
Molecular Signature of Neuroinflammation Induced in Cytokine-Stimulated Human Cortical Spheroids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of H9 Embryonic Stem Cells (ESCs)
2.2. Generation of hCSs
2.3. Culture of Human Microglia (HMC3) and Human Oligodendrocyte (HOG) Cell Lines
2.4. Pro-Inflammatory Cytokine Stimulation
2.5. RNA Isolation
2.6. RNA-Seq Analysis
2.7. Bioinformatics Analysis
2.8. Quantitative PCR (qPCR) Analysis
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
3.1. Neuroinflammation Is the Main Process Elicited in hCSs Stimulated with the Pro-inflammatory Cytokines TNFα and IL-1β
3.2. Genes Dysregulated in TNFα- and IL-1β-Stimulated hCSs Are Involved in Positive and Negative Feedback Loops of Neuroinflammatory Signaling Pathways
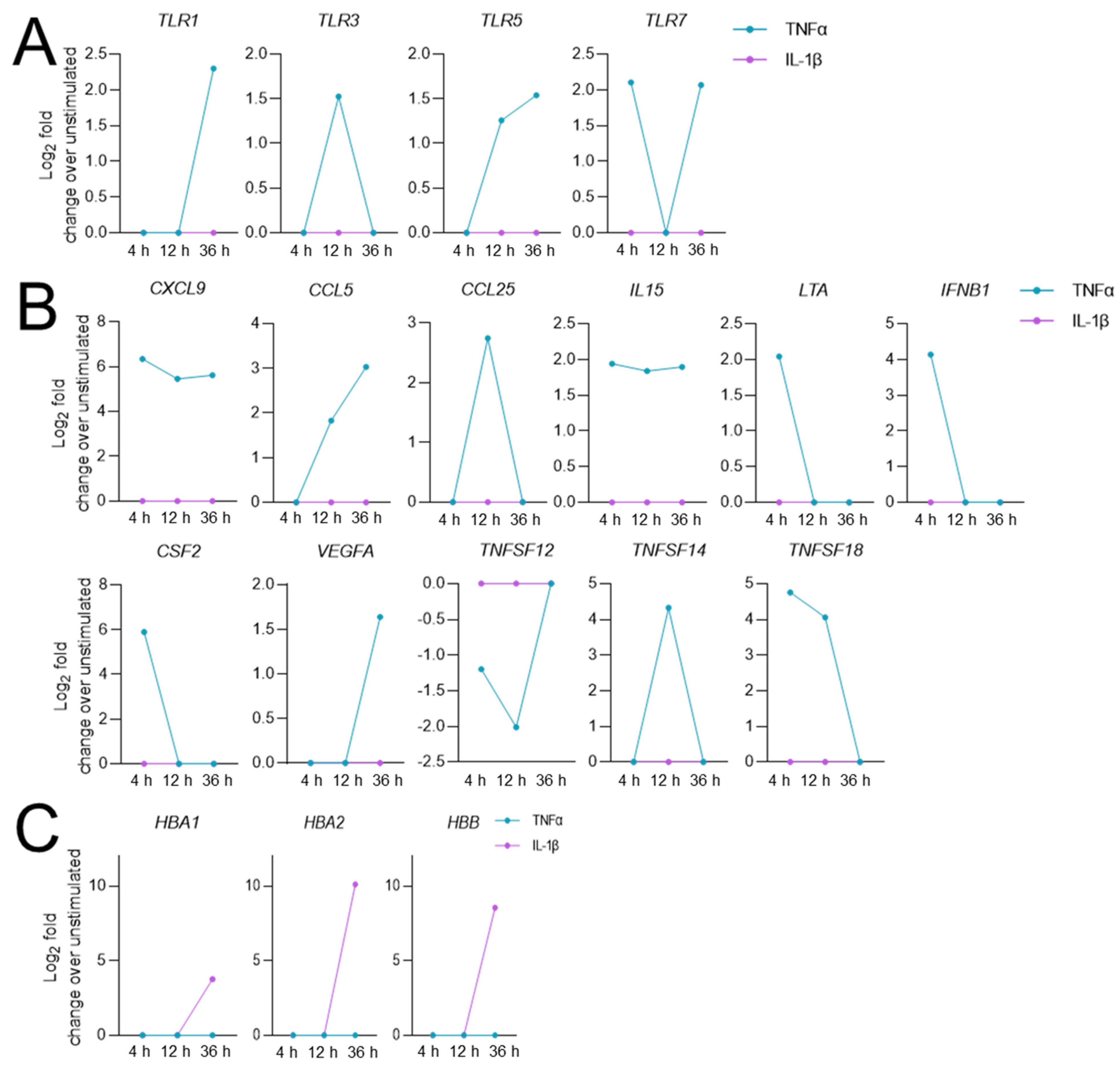
3.3. Stimulation-Time-Dependent Sets of DEGs in TNFα- and IL-1β-Stimulated hCSs
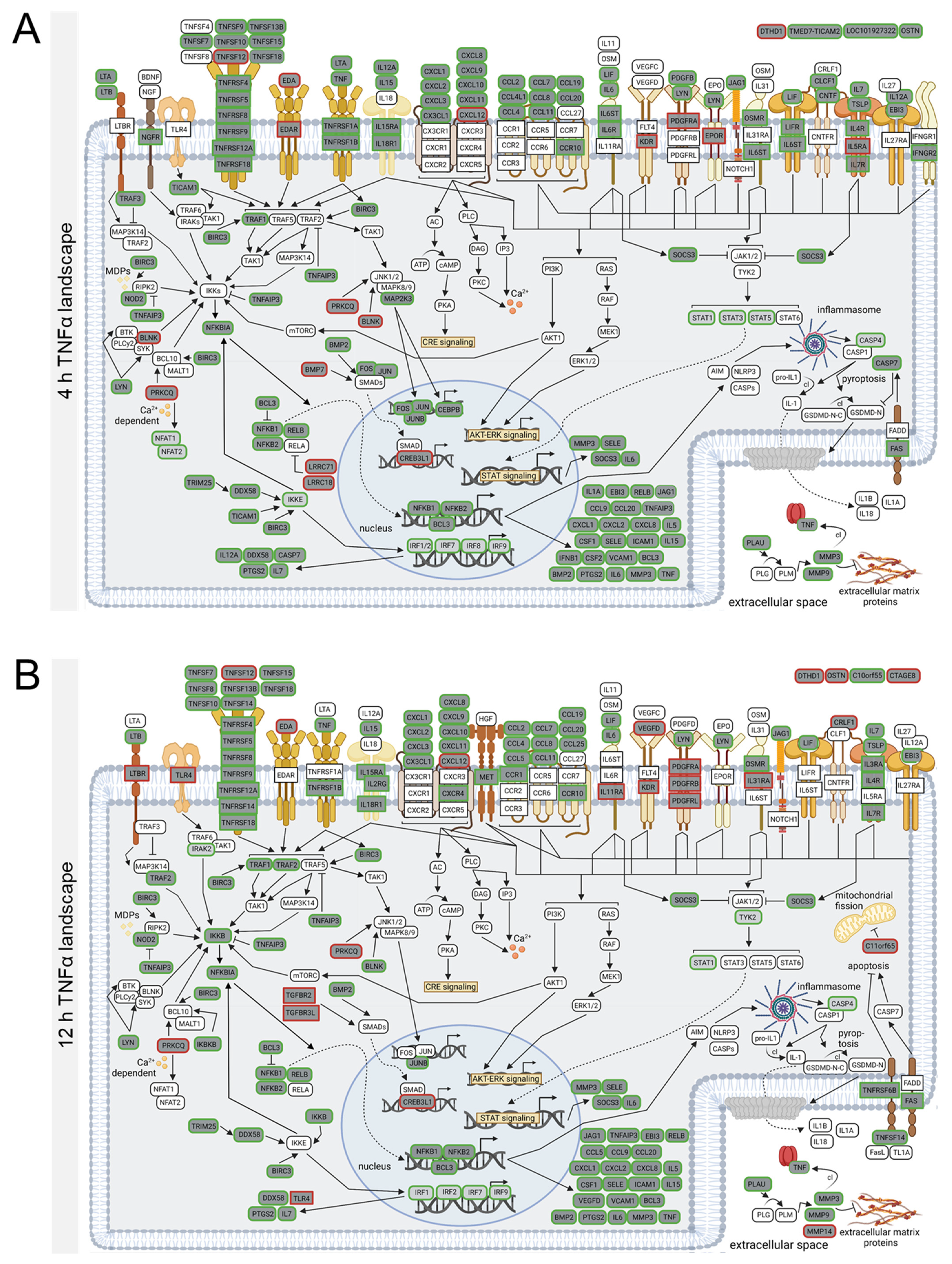
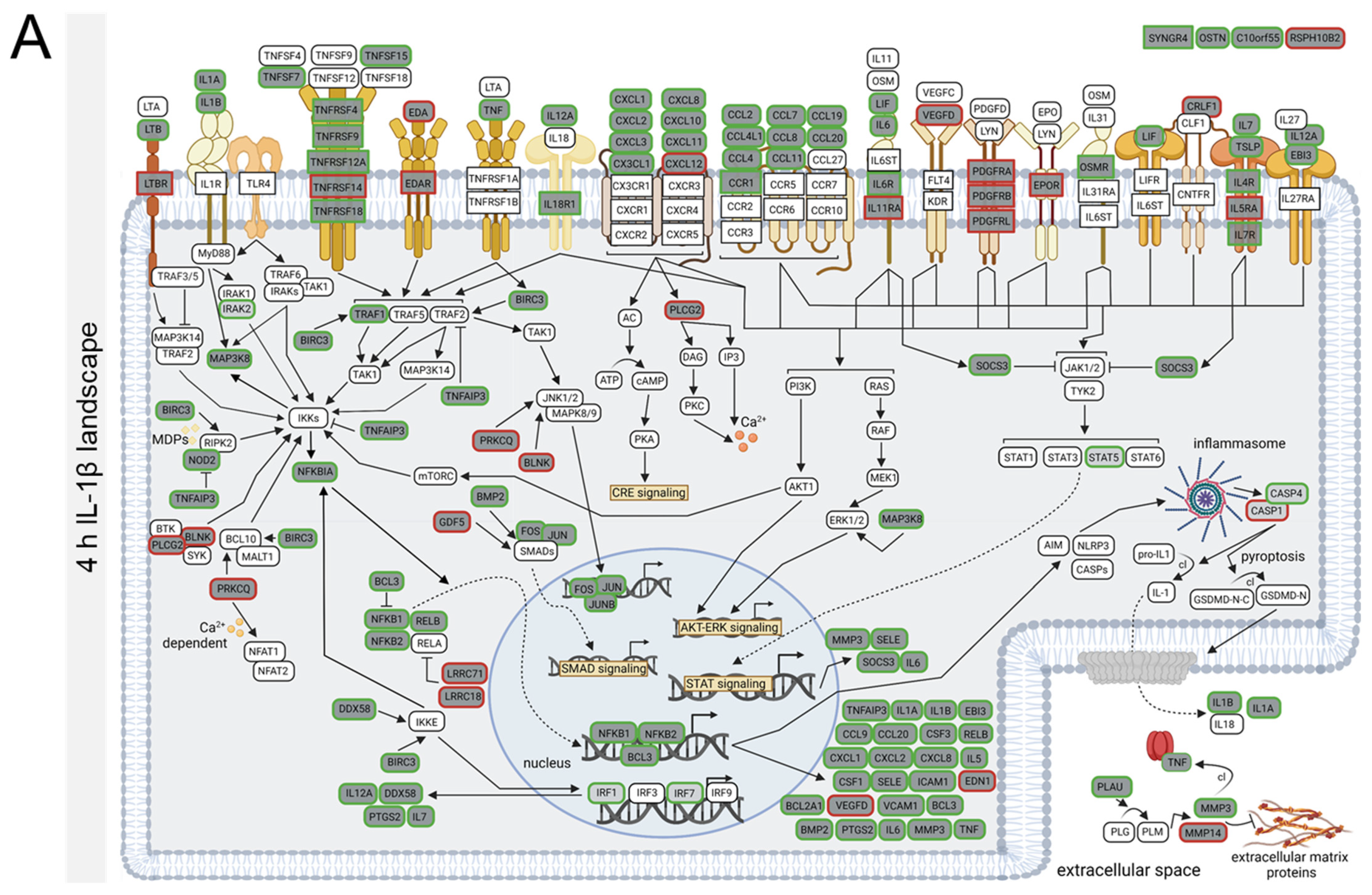
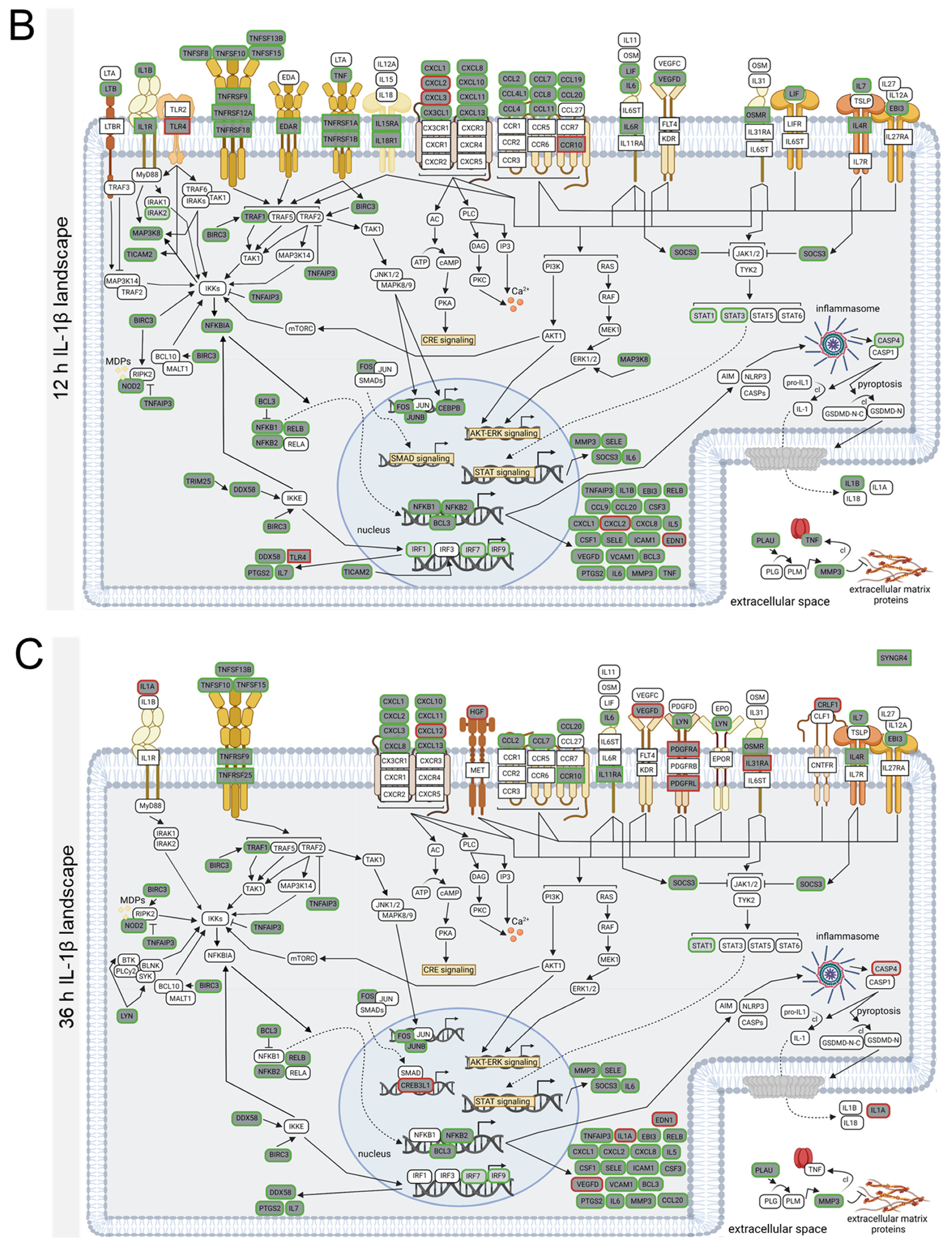
3.4. TNFα- and IL-1β-Specific Effects on Neuroinflammatory Signaling Pathways
3.5. TNFα- and IL-1β-Induced Neuroinflammation Primarily Occurs in Endothelial, Microglia and Astrocyte Cell Populations, and Is More Related to MS Than to AD and PD
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mygind, L.; Bergh, M.S.; Tejsi, V.; Vaitheeswaran, R.; Lambertsen, K.L.; Finsen, B.; Metaxas, A. Tumor Necrosis Factor Tnf Is Required for Spatial Learning and Memory in Male Mice under Physiological, but Not Immune-Challenged Conditions. Cells 2021, 10, 608. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Hewett, S.J.; Jackman, N.A.; Claycomb, R.J. Interleukin-1beta in Central Nervous System Injury and Repair. Eur. J. Neurodegener. Dis. 2012, 1, 195–211. [Google Scholar] [PubMed]
- Portales-Casamar, E.; Thongjuea, S.; Kwon, A.T.; Arenillas, D.; Zhao, X.; Valen, E.; Yusuf, D.; Lenhard, B.; Wasserman, W.W.; Sandelin, A. The Greatly Expanded Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2010, 38, D105–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour Necrosis Factor Signalling in Health and Disease. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The Roles of Tnf in Brain Dysfunction and Disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The Role of Interleukin-1 in Neuroinflammation and Alzheimer Disease: An Evolving Perspective. J. Neuroinflamm. 2008, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Khairova, R.A.; Machado-Vieira, R.; Du, J.; Manji, H.K. A Potential Role for Pro-Inflammatory Cytokines in Regulating Synaptic Plasticity in Major Depressive Disorder. Int. J. Neuropsychopharmacol. 2009, 12, 561–578. [Google Scholar] [CrossRef]
