
Testicular “Inherited Metabolic Memory” of Ancestral High-Fat Diet Is Associated with Sperm sncRNA Content

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Sperm Collection and RNA Extraction
2.3. cDNA Library Preparation and Next Generation Sequencing
2.4. Processing and Annotation of Sequenced Reads
2.5. Annotation of piRNA Clusters and Repeats
2.6. Term Enrichment Analysis
2.7. Statistics
3. Results
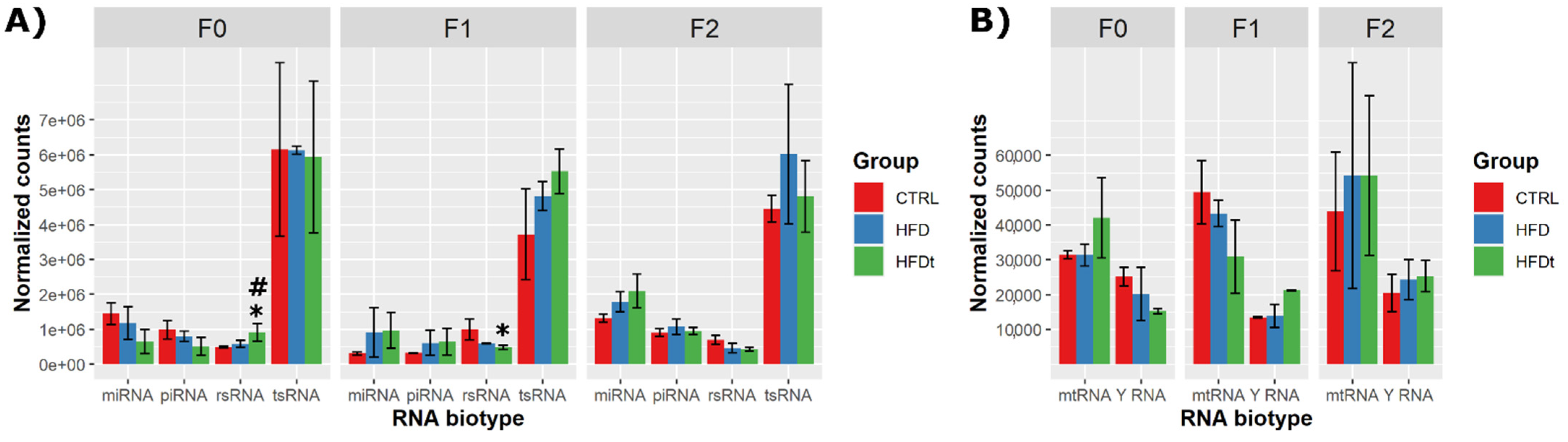
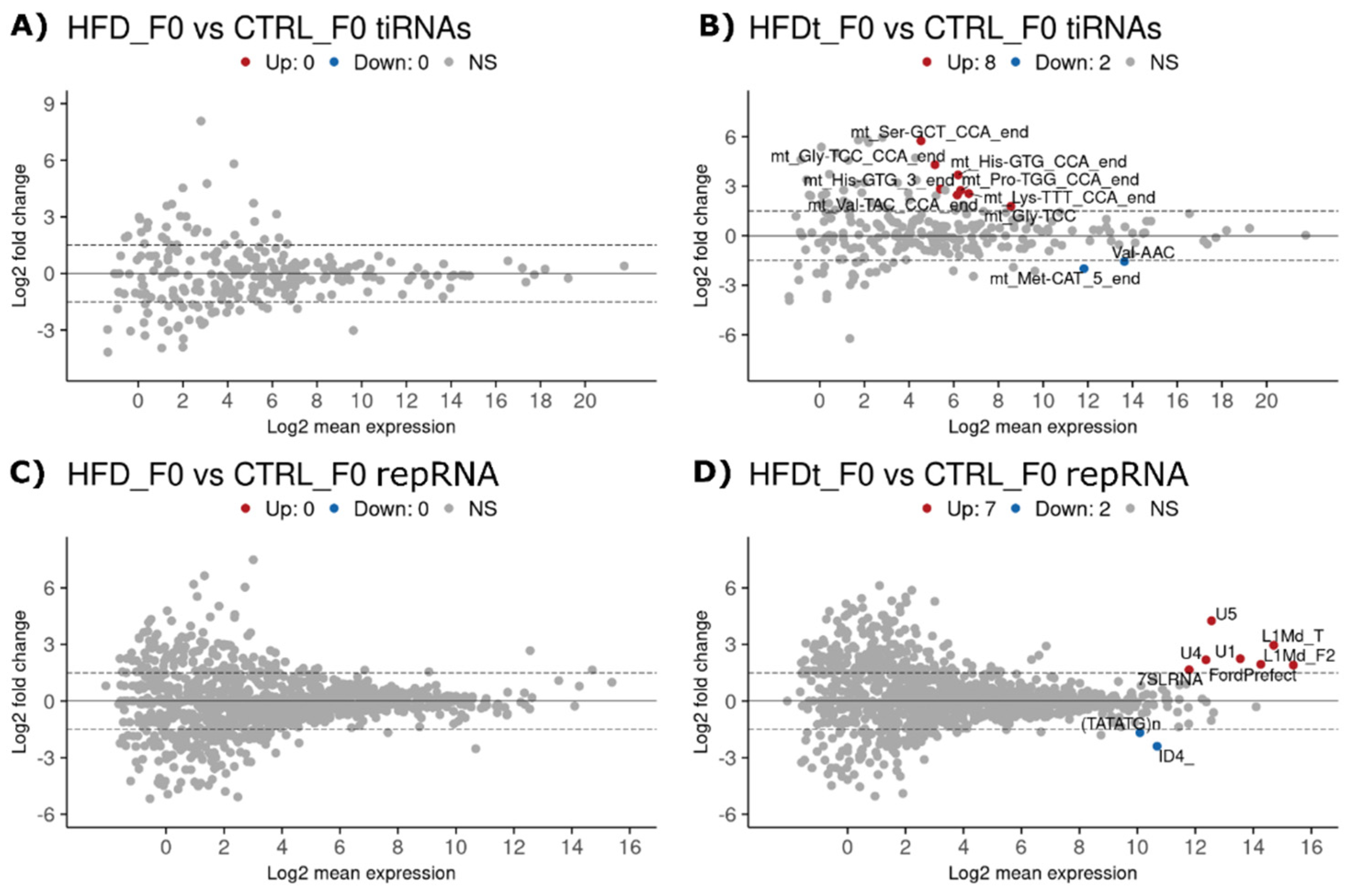
3.1. Dietary Changes Affect Sperm sncRNA Content
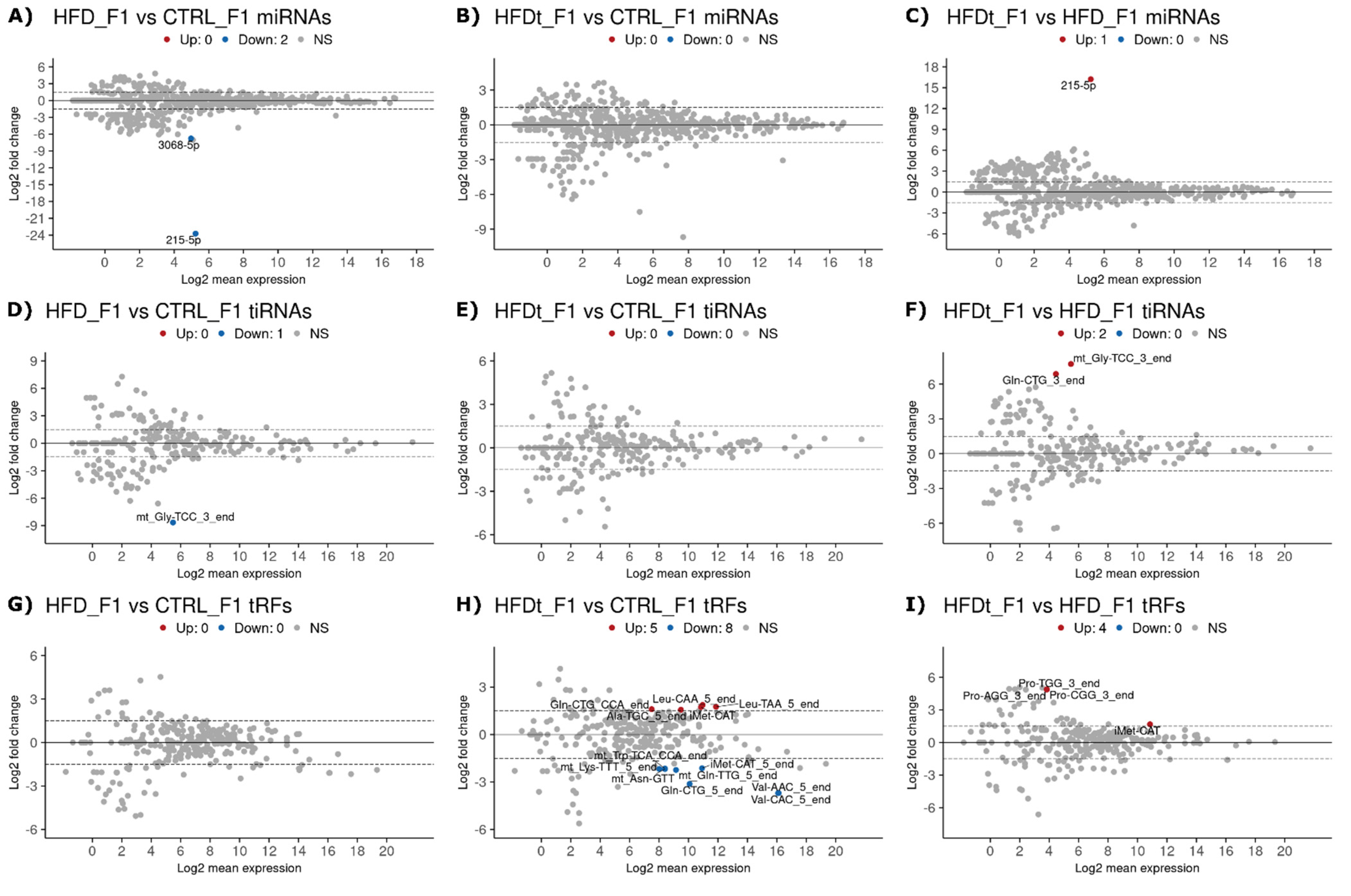
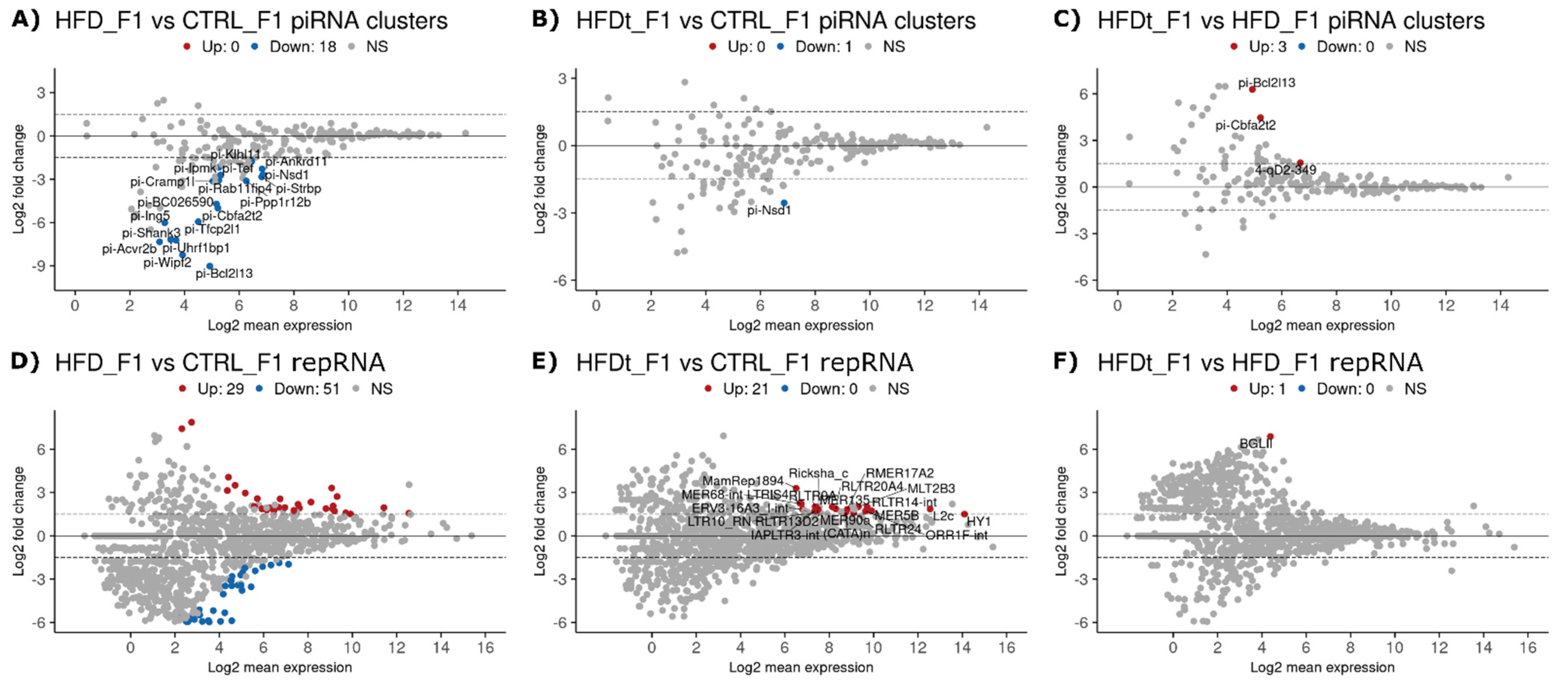
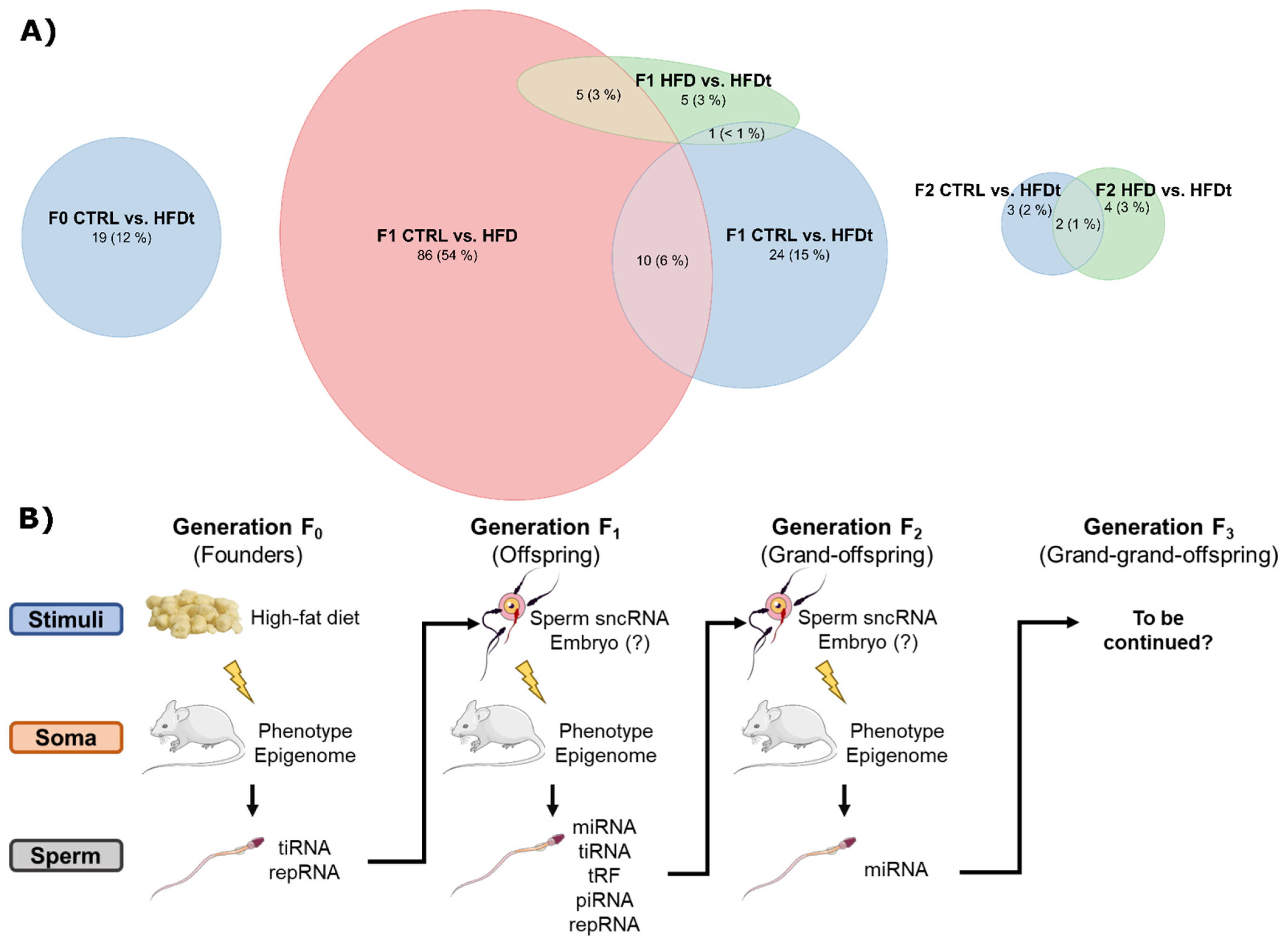
3.2. Dietary Changes Affect Sperm sncRNA Content of the Offspring of Exposed Mice
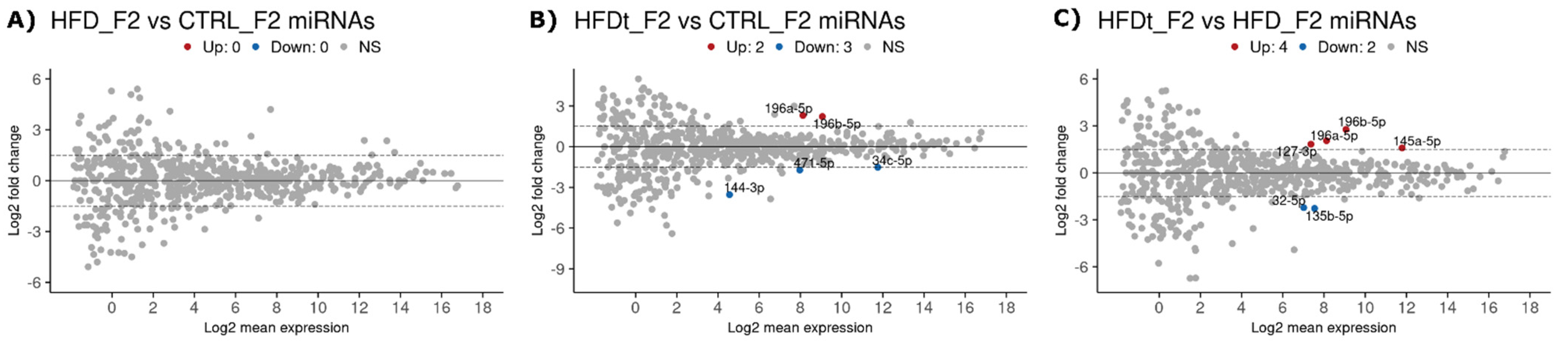
3.3. Ancestral Dietary Changes Can Be Detected in Sperm of the Grand-Offspring
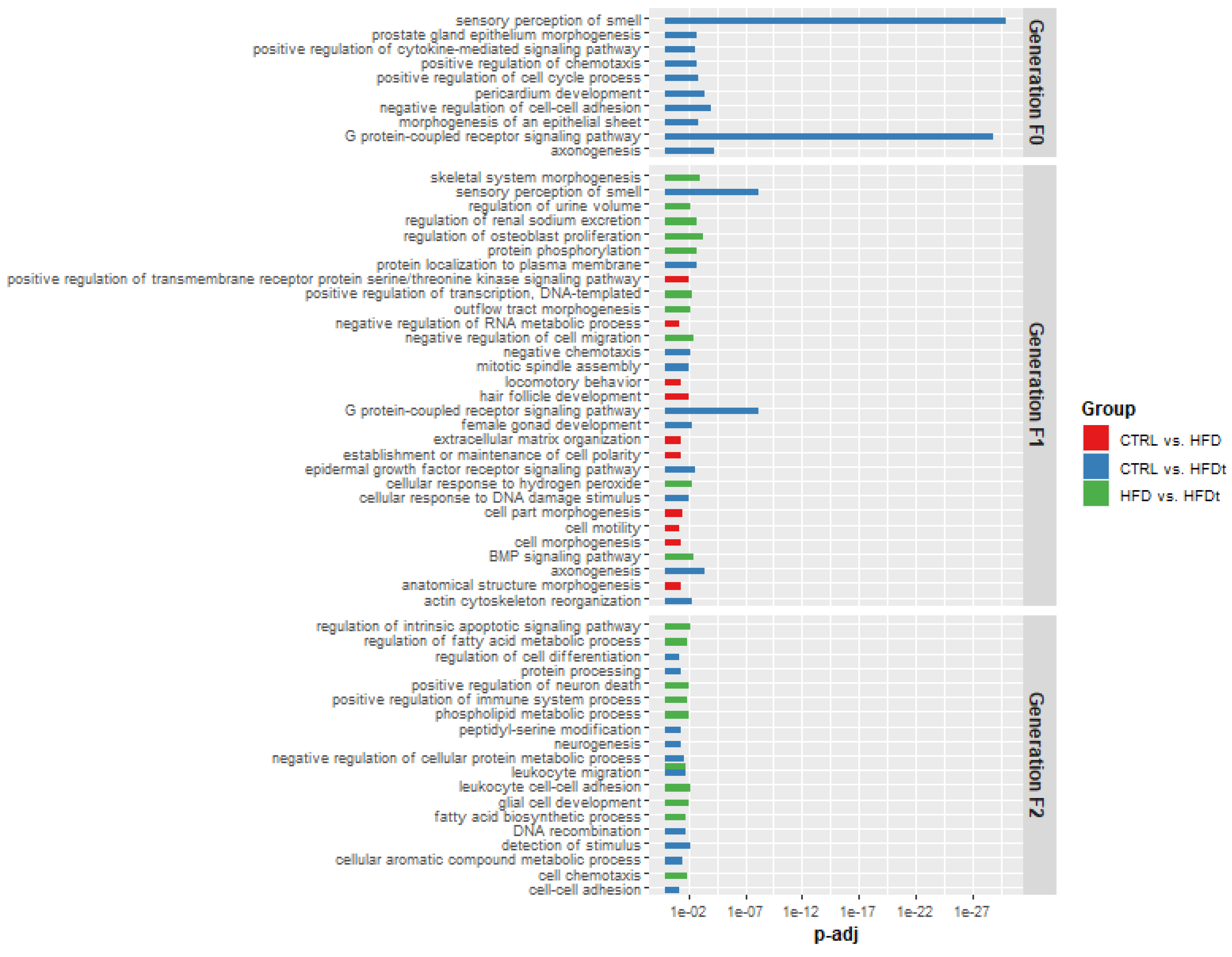
3.4. Targets of Differently Expressed Sperm sncRNAs Are Related with Crucial Biological Processes for Testicular Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; Karuranga, S., Malanda, B., Saeedi, P., Salpea, P., Eds.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2014; WHO Press: Geneva, Switzerland, 2014; p. 280. [Google Scholar]
- Kaati, G.; Bygren, L.O.; Edvinsson, S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 2002, 10, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumey, L.; Stein, A.D.; Kahn, H.S.; van der Pal-de Bruin, K.M.; Blauw, G.; Zybert, P.A.; Susser, E.S. Cohort Profile: The Dutch hunger winter families study. Int. J. Epidemiol. 2007, 36, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.F.; Lehner, B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 2019, 21, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, F.A.; Drake, A.J. Non-genetic inheritance via the male germline in mammals. Philos. Trans. R Soc. B Biol. Sci. 2019, 374, 20180118. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.-h.; Peng, H.; Zhang, X.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Conine, C.C.; Sun, F.; Song, L.; Rivera-Pérez, J.A.; Rando, O.J. Small RNAs gained during epididymal transit of sperm are essential for embryonic development in mice. Dev. Cell 2018, 46, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Z.; Roy, C.K.; Dong, X.; Bolcun-Filas, E.; Wang, J.; Han, B.W.; Xu, J.; Moore, M.J.; Schimenti, J.C.; Weng, Z.; et al. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol. Cell 2013, 50, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Goh, W.S.S.; Falciatori, I.; Tam, O.H.; Burgess, R.; Meikar, O.; Kotaja, N.; Hammell, M.; Hannon, G.J. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 2015, 29, 1032–1044. [Google Scholar] [CrossRef] [Green Version]
- Nätt, D.; Kugelberg, U.; Casas, E.; Nedstrand, E.; Zalavary, S.; Henriksson, P.; Nijm, C.; Jäderquist, J.; Sandborg, J.; Flinke, E. Human sperm displays rapid responses to diet. PloS Biol. 2019, 17, e3000559. [Google Scholar] [CrossRef] [Green Version]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Swanson, G.M.; Estill, M.; Diamond, M.P.; Legro, R.S.; Coutifaris, C.; Barnhart, K.T.; Huang, H.; Hansen, K.R.; Trussell, J.C.; Coward, R.M.; et al. Human chromatin remodeler cofactor, RNA interactor, eraser and writer sperm RNAs responding to obesity. Epigenetics 2020, 15, 32–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullston, T.; Ohlsson Teague, E.M.; Palmer, N.O.; DeBlasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 2013, 27, 4226–4243. [Google Scholar] [CrossRef] [PubMed]
- Pavlinková, G.; Margaryan, H.; Žatecká, E.; Valášková, E.; Elzeinová, F.; Kubátová, A.; Bohuslavová, R.; Pěknicová, J. Transgenerational inheritance of susceptibility to diabetes-induced male subfertility. Sci. Rep. 2017, 7, 4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisóstomo, L.; Rato, L.; Jarak, I.; Silva, B.M.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. A switch from high-fat to normal diet does not restore sperm quality but prevents metabolic syndrome. Reproduction 2019, 158, 377–387. [Google Scholar] [CrossRef]
- Crisóstomo, L.; Videira, R.A.; Jarak, I.; Starčević, K.; Mašek, T.; Rato, L.P.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Diet during early life defines testicular lipid content and sperm quality in adulthood. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1061–E1073. [Google Scholar] [CrossRef]
- Crisóstomo, L.; Jarak, I.; Rato, L.P.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Inheritable testicular metabolic memory of high-fat diet causes transgenerational sperm defects in mice. Sci. Rep. 2021, 11, 9444. [Google Scholar] [CrossRef]
- Crisóstomo, L.; Videira, R.A.; Jarak, I.; Starčević, K.; Mašek, T.; Rato, L.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Inherited metabolic memory of high-fat diet impairs testicular fatty acid content and sperm parameters. Mol. Nutr. Food Res. 2022, 66, 2100680. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Goodrich, R.J.; Anton, E.; Krawetz, S.A. Isolating mRNA and small noncoding RNAs from human sperm. In Spermatogenesis; Springer: Berlin/Heidelberg, Germany, 2013; pp. 385–396. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 3. [Google Scholar] [CrossRef]
- Shi, J.; Ko, E.-A.; Sanders, K.M.; Chen, Q.; Zhou, T. SPORTS1.0: A Tool for Annotating and Profiling Non-coding RNAs Optimized for rRNA- and tRNA-derived Small RNAs. Genom. Proteom. Bioinform. 2018, 16, 144–151. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Sheridan, R.; Marks, D.S.; Sander, C. Computational analysis of mouse piRNA sequence and biogenesis. PLoS Comput. Biol. 2007, 3, e222. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, D. piRNA cluster database: A web resource for piRNA producing loci. Nucleic Acids Res. 2015, 44, D223–D230. [Google Scholar] [CrossRef]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2015, 44, D81–D89. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef]
- Liu, Q.; Ding, C.; Lang, X.; Guo, G.; Chen, J.; Su, X. Small noncoding RNA discovery and profiling with sRNAtools based on high-throughput sequencing. Brief. Bioinform. 2021, 22, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Enright, A.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D. MicroRNA targets in drosophila. Genome Biol. 2003, 4, P8. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; 4.1.0; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology, Version 2.42.0. 2020.
- Carlson, M. GO.db: A Set of Annotation Maps Describing the Entire Gene Ontology, Version 3.8.2. 2019.
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gapp, K.; Jawaid, A.; Sarkies, P.; Bohacek, J.; Pelczar, P.; Prados, J.; Farinelli, L.; Miska, E.; Mansuy, I.M. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014, 17, 667–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Gonskikh, Y.; Gerstl, M.; Kos, M.; Borth, N.; Schosserer, M.; Grillari, J.; Polacek, N. Modulation of mammalian translation by a ribosome-associated tRNA half. RNA Biol. 2020, 17, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Taft, R.J.; Glazov, E.A.; Cloonan, N.; Simons, C.; Stephen, S.; Faulkner, G.J.; Lassmann, T.; Forrest, A.R.R.; Grimmond, S.M.; Schroder, K.; et al. Tiny RNAs associated with transcription start sites in animals. Nat. Genet. 2009, 41, 572–578. [Google Scholar] [CrossRef]
- Park, J.; Ahn, S.H.; Shin, M.G.; Kim, H.K.; Chang, S. tRNA-derived small RNAs: Novel epigenetic regulators. Cancers 2020, 12, 2773. [Google Scholar] [CrossRef]
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-retrotransposon control by tRNA-derived small RNAs. Cell 2017, 170, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef]
- Raad, G.; Serra, F.; Martin, L.; Derieppe, M.-A.; Gilleron, J.; Costa, V.L.; Pisani, D.F.; Amri, E.-Z.; Trabucchi, M.; Grandjean, V. Paternal multigenerational exposure to an obesogenic diet drives epigenetic predisposition to metabolic diseases in mice. eLife 2021, 10, e61736. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, V.; Fourré, S.; De Abreu, D.A.F.; Derieppe, M.-A.; Remy, J.-J.; Rassoulzadegan, M. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci. Rep. 2016, 5, 18193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F.; et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; Kim, R.; Tharmalingam, M.; Matilionyte, G.; Lindskog, C.; et al. The dynamic transcriptional cell atlas of testis development during human puberty. Cell Stem Cell 2020, 26, 262–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spehr, M.; Gisselmann, G.; Poplawski, A.; Riffell, J.A.; Wetzel, C.H.; Zimmer, R.K.; Hatt, H. Identification of a Testicular Odorant Receptor Mediating Human Sperm Chemotaxis. Science 2003, 299, 2054–2058. [Google Scholar] [CrossRef]
- Ali, M.A.; Wang, Y.; Qin, Z.; Yuan, X.; Zhang, Y.; Zeng, C. Odorant and taste receptors in sperm chemotaxis and cryopreservation: Roles and implications in sperm capacitation, motility and fertility. Genes 2021, 12, 488. [Google Scholar] [CrossRef]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Ernst, C.; Odom, D.T.; Kutter, C. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat. Commun. 2017, 8, 1411. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.P.; Mäkelä, J.-A.; Hyssälä, H.; Cisneros-Montalvo, S.; Kotaja, N. DICER regulates the expression of major satellite repeat transcripts and meiotic chromosome segregation during spermatogenesis. Nucleic Acids Res. 2020, 48, 7135–7153. [Google Scholar] [CrossRef]
- Wu, Q.; Song, R.; Ortogero, N.; Zheng, H.; Evanoff, R.; Small, C.L.; Griswold, M.D.; Namekawa, S.H.; Royo, H.; Turner, J.M.; et al. The RNase III enzyme DROSHA is essential for MicroRNA production and spermatogenesis. J. Biol. Chem. 2012, 287, 25173–25190. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian MicroRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Git, A.; Dvinge, H.; Salmon-Divon, M.; Osborne, M.; Kutter, C.; Hadfield, J.; Bertone, P.; Caldas, C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010, 16, 991–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Fang, L.; Shi, B.; Qiu, S.; Ye, Y. Spermatozoa expression of piR-31704, piR-39888, and piR-40349 and their correlation to sperm concentration and fertilization rate after ICSI. Reprod. Sci. 2018, 25, 733–739. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tsRNA | ||||||
|---|---|---|---|---|---|---|
| Generation | Comparison | miRNA | tRF | tiRNA | piRNA | repRNA |
| CTRL vs. HFD | 0 | 0 | 0 | 0 | 0 | |
| F0 | CTRL vs. HFDt | 0 | 0 | 10 | 0 | 9 |
| HFD vs. HFDt | 0 | 0 | 0 | 0 | 0 | |
| CTRL vs. HFD | 2 | 0 | 1 | 18 | 80 | |
| F1 | CTRL vs. HFDt | 0 | 13 | 0 | 1 | 21 |
| HFD vs. HFDt | 1 | 4 | 2 | 3 | 1 | |
| CTRL vs. HFD | 0 | 0 | 0 | 0 | 0 | |
| F2 | CTRL vs. HFDt | 5 | 0 | 0 | 0 | 0 |
| HFD vs. HFDt | 6 | 0 | 0 | 0 | 0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisóstomo, L.; Bourgery, M.; Rato, L.; Raposo, J.F.; Batterham, R.L.; Kotaja, N.; Alves, M.G. Testicular “Inherited Metabolic Memory” of Ancestral High-Fat Diet Is Associated with Sperm sncRNA Content. Biomedicines 2022, 10, 909. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040909
Crisóstomo L, Bourgery M, Rato L, Raposo JF, Batterham RL, Kotaja N, Alves MG. Testicular “Inherited Metabolic Memory” of Ancestral High-Fat Diet Is Associated with Sperm sncRNA Content. Biomedicines. 2022; 10(4):909. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040909
Chicago/Turabian StyleCrisóstomo, Luís, Matthieu Bourgery, Luís Rato, João F. Raposo, Rachel L. Batterham, Noora Kotaja, and Marco G. Alves. 2022. "Testicular “Inherited Metabolic Memory” of Ancestral High-Fat Diet Is Associated with Sperm sncRNA Content" Biomedicines 10, no. 4: 909. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040909