
Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49)

 ,
,  , , and
, , and
Abstract
:1. Introduction
- -
- Blastematous: diffuse, nodular and/or cord-like growth patterns of small, round and huddled blue cells, showing dark nuclei, diminished cytoplasm and frequent mitosis);
- -
- Epithelial (tubules, papillae and glomeruli, similar to structures found in normal nephrogenesis, alongside more primitive rosette-shaped early tubular structures, and cysts lined with immature columnar/cuboidal cells);
- -
- Stromal (resembling embryonal mesenchyme, spindle-shaped cells are seen on a myxoid stromal background, with a variety of patterns of differentiation and heterologous stromal elements, most commonly skeletal muscle in various stages of differentiation, including rhabdomyoblasts, but also cartilage, bone, fat or, more rarely, neuroglia and mature ganglion cells) [22,43].
2. Materials and Methods
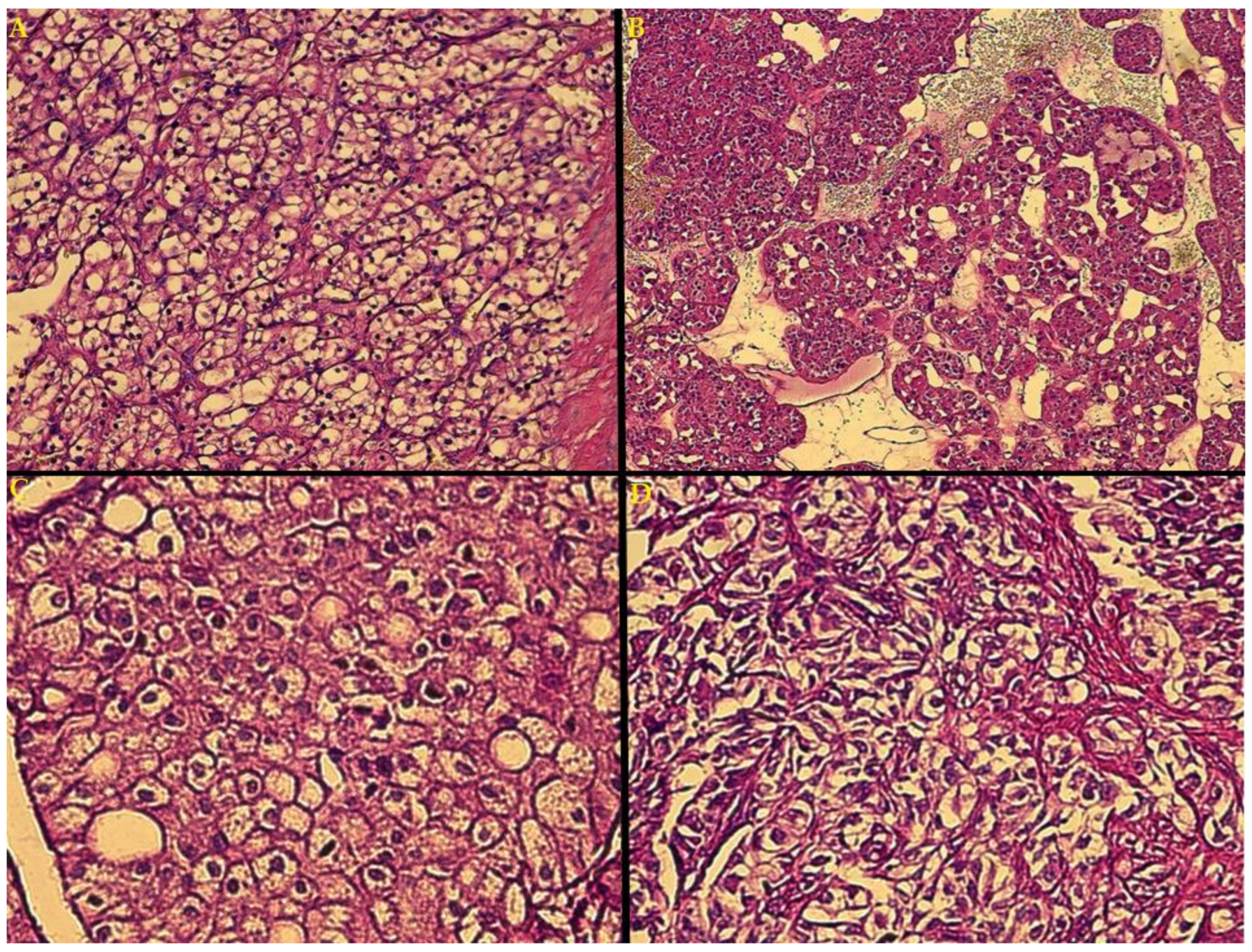
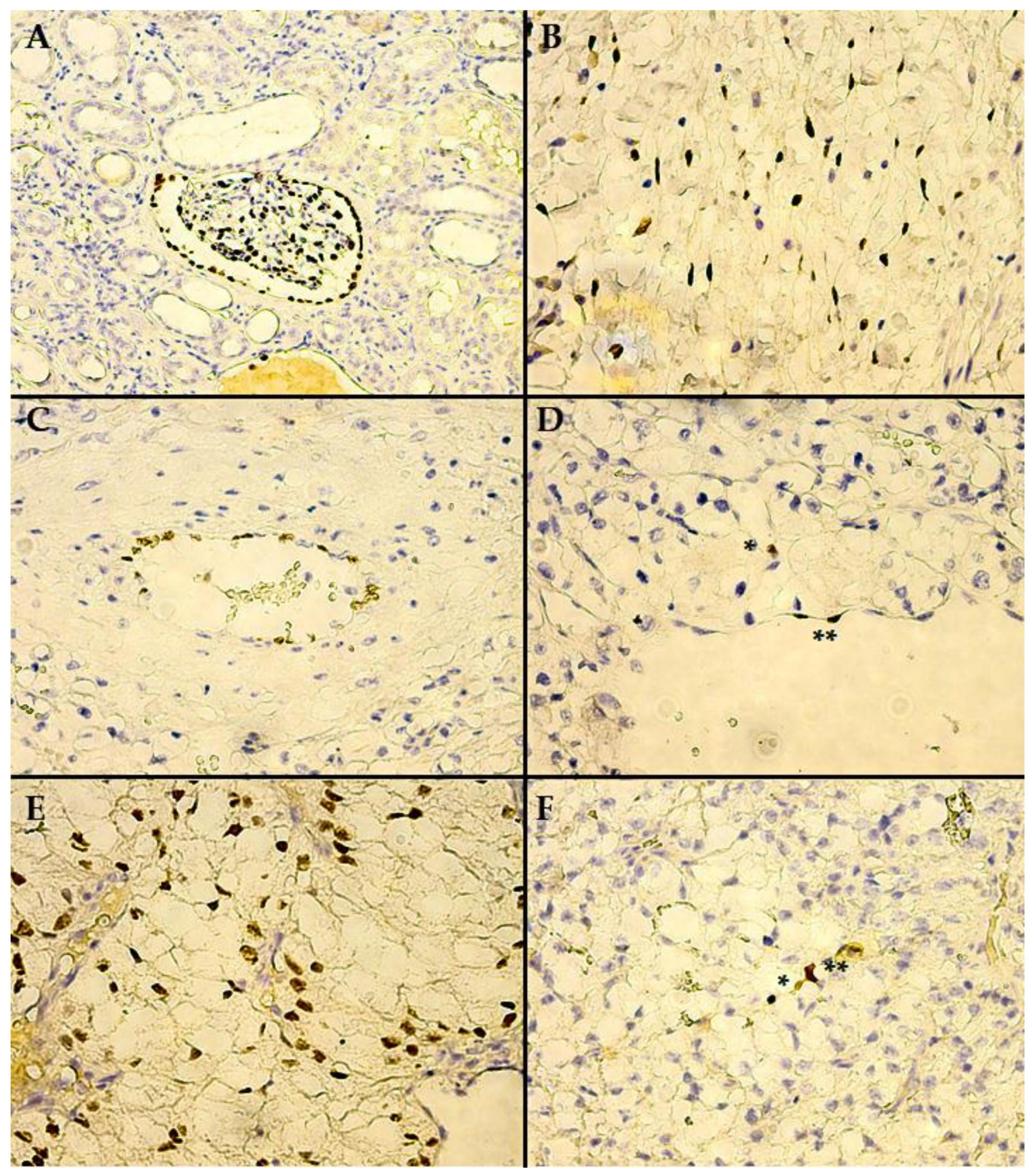
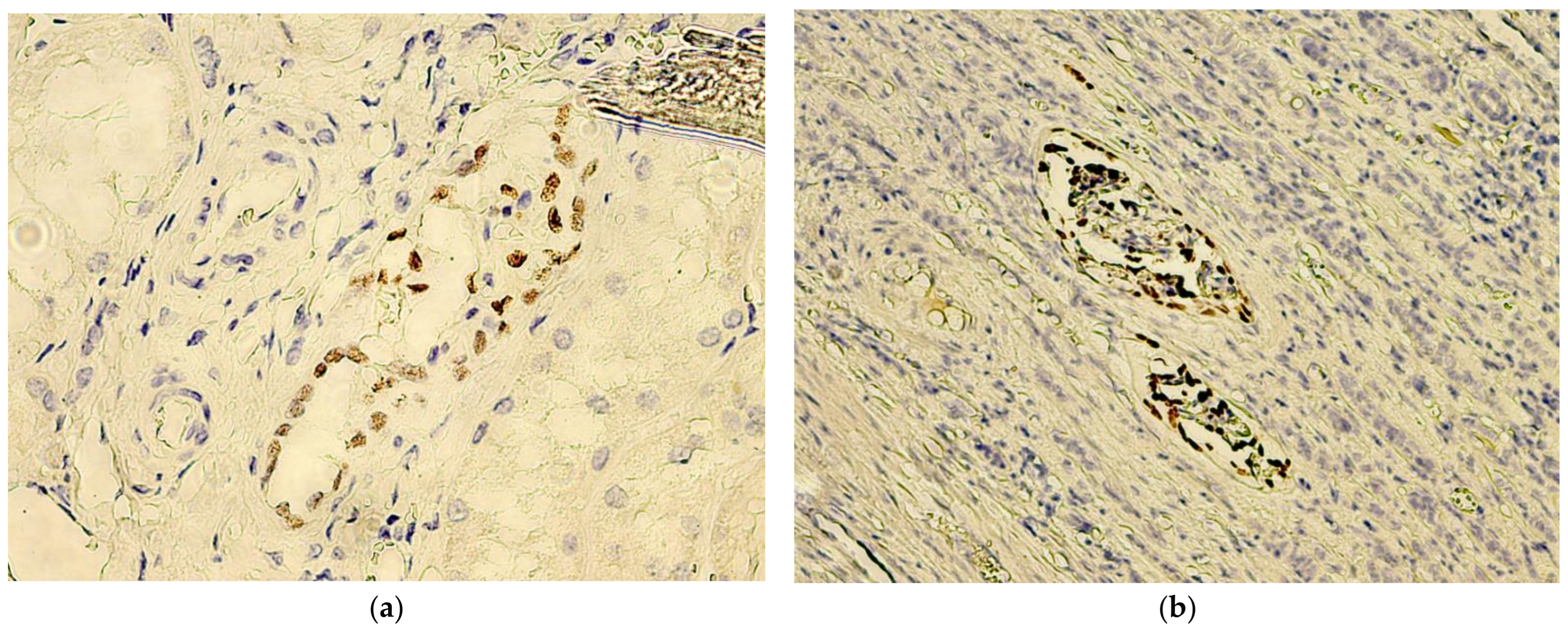
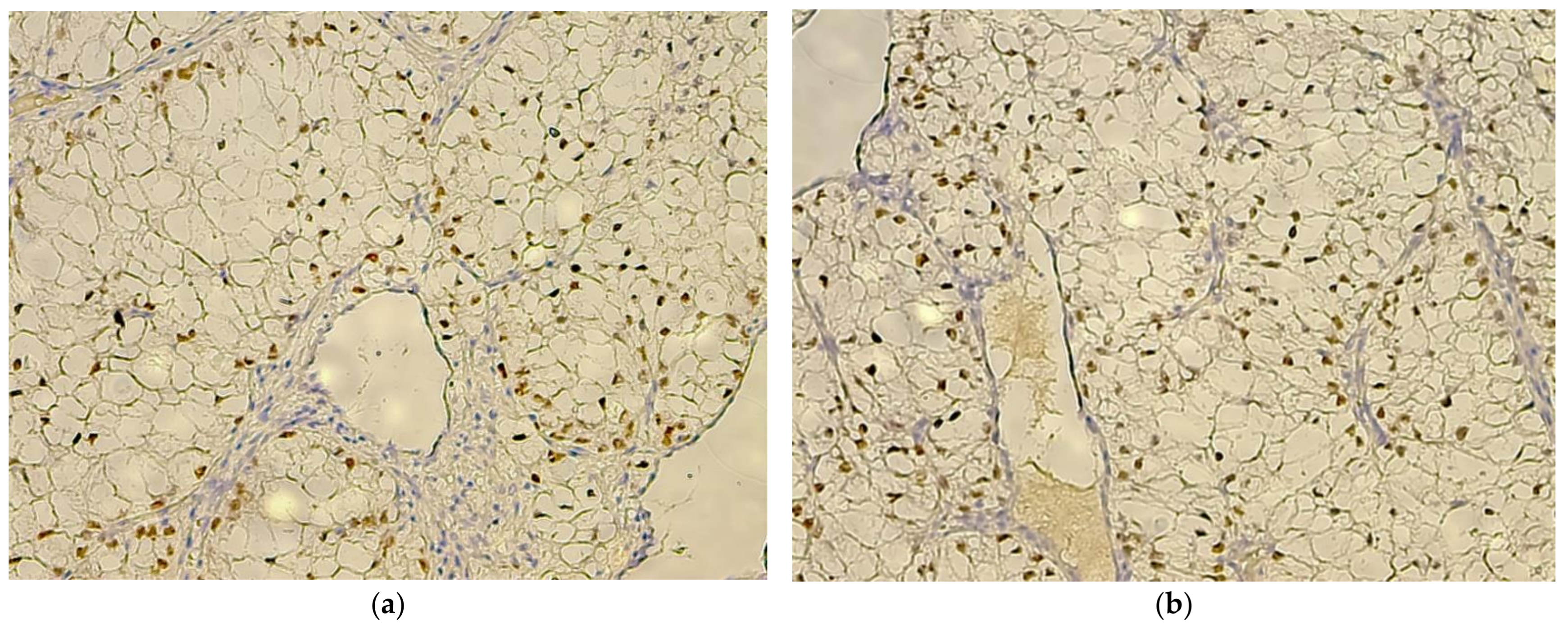
3. Results
4. Discussion
- -
- The development, homeostasis and pathology of intermediate and lateral plate mesoderm derived tissues;
- -
- Transitional cellular events during development, i.e., mesenchyme-to-epithelial transitions (MET)/epithelial-to-mesenchyme transition (EMT);
- -
- Mesenchymal progenitors’ cellular origin in a wide array of human tissues, highlighting the essential role of the mesothelium as a mesenchymal progenitor source;
- -
- Essential inner workings of transcription and epigenetic regulation [25].
- -
- WAGR syndrome—due to characteristic 11p deletions, WT1 haploinsufficiency ensues, generating nephroblastoma (in ~70% of cases), aniridia, genital anomalies and intellectual impairment [88];
- -
- Denys–Drash syndrome (DDS)—due to heterozygous WT1 point variants, mainly involving exons 8 and 9 (zinc finger domain), which are dominant negative in nature (mutant WT1 protein and wild-type protein will dimerize, adversely affecting the functionality of the latter [89]), more severe phenotypes are seen: similarly, high prevalence of nephroblastoma, with an additional constant component of mesangial glomerulosclerosis, which will invariably lead to pediatric end-stage renal disease [90] and genital/gonadal anomalies.
- -
- Meacham Winn Culler syndrome (MWCS)—less well defined genetically, with a phenotype lacking reno-urinary anomalies, characterized by congenital diaphragmatic hernia, genital ambiguity and complex congenital cardiac defects [25].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Decastro, G.J.; McKiernan, J.M. Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma. Urol. Clin. N. Am. 2008, 35, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.; Mallin, K.; Ritchey, J.; Cooperberg, M.R.; Carroll, P.R. Renal Cell Cancer Stage Migration: Analysis of the National Cancer Data Base. Cancer 2008, 113, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Reuter, V.E.; Argani, P.; Zhou, M.; Delahunt, B.; Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best Practices Recommendations in the Application of Immunohistochemistry in the Kidney Tumors: Report from the International Society of Urologic Pathology Consensus Conference. Am. J. Surg. Pathol. 2014, 38, e35–e49. [Google Scholar] [CrossRef]
- Peckova, K.; Martinek, P.; Ohe, C.; Kuroda, N.; Bulimbasic, S.; Condom Mundo, E.; Perez Montiel, D.; Lopez, J.I.; Daum, O.; Rotterova, P.; et al. Chromophobe Renal Cell Carcinoma with Neuroendocrine and Neuroendocrine-like Features. Morphologic, Immunohistochemical, Ultrastructural, and Array Comparative Genomic Hybridization Analysis of 18 Cases and Review of the Literature. Ann. Diagn. Pathol. 2015, 19, 261–268. [Google Scholar] [CrossRef]
- Hes, O.; Condom Mundo, E.; Peckova, K.; Lopez, J.I.; Martinek, P.; Vanecek, T.; Falconieri, G.; Agaimy, A.; Davidson, W.; Petersson, F.; et al. Biphasic Squamoid Alveolar Renal Cell Carcinoma: A Distinctive Subtype of Papillary Renal Cell Carcinoma? Am. J. Surg. Pathol. 2016, 40, 664–675. [Google Scholar] [CrossRef]
- Trpkov, K.; Hes, O.; Bonert, M.; Lopez, J.I.; Bonsib, S.M.; Nesi, G.; Comperat, E.; Sibony, M.; Berney, D.M.; Martinek, P.; et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am. J. Surg. Pathol. 2016, 40, 60–71. [Google Scholar] [CrossRef]
- Falzarano, S.M.; McKenney, J.K.; Montironi, R.; Eble, J.N.; Osunkoya, A.O.; Guo, J.; Zhou, S.; Xiao, H.; Dhanasekaran, S.M.; Shukla, S.; et al. Renal Cell Carcinoma Occurring in Patients with Prior Neuroblastoma: A Heterogenous Group of Neoplasms. Am. J. Surg. Pathol. 2016, 40, 989–997. [Google Scholar] [CrossRef]
- Kusano, H.; Togashi, Y.; Akiba, J.; Moriya, F.; Baba, K.; Matsuzaki, N.; Yuba, Y.; Shiraishi, Y.; Kanamaru, H.; Kuroda, N.; et al. Two Cases of Renal Cell Carcinoma Harboring a Novel STRN-ALK Fusion Gene. Am. J. Surg. Pathol. 2016, 40, 761–769. [Google Scholar] [CrossRef]
- Hes, O.; Compérat, E.M.; Rioux-Leclercq, N. Clear Cell Papillary Renal Cell Carcinoma, Renal Angiomyoadenomatous Tumor, and Renal Cell Carcinoma with Leiomyomatous Stroma Relationship of 3 Types of Renal Tumors: A Review. Ann. Diagn. Pathol. 2016, 21, 59–64. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Tickoo, S.K.; Jacobsen, A.; Sarungbam, J.; Sfakianos, J.P.; Sato, Y.; Morikawa, T.; Kume, H.; Fukayama, M.; Homma, Y.; et al. TCEB1-Mutated Renal Cell Carcinoma: A Distinct Genomic and Morphological Subtype. Mod. Pathol. 2015, 28, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawane, R.; Grindstaff, A.; Parwani, A.V.; Brock, T.; White, W.M.; Nodit, L. Thyroid-like Follicular Carcinoma of the Kidney: One Case Report and Review of the Literature. Am. J. Clin. Pathol. 2015, 144, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.W.; Wein, A.J.; Kavoussi, L.R.; Peters, C.A.; Dmochowski, R.R. Neoplasms of the Upper Urinary Tract. In Campbell-Walsh-Wein Urology, E-Book, 12th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2020; Volume 2, p. 2148. [Google Scholar]
- Amin, M.B.; Epstein, J.I.; Ulbright, T.M.; Humphrey, P.A.; Egevad, L.; Montironi, R.; Grignon, D.; Trpkov, K.; Lopez-Beltran, A.; Zhou, M.; et al. Best Practices Recommendations in the Application of Immunohistochemistry in Urologic Pathology: Report from the International Society of Urological Pathology Consensus Conference. Am. J. Surg. Pathol. 2014, 38, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadie, H.A.; Alden, D.; Fine, S.W.; Gopalan, A.; Touijer, K.A.; Russo, P.; Reuter, V.E.; Tickoo, S.K. Role of Immunohistochemistry in the Evaluation of Needle Core Biopsies in Adult Renal Cortical Tumors: An Ex Vivo Study. Am. J. Surg. Pathol. 2011, 35, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strojan Fležar, M.; Gutnik, H.; Jeruc, J.; Srebotnik Kirbiš, I. Typing of Renal Tumors by Morphological and Immunocytochemical Evaluation of Fine Needle Aspirates. Virchows Arch. 2011, 459, 607–614. [Google Scholar] [CrossRef]
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H. Isolation and Characterization of a Zinc Finger Polypeptide Gene at the Human Chromosome 11 Wilms’ Tumor Locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef]
- Gessler, M.; Poustka, A.; Cavenee, W.; Neve, R.L.; Orkin, S.H.; Bruns, G.A. Homozygous Deletion in Wilms Tumours of a Zinc-Finger Gene Identified by Chromosome Jumping. Nature 1990, 343, 774–778. [Google Scholar] [CrossRef]
- Charlton, J.; Pritchard-Jones, K. WT1 Mutation in Childhood Cancer. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: New York, NY, USA, 2016; Volume 1467, pp. 1–14. [Google Scholar] [CrossRef]
- Hastie, N.D. The Genetics of Wilms’ Tumor—A Case of Disrupted Development. Annu. Rev. Genet. 1994, 28, 523–558. [Google Scholar] [CrossRef]
- Salvatorelli, L.; Parenti, R.; Leone, G.; Musumeci, G.; Vasquez, E.; Magro, G. Wilms Tumor 1 (WT1) Protein: Diagnostic Utility in Pediatric Tumors. Acta Histochem. 2015, 117, 367–378. [Google Scholar] [CrossRef]
- Huff, V. Wilms’ Tumours: About Tumour Suppressor Genes, an Oncogene and a Chameleon Gene. Nat. Rev. Cancer. 2011, 11, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppes, M.J.; Haber, D.A.; Grundy, P.E. Genetic Events in the Development of Wilms’ Tumor. N. Engl. J. Med. 1994, 331, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Hastie, N.D. Wilms’ Tumour 1 (WT1) in Development, Homeostasis and Disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Discenza, M.T.; Pelletier, J. Insights into the Physiological Role of WT1 from Studies of Genetically Modified Mice. Physiol. Genom. 2004, 16, 287–300. [Google Scholar] [CrossRef]
- Hohenstein, P.; Hastie, N.D. The Many Facets of the Wilms’ Tumour Gene, WT1. Hum. Mol. Genet. 2006, 15, R196–R201. [Google Scholar] [CrossRef]
- Roberts, S.G.E. Transcriptional Regulation by WT1 in Development. Curr. Opin. Genet. Dev. 2005, 15, 542–547. [Google Scholar] [CrossRef]
- Chau, Y.-Y.; Hastie, N.D. The Role of Wt1 in Regulating Mesenchyme in Cancer, Development, and Tissue Homeostasis. Trends Genet 2012, 28, 515–524. [Google Scholar] [CrossRef]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor Protein (WT1) in Developing Human Peripheral Sympathetic and Gastroenteric Nervous System. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Vicent, S.; Chen, R.; Sayles, L.C.; Lin, C.; Walker, R.G.; Gillespie, A.K.; Subramanian, A.; Hinkle, G.; Yang, X.; Saif, S.; et al. Wilms Tumor 1 (WT1) Regulates KRAS-Driven Oncogenesis and Senescence in Mouse and Human Models. J. Clin. Investig. 2010, 120, 3940–3952. [Google Scholar] [CrossRef]
- Sugiyama, H. WT1 (Wilms’ Tumor Gene 1): Biology and Cancer Immunotherapy. Jpn. J. Clin. Oncol. 2010, 40, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Saurez Saiz, F.; Minden, M.D. A Tumor Suppressor and Oncogene: The WT1 Story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G.; Strong, L.C. Mutation and Cancer: A Model for Wilms’ Tumor of the Kidney. J. Natl. Cancer Inst. 1972, 48, 313–324. [Google Scholar] [PubMed]
- Huang, L.; Mokkapati, S.; Hu, Q.; Ruteshouser, E.C.; Hicks, M.J.; Huff, V. Nephron Progenitor But Not Stromal Progenitor Cells Give Rise to Wilms Tumors in Mouse Models with β-Catenin Activation or Wt1 Ablation and Igf2 Upregulation. Neoplasia 2016, 18, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Sugiyama, H. Immunotherapy Targeting WT1: Designing a Protocol for WT1 Peptide-Based Cancer Vaccine. Methods Mol. Biol. 2016, 1467, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.K.; Mall, S.; Watson, J.; Berry, P.J. Expression of the Wilms’ Tumour Gene WT1 in the Developing Human and in Paediatric Renal Tumours: An Immunohistochemical Study. Mol. Pathol. 1997, 50, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.E.; Kuriyan, N.P.; Rackley, R.R.; Caulfield, M.J.; Tubbs, R.; Finke, J.; Williams, B.R.G. Constitutive Expression of the Wilms Tumor Suppressor Gene (WT1) in Renal Cell Carcinoma. Int. J. Cancer 1998, 78, 182–188. [Google Scholar] [CrossRef]
- Yeger, H.; Cullinane, C.; Flenniken, A.; Chilton-MacNeil, S.; Campbell, C.; Huang, A.; Bonetta, L.; Coppes, M.J.; Thorner, P.; Williams, B.R. Coordinate Expression of Wilms’ Tumor Genes Correlates with Wilms’ Tumor Phenotypes. Cell Growth Differ. 1992, 3, 855–864. [Google Scholar]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, M.; Miyashita, M.; Ota, S. Vaccination of Urological Cancer Patients with WT1 Peptide-Pulsed Dendritic Cells in Combination with Molecular Targeted Therapy or Conventional Chemotherapy Induces Immunological and Clinical Responses. Ther. Apher. Dial. 2018, 22, 266–277. [Google Scholar] [CrossRef]
- Kashima, S.; Maeda, T.; Masuda, K.; Nagano, S.; Inoue, T.; Takeda, M.; Kono, Y.; Kobayashi, T.; Saito, S.; Higuchi, T.; et al. Cytotoxic T Lymphocytes Regenerated from IPS Cells Have Therapeutic Efficacy in a Patient-Derived Xenograft Solid Tumor Model. iScience 2020, 23, 100998. [Google Scholar] [CrossRef]
- Al-Hussain, T.; Ali, A.; Akhtar, M. Wilms Tumor: An Update. Adv. Anat. Pathol. 2014, 21, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Ramani, P.; Cowell, J.K. The Expression Pattern of Wilms’ Tumour Gene (WT1) Product in Normal Tissues and Paediatric Renal Tumours. J. Pathol. 1996, 179, 162–168. [Google Scholar] [CrossRef]
- Vasei, M.; Moch, H.; Mousavi, A.; Kajbafzadeh, A.M.; Sauter, G. Immunohistochemical Profiling of Wilms Tumor: A Tissue Microarray Study. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Matsumura, L.; Weeks, D.A.; Troxell, M.L. PAX2 Expression in Wilms Tumors and Other Childhood Neoplasms. Am. J. Surg. Pathol. 2011, 35, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Schoenfield, L.; Limketkai, B.; Arnold, C. Diagnostic Pitfalls of Differentiating Desmoplastic Small Round Cell Tumor (DSRCT) From Wilms Tumor (WT): Overlapping Morphologic and Immunohistochemical Features. Am. J. Surg. Pathol. 2014, 38, 1220–1226. [Google Scholar] [CrossRef]
- Sehic, D.; Ciornei, C.D.; Gisselsson, D. Evaluation of CITED1, SIX1, and CD56 Protein Expression for Identification of Blastemal Elements in Wilms Tumor. Am. J. Clin. Pathol. 2014, 141, 828–833. [Google Scholar] [CrossRef] [Green Version]
- Gerald, W.L.; Ladanyi, M.; de Alava, E.; Cuatrecasas, M.; Kushner, B.H.; LaQuaglia, M.P.; Rosai, J. Clinical, Pathologic, and Molecular Spectrum of Tumors Associated with t(11;22)(P13;Q12): Desmoplastic Small Round-Cell Tumor and Its Variants. J. Clin. Oncol. 1998, 16, 3028–3036. [Google Scholar] [CrossRef]
- Shek, T.W.; Luk, I.S.; Peh, W.C.; Chan, K.L.; Chan, G.C. Metanephric Adenofibroma: Report of a Case and Review of the Literature. Am. J. Surg. Pathol. 1999, 23, 727–733. [Google Scholar] [CrossRef]
- Mahmoud, F.; Allen, M.B.; Cox, R.; Davis, R. Wilms Tumor: An Uncommon Entity in the Adult Patient. Perm. J. 2016, 20, e119–e121. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, H.; Aliani, S.; Ruebe, C.; Stöckle, M.; Leuschner, I.; Graf, N. Wilms’ Tumor in Adults: Results of the Society of Pediatric Oncology (SIOP) 93-01/Society for Pediatric Oncology and Hematology (GPOH) Study. J. Clin. Oncol. 2004, 22, 4500–4506. [Google Scholar] [CrossRef]
- Weichert-Jacobsen, K.; Papadopoulos, I.; Skrezek, C.; Wand, H. Adult Wilms’ Tumor: Case Report, Management, Prognosis. Urol. Int. 1995, 54, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Olgac, S.; Hutchinson, B.; Tickoo, S.K.; Reuter, V.E. Alpha-Methylacyl-CoA Racemase as a Marker in the Differential Diagnosis of Metanephric Adenoma. Mod. Pathol. 2006, 19, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordóñez, N.G. The Diagnostic Utility of Immunohistochemistry in Distinguishing between Mesothelioma and Renal Cell Carcinoma: A Comparative Study. Hum. Pathol. 2004, 35, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Bratslavsky, G.; Pinto, P.A.; Schmidt, L.S.; Neckers, L.; Bottaro, D.P.; Srinivasan, R. Molecular Diagnosis and Therapy of Kidney Cancer. Annu. Rev. Med. 2010, 61, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The Genetic Basis of Kidney Cancer: A Metabolic Disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Renshaw, A.A.; Richie, J.P. Subtypes of Renal Cell Carcinoma. Different Onset and Sites of Metastatic Disease. Am. J. Clin. Pathol. 1999, 111, 539–543. [Google Scholar] [CrossRef]
- Stewart, G.D.; O’mahony, F.C.; Powles, T.; Riddick, A.C.; Harrison, D.J.; Faratian, D. What Can Molecular Pathology Contribute to the Management of Renal Cell Carcinoma? Nat. Rev. Urol. 2011, 8, 255–265. [Google Scholar] [CrossRef]
- Kumar-Singh, S.A.; Segers, K.; Rodeck, U.; Backhovens, H.; Bogers, J.; Weyler, J.; Van Broeckhoven, C.H.; Van Marck, E.R. WT1 Mutation in Malignant Mesothelioma and WT1 Immunoreactivity in Relation to P53 and Growth Factor Receptor Expression, Cell-Type Transition, and Prognosis. J. Pathol. 1997, 181, 67–74. [Google Scholar] [CrossRef]
- Ordóñez, N.G. Value of Thyroid Transcription Factor-1, E-Cadherin, BG8, WT1, and CD44S Immunostaining in Distinguishing Epithelial Pleural Mesothelioma from Pulmonary and Nonpulmonary Adenocarcinoma. Am. J. Surg. Pathol. 2000, 24, 598–606. [Google Scholar] [CrossRef]
- Muir, T.E.; Cheville, J.C.; Lager, D.J. Metanephric Adenoma, Nephrogenic Rests, and Wilms’ Tumor: A Histologic and Immunophenotypic Comparison. Am. J. Surg. Pathol. 2001, 25, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zhou, G.X.; Zhou, M.; Chen, L. The Distinction of Clear Cell Carcinoma of the Female Genital Tract, Clear Cell Renal Cell Carcinoma, and Translocation-Associated Renal Cell Carcinoma: An Immunohistochemical Study Using Tissue Microarray. Int. J. Gynecol. Pathol. 2011, 30, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Mishra, K.; Sarkar, U.; Sharma, S.; Kumari, A. Diagnostic Utility of Wilms’ Tumour-1 Protein (WT-1) Immunostaining in Paediatric Renal Tumours. Indian J. Med. Res. 2016, 143 (Suppl. S1), S59–S67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisceglia, M.; Vairo, M.; Galliani, C.; Lastilla, G.; Parafioriti, A.; De Maglio, G. Immunohistochemical Investigation of WT1 Expression in 117 Embryonal Tumors. Pathologica 2011, 103, 182–183. [Google Scholar]
- Parenti, R.; Perris, R.; Vecchio, G.M.; Salvatorelli, L.; Torrisi, A.; Gravina, L.; Magro, G. Immunohistochemical Expression of Wilms’ Tumor Protein (WT1) in Developing Human Epithelial and Mesenchymal Tissues. Acta Histochem. 2013, 115, 70–75. [Google Scholar] [CrossRef]
- Parenti, R.; Cardile, V.; Graziano, A.C.; Parenti, C.; Venuti, A.; Bertuccio, M.P.; Furno, D.L.; Magro, G. Wilms’ Tumor Gene 1 (WT1) Silencing Inhibits Proliferation of Malignant Peripheral Nerve Sheath Tumor SNF96.2 Cell Line. PLoS ONE 2014, 9, e114333. [Google Scholar] [CrossRef] [Green Version]
- Magro, G.; Salvatorelli, L.; Vecchio, G.M.; Musumeci, G.; Rita, A.; Parenti, R. Cytoplasmic Expression of Wilms Tumor Transcription Factor-1 (WT1): A Useful Immunomarker for Young-Type Fibromatoses and Infantile Fibrosarcoma. Acta Histochem. 2014, 116, 1134–1140. [Google Scholar] [CrossRef]
- Magro, G.; Longo, F.; Salvatorelli, L.; Vecchio, G.M.; Parenti, R. Wilms’ Tumor Protein (WT1) in Mammary Myofibroblastoma: An Immunohistochemical Study. Acta Histochem. 2014, 116, 905–910. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Musumeci, G.; Bisceglia, M.; Parenti, R. Oncofetal Expression of Wilms’ Tumor 1 (WT1) Protein in Human Fetal, Adult and Neoplastic Skeletal Muscle Tissues. Acta Histochem. 2015, 117, 492–504. [Google Scholar] [CrossRef]
- Magro, G.; Longo, F.R.; Angelico, G.; Spadola, S.; Amore, F.F.; Salvatorelli, L. Immunohistochemistry as Potential Diagnostic Pitfall in the Most Common Solid Tumors of Children and Adolescents. Acta Histochem. 2015, 117, 397–414. [Google Scholar] [CrossRef]
- Niksic, M.; Slight, J.; Sanford, J.R.; Caceres, J.F.; Hastie, N.D. The Wilms’ Tumour Protein (WT1) Shuttles between Nucleus and Cytoplasm and Is Present in Functional Polysomes. Hum. Mol. Genet. 2004, 13, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuka, S.I.; Oji, Y.; Horiuchi, T.; Kanda, T.; Kitagawa, M.; Takeuchi, T.; Kawano, K.; Kuwae, Y.; Yamauchi, A.; Okumura, M.; et al. Immunohistochemical Detection of WT1 Protein in a Variety of Cancer Cells. Mod. Pathol. 2006, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Pelletier, J. Overlapping RNA and DNA Binding Domains of the Wt1 Tumor Suppressor Gene Product. Nucleic Acids Res. 1998, 26, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.C.; Morris, J.C.; Wang, J.; English, M.A.; Haber, D.A.; Shi, Y.; Licht, J.D. WT1-Mediated Transcriptional Activation Is Inhibited by Dominant Negative Mutant Proteins. J. Biol. Chem. 1995, 270, 10878–10884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffett, P.; Bruening, W.; Nakagama, H.; Bardeesy, N.A.; Housman, D.; Housman, D.E.; Pelletier, J. Antagonism of WT1 Activity by Protein Self-Association. Proc. Natl. Acad. Sci. USA 1995, 92, 11105–11109. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.; Ramsdale, T.; Mattick, J.; Little, M. An RNA Recognition Motif in Wilms’ Tumour Protein (WT1) Revealed by Structural Modelling. Nat. Genet. 1996, 12, 329–331. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 Is Required for Early Kidney Development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Herzer, U.; Crocoll, A.; Barton, D.; Howells, N.; Englert, C. The Wilms Tumor Suppressor Gene Wt1 Is Required for Development of the Spleen. Curr. Biol. 1999, 9, 837–840. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC Complementation Shows a Requirement for Wt1 in the Development of Epicardium, Adrenal Gland and throughout Nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar] [CrossRef]
- Bandiera, R.; Vidal, V.P.; Motamedi, F.J.; Clarkson, M.; Sahut-Barnola, I.; von Gise, A.; Pu, W.T.; Hohenstein, P.; Martinez, A.; Schedl, A. WT1 Maintains Adrenal-Gonadal Primordium Identity and Marks a Population of AGP-like Progenitors within the Adrenal Gland. Dev. Cell 2013, 27, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Barbaux, S.; Niaudet, P.; Gubler, M.C.; Grünfeld, J.P.; Jaubert, F.; Kuttenn, F.; Fékété, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor Splice-Site Mutations in WT1 Are Responsible for Frasier Syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Hammes, A.; Guo, J.K.; Lutsch, G.; Leheste, J.R.; Landrock, D.; Ziegler, U.; Gubler, M.C.; Schedl, A. Two Splice Variants of the Wilms’ Tumor 1 Gene Have Distinct Functions during Sex Determination and Nephron Formation. Cell 2001, 106, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Natoli, T.A.; McDonald, A.; Alberta, J.A.; Taglienti, M.E.; Housman, D.E.; Kreidberg, J.A. A Mammal-Specific Exon of WT1 Is Not Required for Development or Fertility. Mol. Cell. Biol. 2002, 22, 4433–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.J.; Schumacher, V.; Royer-Pokora, B.; Roberts, S.G.E. Par4 Is a Coactivator for a Splice Isoform–Specific Transcriptional Activation Domain in WT1. Genes Dev. 2001, 15, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, V.; Schneider, S.; Figge, A.; Wildhardt, G.; Harms, D.; Schmidt, D.; Weirich, A.; Ludwig, R.; Royer-Pokora, B. Correlation of Germ-Line Mutations and Two-Hit Inactivation of the WT1 Gene with Wilms Tumors of Stromal–Predominant Histology. Proc. Natl. Acad. Sci. USA 1997, 94, 3972–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, V.M.; Sujansky, E.; Smith, A.C.; Francke, U. Chromosomal Imbalance in the Aniridia-Wilms’ Tumor Association: 11p Interstitial Deletion. Pediatrics 1978, 61, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Little, M.; Holmes, G.; Bickmore, W.; van Heyningen, V.; Hastie, N.; Wainwright, B. DNA Binding Capacity of the WT1 Protein Is Abolished by Denys—Drash Syndrome WT1 Point Mutations. Hum. Mol. Genet. 1995, 4, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L.; et al. Germline Mutations in the Wilms’ Tumor Suppressor Gene Are Associated with Abnormal Urogenital Development in Denys-Drash Syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]
- Berry, R.L.; Ozdemir, D.D.; Aronow, B.; Lindström, N.O.; Dudnakova, T.; Thornburn, A.; Perry, P.; Baldock, R.; Armit, C.; Joshi, A.; et al. Deducing the Stage of Origin of Wilms’ Tumours from a Developmental Series of Wt1-Mutant Mice. Dis. Models Mech. 2015, 8, 903–917. [Google Scholar] [CrossRef]
- Sim, E.U.-H.; Smith, A.; Szilagi, E.; Rae, F.; Ioannou, P.; Lindsay, M.H.; Little, M.H. Wnt-4 Regulation by the Wilms’ Tumour Suppressor Gene, WT1. Oncogene 2002, 21, 2948–2960. [Google Scholar] [CrossRef]
- Davies, J.A.; Ladomery, M.; Hohenstein, P.; Michael, L.; Shafe, A.; Spraggon, L.; Hastie, N. Development of an SiRNA-Based Method for Repressing Specific Genes in Renal Organ Culture and Its Use to Show That the Wt1 Tumour Suppressor Is Required for Nephron Differentiation. Hum. Mol. Genet. 2004, 13, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Essafi, A.; Webb, A.; Berry, R.L.; Slight, J.; Burn, S.F.; Spraggon, L.; Velecela, V.; Martinez-Estrada, O.M.; Wiltshire, J.H.; Roberts, S.G.; et al. A Wt1-Controlled Chromatin Switching Mechanism Underpins Tissue-Specific Wnt4 Activation and Repression. Dev. Cell 2011, 21, 559–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial Transformation of Metanephric Mesenchyme in the Developing Kidney Regulated by Wnt-4. Nature 1994, 372, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Gebeshuber, C.A.; Kornauth, C.; Dong, L.; Sierig, R.; Seibler, J.; Reiss, M.; Tauber, S.; Bilban, M.; Wang, S.; Kain, R.; et al. Focal Segmental Glomerulosclerosis Is Induced by MicroRNA-193a and Its Downregulation of WT1. Nat. Med. 2013, 19, 481–487. [Google Scholar] [CrossRef]
- Cano, E.; Carmona, R.; Ruiz-Villalba, A.; Rojas, A.; Chau, Y.-Y.; Wagner, K.D.; Wagner, N.; Hastie, N.D.; Muñoz-Chápuli, R.; Pérez-Pomares, J.M. Extracardiac Septum Transversum/Proepicardial Endothelial Cells Pattern Embryonic Coronary Arterio–Venous Connections. Proc. Natl. Acad. Sci. USA 2016, 113, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 Is Required for Cardiovascular Progenitor Cell Formation through Transcriptional Control of Snail and E-Cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [Green Version]
- von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. WT1 Regulates Epicardial Epithelial to Mesenchymal Transition through β-Catenin and Retinoic Acid Signaling Pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Muñoz-Chápuli, R.; Hastie, N.D.; Pérez-Pomares, J.M.; Martínez-Estrada, O.M. Wt1 Controls Retinoic Acid Signalling in Embryonic Epicardium through Transcriptional Activation of Raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Velecela, V.; Lettice, L.A.; Chau, Y.-Y.; Slight, J.; Berry, R.L.; Thornburn, A.; Gunst, Q.D.; van den Hoff, M.; Reina, M.; Martínez, F.O.; et al. WT1 Regulates the Expression of Inhibitory Chemokines during Heart Development. Hum. Mol. Genet. 2013, 22, 5083–5095. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, T.; Rinkevich, Y. Mesothelial to Mesenchyme Transition as a Major Developmental and Pathological Player in Trunk Organs and Their Cavities. Commun. Biol. 2018, 1, 170. [Google Scholar] [CrossRef] [Green Version]
- Wilm, B.; Ipenberg, A.; Hastie, N.D.; Burch, J.B.E.; Bader, D.M. The Serosal Mesothelium Is a Major Source of Smooth Muscle Cells of the Gut Vasculature. Development 2005, 132, 5317–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, J.; Wilm, B.; Hasegawa, H.; Wang, F.; Bader, D.; Hogan, B.L.M. Mesothelium Contributes to Vascular Smooth Muscle and Mesenchyme during Lung Development. Proc. Natl. Acad. Sci. USA 2008, 105, 16626–16630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, E.; Carmona, R.; Muñoz-Chápuli, R. Wt1-Expressing Progenitors Contribute to Multiple Tissues in the Developing Lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L322–L332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahina, K.; Zhou, B.; Pu, W.T.; Tsukamoto, H. Septum Transversum-Derived Mesothelium Gives Rise to Hepatic Stellate Cells and Perivascular Mesenchymal Cells in Developing Mouse Liver. Hepatology 2011, 53, 983–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, R.; Cano, E.; Mattiotti, A.; Gaztambide, J.; Muñoz-Chápuli, R. Cells Derived from the Coelomic Epithelium Contribute to Multiple Gastrointestinal Tissues in Mouse Embryos. PLoS ONE 2013, 8, e55890. [Google Scholar] [CrossRef] [Green Version]
- Chau, Y.-Y.; Brownstein, D.; Mjoseng, H.; Lee, W.-C.; Buza-Vidas, N.; Nerlov, C.; Jacobsen, S.E.; Perry, P.; Berry, R.; Thornburn, A.; et al. Acute Multiple Organ Failure in Adult Mice Deleted for the Developmental Regulator Wt1. PLoS Genet. 2011, 7, e1002404. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Wagner, N.; Wellmann, S.; Schley, G.; Bondke, A.; Theres, H.; Scholz, H. Oxygen-Regulated Expression of the Wilms’ Tumor Suppressor Wt1 Involves Hypoxia-Inducible Factor-1 (HIF-1). FASEB J. 2003, 17, 1364–1366. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De Novo Cardiomyocytes from within the Activated Adult Heart after Injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Wagner, N.; Bondke, A.; Nafz, B.; Flemming, B.; Theres, H.; Scholz, H. The Wilms’ Tumor Suppressor Wt1 Is Expressed in the Coronary Vasculature after Myocardial Infarction. FASEB J. 2002, 16, 1117–1119. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.-F.; Wagner, N. The Wilms’ Tumour Suppressor Wt1 Is a Major Regulator of Tumour Angiogenesis and Progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [Green Version]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wang, K.; Xiong, Z.; Yuan, C.; Wang, C.; Cao, Q.; Yu, H.; Meng, X.; Xie, K.; Cheng, Z.; et al. Impact of Inflammation and Immunotherapy in Renal Cell Carcinoma. Oncol. Lett. 2020, 20, 272. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Li, L.; Zhang, W.; Wang, W.; Chan, Y.; You, B.; Li, X. Comprehensive Characterization of Immune- and Inflammation-Associated Biomarkers Based on Multi-Omics Integration in Kidney Renal Clear Cell Carcinoma. J. Transl. Med. 2019, 17, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehbi, M.; Hiscott, J.; Pelletier, J. Activation of the Wt1 Wilms’ Tumor Suppressor Gene by NF-KB. 7. Oncogene 1998, 16, 2033–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, M.; Motojima, M.; Miyazaki, Y.; Pastan, I.; Yokoo, T.; Matsusaka, T. Global Polysome Analysis of Normal and Injured Podocytes. Am. J. Physiol. Ren. Physiol. 2019, 316, F241–F252. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Yoshida, M.; Semba, K.; Hunter, T. Inhibition of the DNA-Binding and Transcriptional Repression Activity of the Wilms’ Tumor Gene Product, WT1, by CAMP-Dependent Protein Kinase-Mediated Phosphorylation of Ser-365 and Ser-393 in the Zinc Finger Domain. Oncogene 1997, 15, 2001–2012. [Google Scholar] [CrossRef] [Green Version]
- Bruening, W.; Moffett, P.; Chia, S.; Heinrich, G.; Pelletier, J. Identification of Nuclear Localization Signals within the Zinc Fingers of the WT1 Tumor Suppressor Gene Product. FEBS Lett. 1996, 393, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Raychaudhuri, B.; Gurney, A.; Campbell, C.E.; Williams, B.R. Regulation of WT1 by Phosphorylation: Inhibition of DNA Binding, Alteration of Transcriptional Activity and Cellular Translocation. EMBO J. 1996, 15, 5606–5615. [Google Scholar] [CrossRef]
- Lipska, B.S.; Ranchin, B.; Iatropoulos, P.; Gellermann, J.; Melk, A.; Ozaltin, F.; Caridi, G.; Seeman, T.; Tory, K.; Jankauskiene, A.; et al. PodoNet Consortium. Genotype-Phenotype Associations in WT1 Glomerulopathy. Kidney Int. 2014, 85, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arellano-Rodríguez, M.; Zapata-Benavides, P.; Arellano-Rodríguez, N.C.; Izaguirre-Álvarez, J.M.; Franco-Molina, M.A.; Torres Del Muro, F.D.J.; Mendoza-Gamboa, E.; Soto-Domínguez, A.; Saavedra-Alonso, S.; Rodríguez-Padilla, C. The Inflammatory Process Modulates the Expression and Localization of WT1 in Podocytes Leading to Kidney Damage. In Vivo 2021, 35, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Carlesso, N.; Cheng, T.; Scadden, D.T.; Haber, D.A. The Wilms Tumor Suppressor WT1 Directs Stage-Specific Quiescence and Differentiation of Human Hematopoietic Progenitor Cells. EMBO J. 2001, 20, 1897–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Wang, S.; Xu, C.; Tyler, A.; Li, X.; Andersson, C.; Oji, Y.; Sugiyama, H.; Chen, Y.; Li, A. WT1 Enhances Proliferation and Impedes Apoptosis in KRAS Mutant NSCLC via Targeting CMyc. CPB 2015, 35, 647–662. [Google Scholar] [CrossRef]
- Yamagami, T.; Sugiyama, H.; Inoue, K.; Ogawa, H.; Tatekawa, T.; Hirata, M.; Kudoh, T.; Akiyama, T.; Murakami, A.; Maekawa, T. Growth Inhibition of Human Leukemic Cells by WT1 (Wilms Tumor Gene) Antisense Oligodeoxynucleotides: Implications for the Involvement of WT1 in Leukemogenesis. Blood 1996, 87, 2878–2884. [Google Scholar] [CrossRef] [Green Version]
- Algar, E.M.; Khromykh, T.; Smith, S.I.; Blackburn, D.M.; Bryson, G.J.; Smith, P.J. A WT1 Antisense Oligonucleotide Inhibits Proliferation and Induces Apoptosis in Myeloid Leukaemia Cell Lines. Oncogene 1996, 12, 1005–1014. [Google Scholar]
- Tatsumi, N.; Oji, Y.; Tsuji, N.; Tsuda, A.; Higashio, M.; Aoyagi, S.; Fukuda, I.; Ito, K.; Nakamura, J.; Takashima, S.; et al. Wilms’ Tumor Gene WT1-ShRNA as a Potent Apoptosis-Inducing Agent for Solid Tumors. Int. J. Oncol. 2008, 32, 701–711. [Google Scholar] [CrossRef]
- Nishida, S.; Hosen, N.; Shirakata, T.; Kanato, K.; Yanagihara, M.; Nakatsuka, S.; Hoshida, Y.; Nakazawa, T.; Harada, Y.; Tatsumi, N.; et al. AML1-ETO Rapidly Induces Acute Myeloblastic Leukemia in Cooperation with the Wilms Tumor Gene, WT1. Blood 2006, 107, 3303–3312. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Tamaki, H.; Ogawa, H.; Oka, Y.; Soma, T.; Tatekawa, T.; Oji, Y.; Tsuboi, A.; Kim, E.H.; Kawakami, M.; et al. Wilms’ Tumor Gene (WT1) Competes with Differentiation-Inducing Signal in Hematopoietic Progenitor Cells. Blood 1998, 91, 2969–2976. [Google Scholar] [CrossRef]
- Tsuboi, A.; Oka, Y.; Ogawa, H.; Elisseeva, O.A.; Tamaki, H.; Oji, Y.; Kim, E.H.; Soma, T.; Tatekawa, T.; Kawakami, M.; et al. Constitutive Expression of the Wilms’ Tumor Gene WT1 Inhibits the Differentiation of Myeloid Progenitor Cells but Promotes Their Proliferation in Response to Granulocyte-Colony Stimulating Factor (G-CSF). Leuk. Res. 1999, 23, 499–505. [Google Scholar] [CrossRef]
- Li, H.; Oka, Y.; Tsuboi, A.; Yamagami, T.; Miyazaki, T.; Yusa, S.; Kawasaki, K.; Kishimoto, Y.; Asada, M.; Nakajima, H.; et al. The Lck Promoter-Driven Expression of the Wilms Tumor Gene WT1 Blocks Intrathymic Differentiation of T-Lineage Cells. Int. J. Hematol. 2003, 77, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Jomgeow, T.; Oji, Y.; Tsuji, N.; Ikeda, Y.; Ito, K.; Tsuda, A.; Nakazawa, T.; Tatsumi, N.; Sakaguchi, N.; Takashima, S.; et al. Wilms’ Tumor Gene WT1 17AA(-)/KTS(-) Isoform Induces Morphological Changes and Promotes Cell Migration and Invasion in Vitro. Cancer Sci. 2006, 97, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Oji, Y.; Tatsumi, N.; Shimizu, S.; Kanai, Y.; Nakazawa, T.; Asada, M.; Jomgeow, T.; Aoyagi, S.; Nakano, Y.; et al. Antiapoptotic Function of 17AA(+)WT1 (Wilms’ Tumor Gene) Isoforms on the Intrinsic Apoptosis Pathway. Oncogene 2006, 25, 4217–4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartkamp, J.; Carpenter, B.; Roberts, S.G.E. The Wilms’ Tumor Suppressor Protein WT1 Is Processed by the Serine Protease HtrA2/Omi. Mol. Cell 2010, 37, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, N.; Hojo, N.; Sakamoto, H.; Inaba, R.; Moriguchi, N.; Matsuno, K.; Fukuda, M.; Matsumura, A.; Hayashi, S.; Morimoto, S.; et al. Identification of a Novel C-Terminal Truncated WT1 Isoform with Antagonistic Effects against Major WT1 Isoforms. PLoS ONE 2015, 10, e0130578. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Development and Pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Tun, H.W.; Marlow, L.A.; von Roemeling, C.A.; Cooper, S.J.; Kreinest, P.; Wu, K.; Luxon, B.A.; Sinha, M.; Anastasiadis, P.Z.; Copland, J.A. Pathway Signature and Cellular Differentiation in Clear Cell Renal Cell Carcinoma. PLoS ONE 2010, 5, e10696. [Google Scholar] [CrossRef]
- Zhao, H.; Zongming, M.; Tibshirani, R.; Higgins, J.P.T.; Ljungberg, B.; Brooks, J.D. Alteration of Gene Expression Signatures of Cortical Differentiation and Wound Response in Lethal Clear Cell Renal Cell Carcinomas. PLoS ONE 2009, 4, e6039. [Google Scholar] [CrossRef]
- Banumathy, G.; Cairns, P. Signaling Pathways in Renal Cell Carcinoma. Cancer Biol. Ther. 2010, 10, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Nallandhighal, S.; Vince, R.; Karim, R.; Groves, S.; Stangl-Kremser, J.; Russell, C.; Hu, K.; Pham, T.; Cani, A.K.; Liu, C.-J.; et al. Molecular Characterization of Clear Cell Renal Cell Carcinoma Reveals Prognostic Significance of Epithelial-Mesenchymal Transition Gene Expression Signature. Eur. Urol. Oncol. 2022, 5, 92–99. [Google Scholar] [CrossRef]
- O’Mahony, F.C.; Faratian, D.; Varley, J.; Nanda, J.; Theodoulou, M.; Riddick, A.C.P.; Harrison, D.J.; Stewart, G.D. The Use of Automated Quantitative Analysis to Evaluate Epithelial-to-Mesenchymal Transition Associated Proteins in Clear Cell Renal Cell Carcinoma. PLoS ONE 2012, 7, e31557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iiyama, T.; Udaka, K.; Takeda, S.; Takeuchi, T.; Adachi, Y.C.; Ohtsuki, Y.; Tsuboi, A.; Nakatsuka, S.; Elisseeva, O.A.; Oji, Y.; et al. WT1 (Wilms’ Tumor 1) Peptide Immunotherapy for Renal Cell Carcinoma. Microbiol. Immunol. 2007, 51, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bellantuono, I.; Elsässer, A.; Marley, S.B.; Gordon, M.Y.; Goldman, J.M.; Stauss, H.J. Selective Elimination of Leukemic CD34+ Progenitor Cells by Cytotoxic T Lymphocytes Specific for WT1. Blood 2000, 95, 2198–2203. [Google Scholar] [CrossRef] [PubMed]
- Mailänder, V.; Scheibenbogen, C.; Thiel, E.; Letsch, A.; Blau, I.W.; Keilholz, U. Complete Remission in a Patient with Recurrent Acute Myeloid Leukemia Induced by Vaccination with WT1 Peptide in the Absence of Hematological or Renal Toxicity. Leukemia 2004, 18, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Ohminami, H.; Yasukawa, M.; Fujita, S. HLA Class I-Restricted Lysis of Leukemia Cells by a CD8+ Cytotoxic T-Lymphocyte Clone Specific for WT1 Peptide. Blood 2000, 95, 286–293. [Google Scholar] [CrossRef]
- Oka, Y.; Elisseeva, O.A.; Tsuboi, A.; Ogawa, H.; Tamaki, H.; Li, H.; Oji, Y.; Kim, E.H.; Soma, T.; Asada, M.; et al. Human Cytotoxic T-Lymphocyte Responses Specific for Peptides of the Wild-Type Wilms’ Tumor Gene (WT1) Product. Immunogenetics 2000, 51, 99–107. [Google Scholar] [CrossRef]
- Oka, Y.; Udaka, K.; Tsuboi, A.; Elisseeva, O.A.; Ogawa, H.; Aozasa, K.; Kishimoto, T.; Sugiyama, H. Cancer Immunotherapy Targeting Wilms’ Tumor Gene WT1 Product. J. Immunol. 2000, 164, 1873–1880. [Google Scholar] [CrossRef]
- Scheibenbogen, C.; Letsch, A.; Thiel, E.; Schmittel, A.; Mailaender, V.; Baerwolf, S.; Nagorsen, D.; Keilholz, U. CD8 T-Cell Responses to Wilms Tumor Gene Product WT1 and Proteinase 3 in Patients with Acute Myeloid Leukemia. Blood 2002, 100, 2132–2137. [Google Scholar] [CrossRef]
- Tsuboi, A.; Oka, Y.; Ogawa, H.; Elisseeva, O.A.; Li, H.; Kawasaki, K.; Aozasa, K.; Kishimoto, T.; Udaka, K.; Sugiyama, H. Cytotoxic T-Lymphocyte Responses Elicited to Wilms’ Tumor Gene WT1 Product by DNA Vaccination. J. Clin. Immunol. 2000, 20, 195–202. [Google Scholar] [CrossRef]
- Elisseeva, O.A.; Oka, Y.; Tsuboi, A.; Ogata, K.; Wu, F.; Kim, E.H.; Soma, T.; Tamaki, H.; Kawakami, M.; Oji, Y.; et al. Humoral Immune Responses against Wilms Tumor Gene WT1 Product in Patients with Hematopoietic Malignancies. Blood 2002, 99, 3272–3279. [Google Scholar] [CrossRef] [Green Version]
- Oka, Y.; Tsuboi, A.; Taguchi, T.; Osaki, T.; Kyo, T.; Nakajima, H.; Elisseeva, O.A.; Oji, Y.; Kawakami, M.; Ikegame, K.; et al. Induction of WT1 (Wilms’ Tumor Gene)-Specific Cytotoxic T Lymphocytes by WT1 Peptide Vaccine and the Resultant Cancer Regression. Proc. Natl. Acad. Sci. USA 2004, 101, 13885–13890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, Y.; Tsuboi, A.; Murakami, M.; Hirai, M.; Tominaga, N.; Nakajima, H.; Elisseeva, O.A.; Masuda, T.; Nakano, A.; Kawakami, M.; et al. Wilms Tumor Gene Peptide-Based Immunotherapy for Patients with Overt Leukemia from Myelodysplastic Syndrome (MDS) or MDS with Myelofibrosis. Int. J. Hematol. 2003, 78, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, A.; Oka, Y.; Osaki, T.; Kumagai, T.; Tachibana, I.; Hayashi, S.; Murakami, M.; Nakajima, H.; Elisseeva, O.A.; Fei, W.; et al. WT1 Peptide-Based Immunotherapy for Patients with Lung Cancer: Report of Two Cases. Microbiol. Immunol. 2004, 48, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Gillmore, R.; Xue, S.-A.; Holler, A.; Kaeda, J.; Hadjiminas, D.; Healy, V.; Dina, R.; Parry, S.C.; Bellantuono, I.; Ghani, Y.; et al. Detection of Wilms’ Tumor Antigen–Specific CTL in Tumor-Draining Lymph Nodes of Patients with Early Breast Cancer. Clin. Cancer Res. 2006, 12, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and Persistence of WT1-Specific T-Cell Receptor Gene−transduced Lymphocytes in Patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Hossain, A.; Nixon, M.; Kuo, M.T.; Saunders, G.F. N-Terminally Truncated WT1 Protein with Oncogenic Properties Overexpressed in Leukemia. J. Biol. Chem. 2006, 281, 28122–28130. [Google Scholar] [CrossRef] [Green Version]
- Dechsukhum, C.; Ware, J.L.; Ferreira-Gonzalez, A.; Wilkinson, D.S.; Garrett, C.T. Detection of a Novel Truncated WT1 Transcript in Human Neoplasia. Mol. Diagn. 2000, 5, 117–128. [Google Scholar] [CrossRef]
- Dallosso, A.R.; Hancock, A.L.; Brown, K.W.; Williams, A.C.; Jackson, S.; Malik, K. Genomic Imprinting at the WT1 Gene Involves a Novel Coding Transcript (AWT1) That Shows Deregulation in Wilms’ Tumours. Hum. Mol. Genet. 2004, 13, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Tsuta, K.; Kato, Y.; Tochigi, N.; Hoshino, T.; Takeda, Y.; Hosako, M.; Maeshima, A.M.; Asamura, H.; Kondo, T.; Matsuno, Y. Comparison of Different Clones (WT49 versus 6F-H2) of WT-1 Antibodies for Immunohistochemical Diagnosis of Malignant Pleural Mesothelioma. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 126–130. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grade | Characteristics |
|---|---|
| 1 | Absent or inconspicuous nucleoli and basophilic under 400× magnification. |
| 2 | Conspicuous and eosinophilic nucleoli under 400× magnification, but not prominent under 100× magnification. |
| 3 | Eosinophilic and prominent nucleoli under 100× magnification. |
| 4 | Extreme nuclear pleomorphism and/or giant neoplastic cells and/or any degree of sarcomatoid and/or rhabdoid dedifferentiation. |
| Grade | Definition |
|---|---|
| 0 | Absent WT1 staining. |
| +1 | Rare positive nuclei/cytoplasm, constituting < 2% of total tumor cell population. |
| +2 | Positive nuclei/cytoplasm constitute < 10% of total tumor cell population. |
| +3 | Positive nuclei/cytoplasm constitute > 10% of total tumor cell population. |
| Sex | Age | Subtype | Nuclear Grading | TNM | Quantitative WT1 | Qualitative WT1 | Clinical Outcome |
|---|---|---|---|---|---|---|---|
| Male | 68 | Clear cell RCC | 2 | pT2b | +1 | weak/ moderate | Died 3 years later after 2 lines of treatment for pulmonary metastases. |
| Male | 70 | Clear cell RCC | 2 | pT2a | +3 | moderate | No recurrence at 5 years. |
| Male | 65 | Clear cell RCC | 1 | pT1a | +2 | high | No recurrence at 5 years. |
| Female | 58 | Clear cell RCC | 1 | pT1a | +2 | high | No recurrence at 5 years. |
| Male | 74 | Clear cell RCC | 2 | pT1b | +1 | high | Died of cardiovascular event, but no recurrence at 4 years. |
| Male | 62 | Clear cell RCC | 2 | pT3a | +2 | high | Solitary pulmonary metastasis after 2.5 years, treated, no recurrence at 5 years. |
| Male | 68 | Clear cell RCC | 1 | pT1b | +3 | moderate | No recurrence at 5 years. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novacescu, D.; Cut, T.G.; Cumpanas, A.A.; Latcu, S.C.; Bardan, R.; Ferician, O.; Secasan, C.-C.; Rusmir, A.; Raica, M. Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49). Biomedicines 2022, 10, 912. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040912
Novacescu D, Cut TG, Cumpanas AA, Latcu SC, Bardan R, Ferician O, Secasan C-C, Rusmir A, Raica M. Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49). Biomedicines. 2022; 10(4):912. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040912
Chicago/Turabian StyleNovacescu, Dorin, Talida Georgiana Cut, Alin Adrian Cumpanas, Silviu Constantin Latcu, Razvan Bardan, Ovidiu Ferician, Cosmin-Ciprian Secasan, Andrei Rusmir, and Marius Raica. 2022. "Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49)" Biomedicines 10, no. 4: 912. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040912