- Kim, Y.K.; Na, K.S.; Shin, K.H.; Jung, H.Y.; Choi, S.H.; Kim, J.B. Cytokine Imbalance in the Pathophysiology of Major Depressive Disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 1044–1053. [Google Scholar] [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of Tnf in Ms Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef]
- Lin, C.C.; Edelson, B.T. New Insights into the Role of Il-1beta in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. J. Immunol. 2017, 198, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deverman, B.E.; Patterson, P.H. Cytokines and Cns Development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhaye, J.; Knoblich, J.A. Brain Organoids: An Ensemble of Bioassays to Investigate Human Neurodevelopment and Disease. Cell Death Differ. 2021, 28, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Liang, F.; Xu, S.; Li, X. The Application of Brain Organoids: From Neuronal Development to Neurological Diseases. Front. Cell Dev. Biol. 2020, 8, 579659. [Google Scholar] [CrossRef]
- Agboola, O.S.; Hu, X.; Shan, Z.; Wu, Y.; Lei, L. Brain Organoid: A 3d Technology for Investigating Cellular Composition and Interactions in Human Neurological Development and Disease Models in Vitro. Stem Cell Res. Ther. 2021, 12, 430. [Google Scholar] [CrossRef]
- Chiaradia, I.; Lancaster, M.A. Brain Organoids for the Study of Human Neurobiology at the Interface of in Vitro and in Vivo. Nat. Neurosci. 2020, 23, 1496–1508. [Google Scholar] [CrossRef]
- Saha, R.N.; Ghosh, A.; Palencia, C.A.; Fung, Y.K.; Dudek, S.M.; Pahan, K. Tnf-Alpha Preconditioning Protects Neurons Via Neuron-Specific up-Regulation of Creb-Binding Protein. J. Immunol. 2009, 183, 2068–2078. [Google Scholar] [CrossRef]
- Huang, Y.; Smith, D.E.; Ibanez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-Specific Effects of Interleukin-1beta Are Mediated by a Novel Isoform of the Il-1 Receptor Accessory Protein. J. Neurosci. 2011, 31, 18048–18059. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kang, M.J.; Han, J.S. Interleukin-1 Beta Promotes Neuronal Differentiation through the Wnt5a/Rhoa/Jnk Pathway in Cortical Neural Precursor Cells. Mol. Brain 2018, 11, 39. [Google Scholar] [CrossRef]
- Bras, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. Tnf-Alpha-Induced Microglia Activation Requires Mir-342: Impact on Nf-Kb Signaling and Neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef]
- Hyvarinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-Stimulation with Il-1beta and Tnf-Alpha Induces an Inflammatory Reactive Astrocyte Phenotype with Neurosupportive Characteristics in a Human Pluripotent Stem Cell Model System. Sci. Rep. 2019, 9, 16944. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.A.; Powell, H.R.; Liput, M.; Dinham, S.; Freedman, D.A.; Ignatowski, T.A.; Stachowiak, E.K.; Stachowiak, M.K. Immune Factor, Tnfalpha, Disrupts Human Brain Organoid Development Similar to Schizophrenia-Schizophrenia Increases Developmental Vulnerability to Tnfalpha. Front. Cell Neurosci. 2020, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate Transcript Quantification from Rna-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audic, S.; Claverie, J.M. The Significance of Digital Gene Expression Profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Dong, J.; Zhong, S.; Wei, Y.; Wu, Q.; Yan, L.; Yong, J.; Sun, L.; Wang, X.; Zhao, Y.; et al. Spatial Transcriptomic Survey of Human Embryonic Cerebral Cortex by Single-Cell Rna-Seq Analysis. Cell Res. 2018, 28, 730–745. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Voskuhl, R.R. Cell Specificity Dictates Similarities in Gene Expression in Multiple Sclerosis, Parkinson’s Disease, and Alzheimer’s Disease. PLoS ONE 2017, 12, e0181349. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative Rt-Pcr Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.K. Microglia as a Source and Target of Cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Muhammad, M. Tumor Necrosis Factor Alpha: A Major Cytokine of Brain Neuroinflammation. In Cytokines; Behzadi, P., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Zelova, H.; Hosek, J. Tnf-Alpha Signalling and Inflammation: Interactions between Old Acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation Pathways: A General Review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-Like Receptors in the Pathogenesis of Neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. Nf-Kappab Signaling in Inflammation. Signal Transduct. Target. Ther. Vol. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of Nf-Kappab Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. Jak-Stat Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Tzeng, H.T.; Chyuan, I.T.; Lai, J.H. Targeting the Jak-Stat Pathway in Autoimmune Diseases and Cancers: A Focus on Molecular Mechanisms and Therapeutic Potential. Biochem. Pharmacol. 2021, 193, 114760. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Casteele, N.V. Jak-Stat Pathway Targeting for the Treatment of Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Qing, Y.; Stark, G.R. Alternative Activation of Stat1 and Stat3 in Response to Interferon-Gamma. J. Biol. Chem. 2004, 279, 41679–41685. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, Y.; Nishibori, T.; Su, L.; Arduini, R.M.; Baker, D.P.; David, M. Cutting Edge: Role of Stat1, Stat3, and Stat5 in Ifn-Alpha Beta Responses in T Lymphocytes. J. Immunol. 2005, 174, 609–613. [Google Scholar] [CrossRef]
- Butturini, E.; de Prati, A.C.; Mariotto, S. Redox Regulation of Stat1 and Stat3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, K.; Nogawa, S.; Dembo, T.; Kosakai, A.; Fukuuchi, Y. Phosphorylation of Signal Transducer and Activator of Transcription-3 Stat3 after Focal Cerebral Ischemia in Rats. Exp. Neurol. 2001, 170, 63–71. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G. Extracellular Matrix Regulation of Inflammation in the Healthy and Injured Spinal Cord. Exp. Neurol. 2014, 258, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, A.G.; Iruela-Arispe, M.L. Extracellular Matrix, Inflammation, and the Angiogenic Response. Cardiovasc. Res 2010, 86, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Milner, R.; Campbell, I.L. The Extracellular Matrix and Cytokines Regulate Microglial Integrin Expression and Activation. J. Immunol. 2003, 170, 3850–3858. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, S.; Yong, V.W. The Extracellular Matrix as Modifier of Neuroinflammation and Remyelination in Multiple Sclerosis. Brain 2021, 144, 1958–1973. [Google Scholar] [CrossRef]
- Jang, D.G.; Sim, H.J.; Song, E.K.; Kwon, T.; Park, T.J. Extracellular Matrixes and Neuroinflammation. BMB Rep. 2020, 53, 491–499. [Google Scholar] [CrossRef]
- Ulbrich, P.; Khoshneviszadeh, M.; Jandke, S.; Schreiber, S.; Dityatev, A. Interplay between Perivascular and Perineuronal Extracellular Matrix Remodelling in Neurological and Psychiatric Diseases. Eur. J. Neurosci. 2021, 53, 3811–3830. [Google Scholar] [CrossRef]
- Hanada, T.; Yoshimura, A. Regulation of Cytokine Signaling and Inflammation. Cytokine Growth Factor Rev. 2002, 13, 413–421. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible New Role for Nf-Kappab in the Resolution of Inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef]
- Rex, J.; Lutz, A.; Faletti, L.E.; Albrecht, U.; Thomas, M.; Bode, J.G.; Borner, C.; Sawodny, O.; Merfort, I. Il-1beta and Tnfalpha Differentially Influence Nf-Kappab Activity and Fasl-Induced Apoptosis in Primary Murine Hepatocytes During Lps-Induced Inflammation. Front. Physiol. 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jridi, I.; Cante-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2020, 8, 615131. [Google Scholar] [CrossRef] [PubMed]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Lohman, A.W.; Billaud, M.; Johnstone, S.R.; Dwyer, S.T.; Lee, M.Y.; Bortz, P.S.; Best, A.K.; Columbus, L.; Gaston, B.; et al. Endothelial Cell Expression of Haemoglobin Alpha Regulates Nitric Oxide Signalling. Nature 2012, 491, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangwung, P.; Zhou, G.; Lu, Y.; Liao, X.; Wang, B.; Mutchler, S.M.; Miller, M.; Chance, M.R.; Straub, A.C.; Jain, M.K. Regulation of Endothelial Hemoglobin Alpha Expression by Kruppel-Like Factors. Vasc. Med. 2017, 22, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity 2019, 50, 317–333.e6. [Google Scholar] [CrossRef] [Green Version]
- Fahey, E.; Doyle, S.L. Il-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [Green Version]
- Hauptmann, J.; Johann, L.; Marini, F.; Kitic, M.; Colombo, E.; Mufazalov, I.A.; Krueger, M.; Karram, K.; Moos, S.; Wanke, F.; et al. Interleukin-1 Promotes Autoimmune Neuroinflammation by Suppressing Endothelial Heme Oxygenase-1 at the Blood-Brain Barrier. Acta Neuropathol. 2020, 140, 549–567. [Google Scholar] [CrossRef]
- Chen, J.X.; Chen, Y.; DeBusk, L.; Lin, W.; Lin, P.C. Dual Functional Roles of Tie-2/Angiopoietin in Tnf-Alpha-Mediated Angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H187–H195. [Google Scholar] [CrossRef]
- Sainson, R.C.; Johnston, D.A.; Chu, H.C.; Holderfield, M.T.; Nakatsu, M.N.; Crampton, S.P.; Davis, J.; Conn, E.; Hughes, C.C. Tnf Primes Endothelial Cells for Angiogenic Sprouting by Inducing a Tip Cell Phenotype. Blood 2008, 111, 4997–5007. [Google Scholar] [CrossRef]
- Stahl, J.L.; Cook, E.B.; Graziano, F.M.; Barney, N.P. Differential and Cooperative Effects of Tnfα, Il-1β, and Ifnγ on Human Conjunctival Epithelial Cell Receptor Expression and Chemokine Release. Investig. Opthalmology Vis. Sci. 2003, 44, 2010–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Carroll, S.J.; Kho, D.T.; Wiltshire, R.; Nelson, V.; Rotimi, O.; Johnson, R.; Angel, C.E.; Graham, E.S. Pro-Inflammatory Tnfalpha and Il-1beta Differentially Regulate the Inflammatory Phenotype of Brain Microvascular Endothelial Cells. J. Neuroinflamm. 2015, 12, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.A.; Cunningham, C. A Systematic Analysis of the Peripheral and Cns Effects of Systemic Lps, Il-1beta, [Corrected] Tnf-Alpha and Il-6 Challenges in C57bl/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Ferrari, D.; Wesselborg, S.; Bauer, M.K.; Schulze-Osthoff, K. Extracellular Atp Activates Transcription Factor Nf-Kappab through the P2z Purinoreceptor by Selectively Targeting Nf-Kappab P65. J. Cell Biol. 1997, 139, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of Reactive Oxygen Species and Nf-Kappab Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Quan, N. Microglia and Cns Interleukin-1: Beyond Immunological Concepts. Front. Neurol. 2018, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Frater-Schroder, M.; Risau, W.; Hallmann, R.; Gautschi, P.; Bohlen, P. Tumor Necrosis Factor Type Alpha, a Potent Inhibitor of Endothelial Cell Growth in Vitro, Is Angiogenic in Vivo. Proc. Natl. Acad. Sci. USA 1987, 84, 5277–5281. [Google Scholar] [CrossRef] [Green Version]
- Mohr, T.; Haudek-Prinz, V.; Slany, A.; Grillari, J.; Micksche, M.; Gerner, C. Proteome Profiling in Il-1beta and Vegf-Activated Human Umbilical Vein Endothelial Cells Delineates the Interlink between Inflammation and Angiogenesis. PLoS ONE 2017, 12, e0179065. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells as Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune Crosstalk in the Central Nervous System and Its Significance for Neurological Diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in Cns Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia Crosstalk in Neuroinflammatory Diseases. Front. Cell Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef]
- Li, J.; Ramenaden, E.R.; Peng, J.; Koito, H.; Volpe, J.J.; Rosenberg, P.A. Tumor Necrosis Factor Alpha Mediates Lipopolysaccharide-Induced Microglial Toxicity to Developing Oligodendrocytes When Astrocytes Are Present. J. Neurosci. 2008, 28, 5321–5330. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, C.; Weih, F.; Haenold, R. Role of Nuclear Factor Kappa B in Central Nervous System Regeneration. Neural Regen. Res. 2014, 9, 707–711. [Google Scholar]
- Mincheva-Tasheva, S.; Soler, R.M. Nf-Kappab Signaling Pathways: Role in Nervous System Physiology and Pathology. Neuroscientist 2013, 19, 175–194. [Google Scholar] [CrossRef] [Green Version]
- Ramaglia, V.; Rojas, O.; Naouar, I.; Gommerman, J.L. The Ins and Outs of Central Nervous System Inflammation-Lessons Learned from Multiple Sclerosis. Annu. Rev. Immunol. 2021, 39, 199–226. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The Relation between Inflammation and Neurodegeneration in Multiple Sclerosis Brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Takahashi, N.; Matsui, N.; Tetsuka, T.; Okamoto, T. The Nf-Kappa B Activation in Lymphotoxin Beta Receptor Signaling Depends on the Phosphorylation of P65 at Serine 536. J. Biol. Chem. 2003, 278, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bista, P.; Zeng, W.; Ryan, S.; Bailly, V.; Browning, J.L.; Lukashev, M.E. Traf3 Controls Activation of the Canonical and Alternative Nfkappab by the Lymphotoxin Beta Receptor. J. Biol. Chem. 2010, 285, 12971–12978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, P.D.; VanArsdale, T.L.; Walter, B.N.; Ware, C.F.; Hession, C.; Ehrenfels, B.; Browning, J.L.; Din, W.S.; Goodwin, R.G.; Smith, C.A. A Lymphotoxin-Beta-Specific Receptor. Science 1994, 264, 707–710. [Google Scholar] [CrossRef]
- Wesche, H.; Henzel, W.J.; Shillinglaw, W.; Li, S.; Cao, Z. Myd88: An Adapter That Recruits Irak to the Il-1 Receptor Complex. Immunity 1997, 7, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Chen, S.; Huang, H.; Peng, T.; Lan, M.; Yang, X.; Dong, M.; Chen, S.; Xu, A.; Huang, S. Functional Variation of Il-1r-Associated Kinases in the Conserved Myd88-Traf6 Pathway During Evolution. J. Immunol. 2020, 204, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Muroi, M.; Tanamoto, K. Traf6 Distinctively Mediates Myd88- and Irak-1-Induced Activation of Nf-Kappab. J. Leukoc. Biol. 2008, 83, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Ni, J.; Feng, P.; Dixit, V.M. Irak Pelle Family Member Irak-2 and Myd88 as Proximal Mediators of Il-1 Signaling. Science 1997, 278, 1612–1615. [Google Scholar] [CrossRef]
- Zheng, C.; Chen, J.; Chu, F.; Zhu, J.; Jin, T. Inflammatory Role of Tlr-Myd88 Signaling in Multiple Sclerosis. Front. Mol. Neurosci. 2019, 12, 314. [Google Scholar] [CrossRef]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical Assembly in the Myd88-Irak4-Irak2 Complex in Tlr/Il-1r Signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by Ikkalpha of a Second, Evolutionary Conserved, Nf-Kappa B Signaling Pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kishimoto, T.; Minamoto, S. Nf-Kappab Activation in Cd27 Signaling: Involvement of Tnf Receptor-Associated Factors in Its Signaling and Identification of Functional Region of Cd27. J. Immunol. 1998, 161, 4753–4759. [Google Scholar] [PubMed]
- Arch, R.H.; Thompson, C.B. 4-1bb and Ox40 Are Members of a Tumor Necrosis Factor Tnf-Nerve Growth Factor Receptor Subfamily That Bind Tnf Receptor-Associated Factors and Activate Nuclear Factor Kappab. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, I.K.; Lee, Z.H.; Kim, Y.J.; Kim, S.H.; Kwon, B.S. Human 4-1bb Cd137 Signals Are Mediated by Traf2 and Activate Nuclear Factor-Kappa B. Biochem. Biophys. Res. Commun. 1998, 242, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-Canonical Nf-Kappab Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsters, S.A.; Ayres, T.M.; Skubatch, M.; Gray, C.L.; Rothe, M.; Ashkenazi, A. Herpesvirus Entry Mediator, a Member of the Tumor Necrosis Factor Receptor Tnfr Family, Interacts with Members of the Tnfr-Associated Factor Family and Activates the Transcription Factors Nf-Kappab and Ap-1. J. Biol. Chem. 1997, 272, 14029–14032. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.; Solovyev, I.; Colombero, A.; Elliott, R.; Kelley, M.; Boyle, W.J. Atar, a Novel Tumor Necrosis Factor Receptor Family Member, Signals through Traf2 and Traf5. J. Biol. Chem. 1997, 272, 13471–13474. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, G.; Riccardi, C. Gitr: A Multifaceted Regulator of Immunity Belonging to the Tumor Necrosis Factor Receptor Superfamily. Eur. J. Immunol. 2005, 35, 1016–1022. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Donahue, C.J.; Pitti, R.M.; Gray, C.L.; Goddard, A.D.; Bauer, K.D.; Ashkenazi, A. Apo-3, a New Member of the Tumor Necrosis Factor Receptor Family, Contains a Death Domain and Activates Apoptosis and Nf-Κb. Curr. Biol. 1996, 6, 1669–1676. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Eby, M.T.; Sinha, S.; Jasmin, A.; Chaudhary, P.M. The Ectodermal Dysplasia Receptor Activates the Nuclear Factor-Kappab, Jnk, and Cell Death Pathways and Binds to Ectodysplasin A. J. Biol. Chem. 2001, 276, 2668–2677. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Canning, P.; Ruan, Q.; Schwerd, T.; Hrdinka, M.; Maki, J.L.; Saleh, D.; Suebsuwong, C.; Ray, S.; Brennan, P.E.; Cuny, G.D.; et al. Inflammatory Signaling by Nod-Ripk2 Is Inhibited by Clinically Relevant Type Ii Kinase Inhibitors. Chem. Biol. 2015, 22, 1174–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T.; Rennert, P. Nf-Kappab Activation by Protein Kinase C Isoforms and B-Cell Function. EMBO Rep. 2003, 4, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiai, M.; Kurosaki, M.; Pappu, R.; Okawa, K.; Ronko, I.; Fu, C.; Shibata, M.; Iwamatsu, A.; Chan, A.C.; Kurosaki, T. Blnk Required for Coupling Syk to Plcγ2 and Rac1-Jnk in B Cells. Immunity 1999, 10, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.K.; Hu, J.; Meng, Q.; Xia, Y.; Shapiro, P.S.; Reddy, S.P.; Platanias, L.C.; Lindner, D.J.; Johnson, P.F.; Pritchard, C.; et al. Mekk1 Plays a Critical Role in Activating the Transcription Factor C/Ebp-Beta-Dependent Gene Expression in Response to Ifn-Gamma. Proc. Natl. Acad. Sci. USA 2002, 99, 7945–7950. [Google Scholar] [CrossRef] [Green Version]
- Scapini, P.; Pereira, S.; Zhang, H.; Lowell, C.A. Multiple Roles of Lyn Kinase in Myeloid Cell Signaling and Function. Immunol. Rev. 2009, 228, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Didonato, J.A.; Karin, M.; McKeithan, T.W. Bcl3 Encodes a Nuclear Protein Which Can Alter the Subcellular Location of Nf-Kappa B Proteins. Mol. Cell. Biol. 1994, 14, 3915–3926. [Google Scholar]
- Wang, V.Y.; Li, Y.; Kim, D.; Zhong, X.; Du, Q.; Ghassemian, M.; Ghosh, G. Bcl3 Phosphorylation by Akt, Erk2, and Ikk Is Required for Its Transcriptional Activity. Mol. Cell 2017, 67, 484–497.e5. [Google Scholar] [CrossRef]
- Dimitrakopoulos, F.D.; Antonacopoulou, A.G.; Kottorou, A.E.; Panagopoulos, N.; Kalofonou, F.; Sampsonas, F.; Scopa, C.; Kalofonou, M.; Koutras, A.; Makatsoris, T.; et al. Expression of Intracellular Components of the Nf-Kappab Alternative Pathway Nf-Kappab2, Relb, Nik and Bcl3 Is Associated with Clinical Outcome of Nsclc Patients. Sci. Rep. 2019, 9, 14299. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; O’Mahony, A.; Mu, Y.; Geleziunas, R.; Greene, W.C. Protein Kinase C-Theta Participates in Nf-Kappab Activation Induced by Cd3-Cd28 Costimulation through Selective Activation of Ikappab Kinase Beta. Mol. Cell. Biol. 2000, 20, 2933–2940. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Arendt, C.W.; Ellmeier, W.; Schaeffer, E.M.; Sunshine, M.J.; Gandhi, L.; Annes, J.; Petrzilka, D.; Kupfer, A.; Schwartzberg, P.L.; et al. Pkc-Theta Is Required for Tcr-Induced Nf-Kappab Activation in Mature but Not Immature T Lymphocytes. Nature 2000, 404, 402–407. [Google Scholar] [CrossRef]
- Ghaffari-Tabrizi, N.; Bauer, B.; Villunger, A.; Baier-Bitterlich, G.; Altman, A.; Utermann, G.; Überall, F.; Baier, G. