- Santi, L.; De Ponti, G.; Dina, G.; Pievani, A.; Corsi, A.; Riminucci, M.; Khan, S.; Sawamoto, K.; Antolini, L.; Gregori, S.; et al. Neonatal combination therapy improves some of the clinical manifestations in the Mucopolysaccharidosis type I murine model. Mol. Genet. Metab. 2020, 130, 197–208. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Törnquist, A.L.; Malm, G.; Winiarski, J. Ocular findings in four children with mucopolysaccharidosis I-Hurler (MPS I-H) treated early with haematopoietic stem cell transplantation. Acta Ophthalmol. Scand. 2006, 84, 781–785. [Google Scholar] [CrossRef]
- Summers, C.G.; Purple, R.L.; Krivit, W.; Pineda, R.; Copland, G.T.; Ramsay, N.K.C.; Kersey, J.H.; Whitley, C.B. Ocular changes in the mucopolysaccharidoses after bone marrow transplantation: A preliminary report. Ophthalmology 1989, 96, 977–985. [Google Scholar] [CrossRef]
- Coletti, H.Y.; Aldenhoven, M.; Yelin, K.; Poe, M.D.; Kurtzberg, J.; Escolar, M.L. Long-term functional outcomes of children with hurler syndrome treated with unrelated umbilical cord blood transplantation. JIMD Rep. 2015, 20, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, A.; Aslam, T.; Jones, S.A.; Ashworth, J. Objective quantification of changes in corneal clouding over time in patients with mucopolysaccharidosis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 954–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guffon, N.; Souillet, G.; Maire, I.; Straczek, J.; Guibaud, P. Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J. Pediatr. 1998, 133, 119–125. [Google Scholar] [CrossRef]
- Van Den Broek, B.T.A.; Van Doorn, J.; Hegeman, C.V.; Nierkens, S.; Lindemans, C.A.; Verhoeven-Duif, N.; Boelens, J.J.; Van Hasselt, P.M. Hurdles in treating Hurler disease: Potential routes to achieve a “real” cure. Blood Adv. 2020, 4, 2837–2849. [Google Scholar] [CrossRef]
- Yuan, C.; Bothun, E.; Hardten, D.; Tolar, J.; McLoon, L. A novel explanation of corneal clouding in a bone marrow transplant-treated patient with Hurler syndrome. Exp. Eye Res. 2016, 148, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Gullingsrud, E.O.; Krivit, W.; Summers, C.G. Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer follow-up. Ophthalmology 1998, 105, 1099–1105. [Google Scholar] [CrossRef]
- Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Breider, M.A.; Shull, R.M.; Constantopoulos, G. Long-term effects of bone marrow transplantation in dogs with mucopolysaccharidosis I. Am. J. Pathol. 1989, 134, 677–692. [Google Scholar]
- Constantopoulos, G.; Scott, J.A.; Shull, R.M. Corneal opacity in canine MPS I. Changes after bone marrow transplantation. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1802–1807. [Google Scholar]
- Newkirk, K.M.; Atkins, R.M.; Dickson, P.I.; Rohrbach, B.W.; McEntee, M.F. Ocular lesions in canine mucopolysaccharidosis I and response to enzyme replacement therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5130–5135. [Google Scholar] [CrossRef]
- Pitz, S.; Ogun, O.; Bajbouj, M.; Arash, L.; Schulze-Frenking, G.; Beck, M. Ocular changes in patients with mucopolysaccharidosis I receiving enzyme replacement therapy: A 4-year experience. Arch. Ophthalmol. 2007, 125, 1353–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.A.; Visioli, F.; Pasqualim, G.; de Souza, C.F.M.; Marinho, D.R.; Giugliani, R.; Matte, U.; Baldo, G. Progressive eye pathology in mucopolysaccharidosis type I mice and effects of enzyme replacement therapy. Clin. Exp. Ophthalmol. 2020, 48, 334–342. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, J.; van den Broek, B.T.A.; Geboers, A.J.; Kuiper, G.A.; Boelens, J.J.; van Hasselt, P.M. Salivary α-Iduronidase activity as a potential new biomarker for the diagnosis and monitoring the effect of therapy in mucopolysaccharidosis type I. Biol. Blood Marrow Transplant. 2018, 24, 1808–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sornalingam, K.; Javed, A.; Aslam, T.; Sergouniotis, P.; Jones, S.; Ghosh, A.; Ashworth, J. Variability in the ocular phenotype in mucopolysaccharidosis. Br. J. Ophthalmol. 2019, 103, 504–510. [Google Scholar] [CrossRef]
- Ferrari, S.; Ponzin, D.; Ashworth, J.L.; Fahnehjelm, K.T.; Summers, C.G.; Harmatz, P.R.; Scarpa, M. Diagnosis and management of ophthalmological features in patients with mucopolysaccharidosis. Br. J. Ophthalmol. 2011, 95, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Belani, K.G.; Krivit, W.; Carpenter, B.L.; Braunlin, E.; Buckley, J.J.; Liao, J.C.; Floyd, T.; Leonard, A.S.; Summers, C.G.; Levine, S.; et al. Children with mucopolysaccharidosis: Perioperative care, morbidity, mortality, and new findings. J. Pediatr. Surg. 1993, 28, 403–410. [Google Scholar] [CrossRef]
- Yeung, A.H.; Cowan, M.J.; Horn, B.; Rosbe, K.W. Airway management in children with mucopolysaccharidoses. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Koehne, T.; Müller-Stöver, S.; Köhn, A.; Stumpfe, K.; Lezius, S.; Schmid, C.; Lukacs, Z.; Kahl-Nieke, B.; Muschol, N. Obstructive sleep apnea and craniofacial appearance in MPS type I-Hurler children after hematopoietic stem cell transplantation. Sleep Breath. 2019, 23, 1315–1321. [Google Scholar] [CrossRef]
- Malone, B.N.; Whitley, C.B.; Duvall, A.J.; Belani, K.; Sibley, R.K.; Ramsay, N.K.; Kersey, J.H.; Krivit, W.; Berlinger, N.T. Resolution of obstructive sleep apnea in Hurler syndrome after bone marrow transplantation. ; Int. J. Pediatr. Otorhinolaryngol. 1988, 15, 23–31. [Google Scholar] [CrossRef]
- Moreau, J.; Brassier, A.; Amaddeo, A.; Neven, B.; Caillaud, C.; Chabli, A.; Fernandez-Bolanos, M.; Olmo, J.; Valayannopoulos, V.; Fauroux, B. Obstructive sleep apnea syndrome after hematopoietic stem cell transplantation in children with mucopolysaccharidosis type I. Mol. Genet. Metab. 2015, 116, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, K.; Ellwood, J.; Walker, R.W. Mucopolysaccharidosis type I (Hurler syndrome) and anesthesia: The impact of bone marrow transplantation, enzyme replacement therapy, and fiberoptic intubation on airway management. Paediatr. Anaesth. 2012, 22, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Frawley, G.; Fuenzalida, D.; Donath, S.; Yaplito-Lee, J.; Peters, H. A retrospective audit of anesthetic techniques and complications in children with mucopolysaccharidoses. Paediatr. Anaesth. 2012, 22, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Megens, J.H.; de Wit, M.; van Hasselt, P.M.; Boelens, J.J.; van der Werff, D.B.; de Graaff, J.C. Perioperative complications in patients diagnosed with mucopolysaccharidosis and the impact of enzyme replacement therapy followed by hematopoietic stem cell transplantation at early age. Paediatr. Anaesth. 2014, 24, 521–527. [Google Scholar] [CrossRef]
- Madoff, L.U.; Kordun, A.; Cravero, J.P. Airway management in patients with mucopolysaccharidoses: The progression toward difficult intubation. Paediatr. Anaesth. 2019, 29, 620–627. [Google Scholar] [CrossRef]
- Laraway, S.; Mercer, J.; Jameson, E.; Ashworth, J.; Hensman, P.; Jones, S.A. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J. Pediatr. 2016, 178, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Jezela-Stanek, A.; Chorostowska-Wynimko, J.; Tylki-Szymańska, A. Pulmonary involvement in selected lysosomal storage diseases and the impact of enzyme replacement therapy: A state-of-the art review. Clin. Respir. J. 2020, 14, 422–429. [Google Scholar] [CrossRef]
- Dualibi, A.P.; Martins, A.M.; Moreira, G.A.; de Azevedo, M.F.; Fujita, R.R.; Pignatari, S.S. The impact of laronidase treatment in otolaryngological manifestations of patients with mucopolysaccharidosis. Braz. J. Otorhinolaryngol. 2016, 82, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the mucopolysaccharidoses. Rheumatology 2011, 50, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Broomfield, A.; Sims, J.; Mercer, J.; Hensman, P.; Ghosh, A.; Tylee, K.; Stepien, K.M.; Oldham, A.; Prathivadi Bhayankaram, N.; Wynn, R.; et al. The evolution of pulmonary function in childhood onset Mucopolysaccharidosis type I. Mol. Genet. Metab. 2020, 1. [Google Scholar] [CrossRef]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Gonzalez, E.A.; Tavares, A.M.V.; Seolin, B.G.; Elias, L.; de Sr. Vera, L.N.P.; Kubaski, F.; Poletto, E.; Giugliani, R.; Teixeira, H.F.; et al. Neonatal nonviral gene editing with the CRISPR/Cas9 system improves some cardiovascular, respiratory, and bone disease features of the mucopolysaccharidosis I phenotype in mice. Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Papsin, B.C.; Vellodi, A.; Bailey, C.M.; Racliffe, P.C.; Leighton, S.E.J. Otologic and laryngologic manifestations of mucopolysaccharidoses after bone marrow transplantation. Otolaryngol. Head Neck Surg. 1998, 118, 30–36. [Google Scholar] [CrossRef]
- Da Costa, V.; O’Grady, G.; Jackson, L.; Kaylie, D.; Raynor, E. Improvements in sensorineural hearing loss after cord blood transplant in patients with mucopolysaccharidosis. Arch. Otolaryngol. Head Neck Surg. 2012, 138, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis—a ten-year report from the China children transplant group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef]
- van den Broek, B.T.A.; Smit, A.L.; Boelens, J.J.; van Hasselt, P.M. Hearing loss in patients with mucopolysaccharidoses-1 and -6 after hematopoietic cell transplantation: A longitudinal analysis. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Tokic, V.; Barisic, I.; Huzjak, N.; Petkovic, G.; Fumic, K.; Paschke, E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur. J. Pediatr. 2007, 166, 727–732. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Reilly, C.; Human, D.; Waters, P.J.; Stoeckler-Ipsiroglu, S. Progression of organ manifestations upon enzyme replacement therapy in a patient with mucopolysaccharidosis type I/Hurler. World J. Pediatr. 2009, 5, 319–321. [Google Scholar] [CrossRef]
- Dornelles, A.D.; Lapagesse, L.; Pinto, D.C.; Paula, A.C.; de Eduardo, C.; Lourenço, C.M.; Kim, C.A.; Dain, D.; Horovitz, G.; Marques, E.; et al. Enzyme replacement therapy for Mucopolysaccharidosis Type I among patients followed within the MPS Brazil Network. Genet. Mol. Biol. 2014, 37, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Hugh-Jones, K. Psychomotor development of children with mucopolysaccharidosis type 1-H following bone marrow transplantation. Birth Defects Orig. Artic. Ser. 1986, 22, 25–29. [Google Scholar]
- Field, R.; Buchanan, J.; Copplemans, M.; Aichroth, P. Bone-marrow transplantation in Hurler’s syndrome. Effect on skeletal development. J. Bone Jt. Surg. Br. 1994, 76, 975–981. [Google Scholar] [CrossRef]
- Vellodi, A.; Young, E.P.; Cooper, A.; Wraith, J.E.; Winchester, B.; Meaney, C.; Ramaswami, U.; Will, A.; Marrow, B. Bone marrow transplantation for mucopolysaccharidosis type I: Experience of two British centres. Arch. Dis. Child 1997, 76, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polgreen, L.; Plog, M.; Schwender, J.; Tolar, J.; Thomas, W.; Orchard, P.; Miller, B.; Petryk, A. Short-term growth hormone treatment in children with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant. 2009, 44, 279–285. [Google Scholar] [CrossRef]
- Polgreen, L.; Tolar, J.; Plog, M.; Himes, J.; Orchard, P.; Whitley, C.; Miller, B.; Petryk, A. Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008, 41, 1005–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, V.; Williamson, J.B.; Cowie, R.A.; Wraith, J.E. Spinal problems in mucopolysaccharidosis I (Hurler syndrome). J. Bone Jt. Surg. Br. 1996, 78, 938–944. [Google Scholar] [CrossRef]
- Langereis, E.J.; den Os, M.M.; Breen, C.; Jones, S.A.; Knaven, O.C.; Mercer, J.; Miller, W.P.; Kelly, P.M.; Kennedy, J.; Ketterl, T.G.; et al. Progression of hip dysplasia in mucopolysaccharidosis type I hurler after successful hematopoietic stem cell transplantation. J. Bone Jt. Surg. 2016, 98, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Thawrani, D.P.; Walker, K.; Polgreen, L.; Tolar, J.; Orchard, P.J. Hip dysplasia in patients with Hurler syndrome (mucopolysaccharidosis type 1H). J. Pediatr. Orthop. 2013, 33, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Stoop, F.; Kruyt, M.; van der Linden, M.; Sakkers, R.; van Hasselt, P.; Casterlein, R. Prevalence and development of orthopaedic symptoms in the Dutch hurler patient population after haematopoietic stem cell transplantation. JIMD Rep. 2012, 17–29. [Google Scholar] [CrossRef]
- Yasin, M.N.; Sacho, R.; Oxborrow, N.J.; Wraith, J.E.; Williamson, J.B.; Siddique, I. Thoracolumbar kyphosis in treated mucopolysaccharidosis 1 (hurler syndrome). Spine 2014, 39, 381–387. [Google Scholar] [CrossRef]
- Abelin Genevois, K.; Garin, C.; Solla, F.; Guffon, N.; Kohler, R. Surgical management of thoracolumbar kyphosis in mucopolysaccharidosis type 1 in a reference center. J. Inherit. Metab. Dis. 2014, 37, 69–78. [Google Scholar] [CrossRef]
- Tomatsu, S.; Alméciga-Díaz, C.J.; Montaño, A.; Yabe, H.; Tanaka, A.; Dung, V.; Giugliani, R.; Kubaski, F.; Mason, R.W.; Yasuda, E.; et al. Therapies for the bone in mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukimura, T.; Tajima, Y.; Kawashima, I.; Fukushige, T.; Kanzaki, T.; Kanekura, T.; Ikekita, M.; Sugawara, K.; Suzuki, T.; Togawa, T.; et al. Uptake of a recombinant human α-L-iduronidase (laronidase) by cultured fibroblasts and osteoblasts. Biol. Pharm. Bull. 2008, 31, 1691–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hite, S.; Peters, C.; Krivit, W. Correction of odontoid dysplasia following bone-marrow transplantation and engraftment (in Hurler syndrome MPS 1H). Pediatr. Radiol. 2000, 30, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Miebach, E. Management of infusion-related reactions to enzyme replacement therapy in a cohort of patients with mucopolysaccharidosis disorders. Int. J. Clin. Pharmacol. Ther. 2009, 47 (Suppl. 1), S100–S106. [Google Scholar] [CrossRef] [PubMed]
- Chiaro, J.A.; O’Donnell, P.; Shore, E.M.; Malhotra, N.R.; Ponder, K.P.; Haskins, M.E.; Smith, L.J. Effects of neonatal enzyme replacement therapy and simvastatin treatment on cervical spine disease in mucopolysaccharidosis I dogs. J. Bone Min. Res. 2014, 29, 2610–2617. [Google Scholar] [CrossRef] [Green Version]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymanska, A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteroarthritis Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Gabrielli, O.; Clarke, L.A.; Ficcadenti, A.; Santoro, L.; Zampini, L.; Volpi, N.; Coppa, G. V 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: The important role of early treatment. BMC Med. Genet. 2016, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Guelbert, N.; Jones, S.A.; Kyosen, S.O.; Martins, A.M.; Phornphutkul, C.; et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: A retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, S.C.; Koehne, T.; Cornils, K.; Markmann, S.; Riedel, C.; Pestka, J.M.; Schweizer, M.; Baldauf, C.; Yorgan, T.A.; Krause, M.; et al. Impaired bone remodeling and its correction by combination therapy in a mouse model of mucopolysaccharidosis-I. Hum. Mol. Genet. 2015, 24, 7075–7086. [Google Scholar] [CrossRef]
- Baldo, G.; Mayer, F.Q.; Martinelli, B.Z.; de Carvalho, T.G.; Meyer, F.S.; de Oliveira, P.G.; Meurer, L.; Tavares, Â.; Matte, U.; Giugliani, R. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2013, 109, 33–40. [Google Scholar] [CrossRef]
- Clarke, L.A. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: Glycosaminoglycan storage is merely the instigator. Rheumatology 2011, 50, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of glycosaminoglycan-mediated bone and joint disease: Implications for the mucopolysaccharidoses and other connective tissue diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.F.; Muenzer, I. The Metabolic & Molecular Basis of Inherited Disease, 8th ed; Scriver, C.R., Beudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 3. [Google Scholar]
- Weisstein, J.S.; Delgado, E.; Steinbach, L.S.; Hart, K.; Packman, S. Musculoskeletal manifestations of Hurler syndrome: Long-term follow-up after bone marrow transplantation. J. Pediatr. Orthop. 2004, 24, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.A.; Barreto, R.; Acosta, A.X. Evaluation of motor response in mucopolysaccharidosis patients treated with enzyme replacement therapy. Ortop. Traumatol. Rehabil. 2013, 15, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, G.; Van Heest, A.E.; Agel, J.; Bjoraker, K.; Grewal, S.; Abel, S.; Krivit, W.; Peters, C.; Orchard, P.J. Analysis of factors affecting development of carpal tunnel syndrome in patients with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant. 2007, 39, 331–334. [Google Scholar] [CrossRef] [Green Version]
- Wyffels, M.L.; Orchard, P.J.; Shanley, R.M.; Miller, W.P.; Van Heest, A.E. The frequency of carpal tunnel syndrome in hurler syndrome after peritransplant enzyme replacement therapy: A retrospective comparison. J. Hand Surg. Am. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guffon, N.; Bertrand, Y.; Forest, I.; Fouilhoux, A.; Froissart, R. Bone marrow transplantation in children with hunter syndrome: Outcome after 7 to 17 Years. J. Pediatr. 2009, 154, 733–737. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Vellodi, A.; Redington, A.N. Cardiac involvement in mucopolysaccharidoses: Effects of allogeneic bone marrow transplantation. Arch. Dis. Child. 1995, 73, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Viñallonga, X.; Sanz, N.; Balaguer, A.; Miro, L.; Ortega, J.J.; Casaldaliga, J. Hypertrophic cardiomyopathy in mucopolysaccharidoses: Regression after bone marrow transplantation. Pediatr. Cardiol. 1992, 13, 107–109. [Google Scholar] [CrossRef]
- Orchard, P.J.; Blazar, B.R.; Wagner, J.; Charnas, L.; Krivit, W.; Tolar, J. Hematopoietic cell therapy for metabolic disease. J. Pediatr. 2007, 151, 340–346. [Google Scholar] [CrossRef]
- Malm, G.; Gustafsson, B.; Berglund, G.; Lindström, M.; Naess, K.; Borgström, B.; Von Döbeln, U.; Ringdén, O. Outcome in six children with mucopolysaccharidosis type IH, hurler syndrome, after haematopoietic stem cell transplantation (HSCT). Acta Paediatr. Int. J. Paediatr. 2008, 97, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Gompf, R.E.; Shull, R.; Breider, M.A.; Scott, J.A.; Constantopoulos, G.C. Cardiovascular changes after bone marrow transplantation in dogs with mucopolysaccharidosis I. Am. J. Vet. Res. 1990, 51, 2054–2060. [Google Scholar] [PubMed]
- Lin, H.Y.; Chan, W.C.; Chen, L.J.; Lee, Y.C.; Yeh, S.I.; Niu, D.M.; Chiu, P.C.; Tsai, W.H.; Hwu, W.L.; Chuang, C.K.; et al. Ophthalmologic manifestations in Taiwanese patients with mucopolysaccharidoses. Mol. Genet. Genomic Med. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunlin, E.A.; Berry, J.M.; Whitley, C.B. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am. J. Cardiol. 2006, 98, 416–418. [Google Scholar] [CrossRef]
- Okuyama, T.; Tanaka, A.; Suzuki, Y.; Ida, H.; Tanaka, T.; Cox, G.F.; Eto, Y.; Orii, T. Japan Elaprase Treatment (JET) study: Idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol. Genet. Metab. 2010, 99, 18–25. [Google Scholar] [CrossRef]
- Brands, M.M.; Frohn-Mulder, I.M.; Hagemans, M.L.; Hop, W.C.; Oussoren, E.; Helbing, W.A.; van der Ploeg, A.T. Mucopolysaccharidosis: Cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI. J. Inherit. Metab. Dis. 2013, 36, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef]
- Braunlin, E.; Miettunen, K.; Lund, T.; Luquette, M.; Orchard, P. Hematopoietic cell transplantation for severe MPS I in the first six months of life: The heart of the matter. Mol. Genet. Metab. 2019, 126, 117–120. [Google Scholar] [CrossRef]
- Pasqualim, G.; Baldo, G.; de Carvalho, T.G.; Tavares, A.M.V.; Giugliani, R.; Matte, U. Effects of enzyme replacement therapy started late in a murine model of mucopolysaccharidosis type I. PLoS ONE 2015, 10, e0117271. [Google Scholar] [CrossRef]
- Dickson, P.; Peinovich, M.; McEntee, M.; Lester, T.; Le, S.; Krieger, A.; Manuel, H.; Jabagat, C.; Passage, M.; Kakkis, E.D. Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I. J. Clin. Investig. 2008, 118, 2868–2876. [Google Scholar] [CrossRef] [Green Version]
- Bjoraker, K.J.; Delaney, K.; Peters, C.; Krivit, W.; Shapiro, E.G. Long-term outcomes of adaptive functions for children with mucopolysaccharidosis I (Hurler syndrome) treated with hematopoietic stem cell transplantation. J. Dev. Behav. Pediatr. 2006, 27, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in mucopolysaccharidosis type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early treatment is associated with improved cognition in Hurler syndrome. Ann. Neurol. 2014, 76, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Herzog, T.L.; Wilmot, C.M.; Whitley, C.B. Standardization of α-L-iduronidase enzyme assay with Michaelis-Menten kinetics. Mol. Genet. Metab. 2014, 111, 113–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Amouri, S.S.; Dai, M.; Han, J.F.; Brady, R.O.; Pan, D. Normalization and improvement of CNS deficits in mice with hurler syndrome after long-term peripheral delivery of BBB-targeted iduronidase. Mol. Ther. 2014, 22, 2028–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogler, C.; Levy, B.; Grubb, J.H.; Galvin, N.; Tan, Y.; Kakkis, E.; Pavloff, N.; Sly, W.S. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA 2005, 102, 14777–14782. [Google Scholar] [CrossRef] [Green Version]
- Derrick-Roberts, A.; Jackson, M.; Pyragius, C.; Byers, S. Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice. Diseases 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Rintz, E.; Pierzynowska, K.; Podlacha, M.; Węgrzyn, G. Has resveratrol a potential for mucopolysaccharidosis treatment? Eur. J. Pharmacol. 2020, 888. [Google Scholar] [CrossRef]
- JOTROLTM Pre-Clinical Studies. Available online: http://www.jupiterorphan.com/jotroltrade8203-pre-clinical-studies.html (accessed on 15 December 2020).
- Simonaro, C.M.; Tomatsu, S.; Sikora, T.; Kubaski, F.; Frohbergh, M.; Guevara, J.M.; Wang, R.Y.; Vera, M.; Kang, J.L.; Smith, L.J.; et al. Pentosan polysulfate: Oral versus subcutaneous injection in mucopolysaccharidosis type I dogs. PLoS ONE 2016, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hennermann, J.B.; Gökce, S.; Solyom, A.; Mengel, E.; Schuchman, E.H.; Simonaro, C.M. Treatment with pentosan polysulphate in patients with MPS I: Results from an open label, randomized, monocentric phase II study. J. Inherit. Metab. Dis. 2016, 39, 831–837. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Kunin-Batson, A.; Rudser, K.; Vehe, R.K.; Utz, J.J.; Whitley, C.B.; Dickson, P. Pilot study of the safety and effect of adalimumab on pain, physical function, and musculoskeletal disease in mucopolysaccharidosis types I and II. Mol. Genet. Metab. Rep. 2017, 10, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Vite, C.H.; Nestrasil, I.; Mlikotic, A.; Jens, J.K.; Snella, E.; Gross, W.; Shapiro, E.G.; Kovac, V.; Provenzale, J.M.; Chen, S.; et al. Features of brain MRI in dogs with treated and untreated mucopolysaccharidosis type I. Comp. Med. 2013, 63, 163–173. [Google Scholar] [PubMed]
- Nan, Z.; Shekels, L.; Ryabinin, O.; Evavold, C.; Nelson, M.S.; Khan, S.A.; Deans, R.J.; Mays, R.W.; Low, W.C.; Gupta, P. Intracerebroventricular transplantation of human bone marrow-derived multipotent progenitor cells in an immunodeficient mouse model of mucopolysaccharidosis type I (MPS-I). Cell Transplant. 2012, 21, 1577–1593. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Rojas, M.V.; Vieira, T.; Costa, R.; Fagondes, S.; John, A.; Jardim, L.B.; Vedolin, L.M.; Raymundo, M.; Dickson, P.I.; Kakkis, E.; et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am. J. Med. Genet. A 2008, 146A, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.I.; Kaitila, I.; Harmatz, P.; Mlikotic, A.; Chen, A.H.; Victoroff, A.; Passage, M.B.; Madden, J.; Le, S.Q.; Naylor, D.E.; et al. Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mucopolysaccharidosis I. Mol. Genet. Metab. 2015, 116, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.H.; Harmatz, P.; Nestrasil, I.; Eisengart, J.B.; King, K.E.; Rudser, K.; Kaizer, A.M.; Svatkova, A.; Wakumoto, A.; Le, S.Q.; et al. Intrathecal enzyme replacement for cognitive decline in mucopolysaccharidosis type I, a randomized, open-label, controlled pilot study. Mol. Genet. Metab. 2020, 129, 80–90. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Pierpont, E.I.; Kaizer, A.M.; Rudser, K.D.; King, K.E.; Pasquali, M.; Polgreen, L.E.; Dickson, P.I.; Le, S.Q.; Miller, W.P.; et al. Intrathecal enzyme replacement for Hurler syndrome: Biomarker association with neurocognitive outcomes. Genet. Med. 2019, 21, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Lutzko, C.; Omori, F.; Abrams-Ogg, A.C.G.; Shull, R.; Li, L.; Lau, K.; Ruedy, C.; Nanji, S.; Gartley, C.; Dobson, H.; et al. Gene therapy for canine α-L-iduronidase deficiency: In utero adoptive transfer of genetically corrected hematopoietic progenitors results in engraftment but not amelioration of disease. Hum. Gene Ther. 1999, 10, 1521–1532. [Google Scholar] [CrossRef]
- Meertens, L.; Kohn, D.; Kruth, S.; Hough, M.R.; Dubé, I.D.; Zhao, Y.; Rosic-Kablar, S.; Li, L.; Chan, K.; Dobson, H.; et al. In utero injection of α-L-iduronidase-carrying retrovirus in canine mucopolysaccharidosis type I: Infection of multiple tissues and neonatal gene expression. Hum. Gene Ther. 2002, 13, 1809–1820. [Google Scholar] [CrossRef]
- Wang, D.; El-Amouri, S.S.; Dai, M.; Kuan, C.-Y.; Hui, D.; Brady, R.O.; Pan, D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2013, 110, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Przybilla, M.; Koniar, B.; Whitley, C.B. RTB lectin-mediated delivery of lysosomal α-L-iduronidase mitigates disease manifestations systemically including the central nervous system. Mol. Genet. Metab. 2018, 123, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.Y.; Xia, C.; Wang, Y.; Pardridge, W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 99, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Hui, E.K.-W.; Lu, J.Z.; Zhou, Q.-H.; Pardridge, W.M. Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol. Pharm. 2011, 8, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.-W.; Lin, H.; Pardridge, W.M. Insulin receptor antibody-alpha-N-acetylglucosaminidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in sanfilippo type B fibroblasts. Mol. Pharm. 2016, 13, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Dalla Corte, A.; Poswar, F.; Vanzella, C.; Horovitz, D.; Riegel, M.; Baldo, G.; Vairo, F. Intrathecal/Intracerebroventricular enzyme replacement therapy for the mucopolysaccharidoses: Efficacy, safety, and prospects. Expert Opin. Orphan Drugs 2018, 6, 403–411. [Google Scholar] [CrossRef]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: A phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.J.; McElmurry, R.T.; Peacock, B.; Tolar, J.; Blazar, B.R. Targeting of the CNS in MPS-IH using a nonviral transferrin-α-L-iduronidase fusion gene product. Mol. Ther. 2008, 16, 1459–1466. [Google Scholar] [CrossRef]
- Lee, H.L.R.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef]
- Gunn, G.; Dai, Y.; Du, M.; Belakhov, V.; Kandasamy, J.; Schoeb, T.R.; Baasov, T.; Bedwell, D.M.; Keeling, K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014, 111, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Liu, N. A novel oligonucleotide-based RNA base editing therapeutic approach for the treatment of Hurler syndrome. Mol. Ther. 2020, 28, 18. [Google Scholar]
- Wang, J.; Ren, L.; Li, J.; Gao, G.; Wang, D. dCas13-Mediated Therapeutic RNA Base Editing for In Vivo Gene Therapy. Mol. Ther. 2020, 28, R3–R14. [Google Scholar]
- Osborn, M.J.; Levy, J.M.; Newby, G.A.; McElroy, A.N.; Nielsen, S.A.; Liu, D.R.; Tolar, J. In vivo base editing to correct a murine model of mucopolysaccharidosis type IH. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar]
- Mendonca, C.; Ren, L.; Li, J.; Zhang, Y.; Min, J.; Wang, J.; Su, Q.; Gao, G.; Wang, D. In vivo suppressor tRNA mediated readthrough therapy for nonsense mutations. Mol. Ther. 2020, 28, 120. [Google Scholar]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, W.; Kalfa, T.; Grabowski, G.; Davies, S.; Malik, P.; Pan, D. Reprogramming erythroid cells for lysosomal enzyme production leads to visceral and CNS cross-correction. Proc. Natl. Acad. Sci. USA 2009, 106, 19958–19963. [Google Scholar] [CrossRef] [Green Version]
- Shull, R.; Lu, X.; Dubé, I.; Lutzko, C.; Kruth, S.; Abrams-Ogg, A.; Kiem, H.P.; Goehle, S.; Schuening, F.; Millan, C.; et al. Humoral immune response limits gene therapy in canine MPS I. Blood 1996, 88, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Lutzko, C.; Kruth, S.; Abrams-Ogg, A.; Lau, K.; Clark, B.; Ruedy, C.; Nanji, S.; Foster, R.; Kohn, D.; Shull, R.; et al. Genetically corrected autologous stem cells engraft, but host immune responses limit their utility in canine alpha-L-iduronidase deficiency. Blood 1999, 93, 1895–1905. [Google Scholar]
- Gentner, B.; Bernardo, M.E.; Tucci, F.; Zonari, E.; Fumagalli, F.; Pontesilli, S.; Acquati, S.; Silvani, P.; Ciceri, F.; Rovelli, A.; et al. Extensive metabolic correction of hurler disease by hematopoietic stem cell-based gene therapy: Preliminary results from a phase I/II trial. Blood 2019, 134, 607. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Aronovich, E.L.; Bell, J.B.; Belur, L.R.; Gunther, R.; Erickson, D.C.C.; Schachern, P.A.; Matise, I.; McIvor, R.S.; Whitley, C.B.; Hackett, P.B.; et al. Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: Implications for non-viral gene therapy of mucopolysaccharidoses. J. Gene Med. 2007, 9, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Aronovich, E.L.; Hyland, K.A.; Hall, B.C.; Bell, J.B.; Olson, E.R.; Rusten, M.U.; Hunter, D.W.; Ellinwood, N.M.; McIvor, R.S.; Hackett, P.B. Prolonged expression of secreted enzymes in dogs after liver-directed delivery of sleeping beauty transposons: Implications for non-viral gene therapy of systemic disease. Hum. Gene Ther. 2017, 28, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronovich, E.L.; Bell, J.B.; Khan, S.A.; Belur, L.R.; Gunther, R.; Koniar, B.; Schachern, P.A.; Parker, J.B.; Carlson, C.S.; Whitley, C.B.; et al. Systemic correction of storage disease in MPS I NOD/SCID mice using the sleeping beauty transposon system. Mol. Ther. 2009, 17, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Olson, E.; de Laat, R.; Meeker, K.; Lund, T.C.; Swietlicka, M.; Wesley, J.; Xu, M.; Grandea, G.; Scholz, M.; et al. Sleeping beauty IDUA transposed human plasma cells for long-term treatment of an immunodeficient murine model of mucopolysaccharidosis Type I. Mol. Ther. 2020, 28, 580–589. [Google Scholar]
- Laoharawee, K.; Johnson, M.; Peterson, J.; Yamamoto, K.; Lahr, W.; Webber, B.R.; Moriarity, B.S. Engineered B cells as a novel and sustainable cell-based enzyme replacement therapy for Hurler/Scheie syndrome. Mol. Ther. 2020, 28, 178–187. [Google Scholar]
- Donovan, M.; Makino, E.; Fluharty, B.; Tietz, D.; Pearson, E.; Jansen, L.; Barney, L.; Sewel, J.; Huang, J.; Corzo, D.; et al. Preclinical development of SIG-005 for treatment of MPS I. Mol. Genet. Metab. 2019, 129, S50. [Google Scholar] [CrossRef]
- Miyadera, K.; Conatser, L.; Llanga, T.A.; Carlin, K.; O’Donnell, P.; Bagel, J.; Song, L.; Kurtzberg, J.; Samulski, R.J.; Gilger, B.; et al. Intrastromal gene therapy prevents and reverses advanced corneal clouding in a canine model of mucopolysaccharidosis I. Mol. Ther. 2020, 28, 1455–1463. [Google Scholar] [CrossRef]
- Hartung, S.D.; Frandsen, J.L.; Pan, D.; Koniar, B.L.; Graupman, P.; Gunther, R.; Low, W.C.; Whitley, C.B.; McIvor, R.S. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol. Ther. 2004, 9, 866–875. [Google Scholar] [CrossRef]
- Wolf, D.A.; Lenander, A.W.; Nan, Z.; Belur, L.R.; Whitley, C.B.; Gupta, P.; Low, W.C.; McIvor, R.S. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol. Dis. 2011, 43, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Belur, L.R.; Temme, A.; Podetz-Pedersen, K.M.; Riedl, M.; Vulchanova, L.; Robinson, N.; Hanson, L.R.; Kozarsky, K.F.; Orchard, P.J.; Frey, W.H., 2nd; et al. Intranasal adeno-associated virus mediated gene delivery and expression of human iduronidase in the central nervous system: A noninvasive and effective approach for prevention of neurologic disease in mucopolysaccharidosis type I. Hum. Gene Ther. 2017, 28, 576–587. [Google Scholar] [CrossRef]
- Hinderer, C.; Bell, P.; Gurda, B.L.; Wang, Q.; Louboutin, J.-P.; Zhu, Y.; Bagel, J.; O’Donnell, P.; Sikora, T.; Ruane, T.; et al. Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc. Natl. Acad. Sci. USA 2014, 111, 14894–14899. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Hinderer, C.; Buza, E.L.; Louboutin, J.P.; Jahan, T.; Bell, P.; Chichester, J.A.; Tarantal, A.F.; Wilson, J.M. Safe and sustained expression of human iduronidase after intrathecal administration of adeno-associated virus serotype 9 in infant rhesus monkeys. Hum. Gene Ther. 2019, 30, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, L.; Hennig, A.K.; Kovacs, A.; Fu, A.; Chung, S.; Lee, D.; Wang, B.; Herati, R.S.; Mosinger Ogilvie, J.; et al. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol. Ther. 2005, 11, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Tittiger, M.; Hennig, A.; Kovacs, A.; Popelka, S.; Wang, B.; Herati, R.; Bigg, M.; Ponder, K.P. Improvements in mucopolysaccharidosis I mice after adult retroviral Vector-mediated gene therapy with immunomodulation. Mol. Ther. 2007, 15, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; De Kelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; St. Martin, S.J.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol. Ther. [CrossRef] [PubMed]

{kind=link}
| Clinical Manifestation | HSCT | ERT | HSCT + ERT |
|---|---|---|---|
| Partial improvement with added benefit of combination therapy | |||
| Cognitive function | Stabilization | No effect | Improvement |
| Pulmonary function | Limited improvement | Improvement | Improvement |
| Skeletal manifestations |
| No effect | Improved growth rate |
| Partial improvement | |||
| Upper respiratory | Improvement | Improvement | Improvement |
| Joint mobility | Improved range of motion | Improved range of motion (shoulder) | NA |
| Cardiac function |
|
| Improvement |
| Limited effect | |||
| Hearing loss | Improvement/ stabilization | No effect | NA |
| Corneal clouding | Limited stabilization | No effect | NA |
| Retinal dysfunction | No effect | No effect | NA |
| Hearing loss | Improvement/ stabilization | No effect | NA |
| Drug | Clinical Trials |
|---|---|
| Anti-inflammatory therapy | |
| Adalimumab | Phase I/II: NCT02437253: completed NCT03153319: recruiting |
| In utero ERT | |
| laronidase | Phase I: NCT04532047: not yet recruiting |
| Intrathecal delivery | |
| laronidase | Phase I: NCT00215527, NCT00786968: terminated due to slow enrolment) Phase not applicable: NCT00852358: completed NCT02232477: terminated due to COVID-19 |
| laronidase with HSCT | Phase I: NCT00638547: completed |
| BBB-crossing IDUA-fusion proteins | |
| AGT-181 (fusion to Insulin receptor monoclonal antibody) | Phase I/II: NCT03071341, NCT03053089, NCT02597114: completed |
| JR-171 (Undisclosed fusion partner) | Phase I/II: NCT04227600, NCT04453085: not yet recruiting |
| Ex vivo gene transfer | |
| Autologous HSPC transduced with IDUA | Phase I/II: NCT03488394: recruiting |
| ISP-001 (B cells transposed with IDUA) | Phase I/II: NCT04284254: not yet recruiting |
| In vivo gene transfer | |
| RGX-111 (AAV9-mediated) | Phase I/II: NCT03580083: recruiting |
| SB-318 (Genome editing ZFN) | Phase I/II: NCT02702115: active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hampe, C.S.; Wesley, J.; Lund, T.C.; Orchard, P.J.; Polgreen, L.E.; Eisengart, J.B.; McLoon, L.K.; Cureoglu, S.; Schachern, P.; McIvor, R.S. Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement. Biomolecules 2021, 11, 189. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020189
Hampe CS, Wesley J, Lund TC, Orchard PJ, Polgreen LE, Eisengart JB, McLoon LK, Cureoglu S, Schachern P, McIvor RS. Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement. Biomolecules. 2021; 11(2):189. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020189
Chicago/Turabian StyleHampe, Christiane S., Jacob Wesley, Troy C. Lund, Paul J. Orchard, Lynda E. Polgreen, Julie B. Eisengart, Linda K. McLoon, Sebahattin Cureoglu, Patricia Schachern, and R. Scott McIvor. 2021. "Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement" Biomolecules 11, no. 2: 189. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020189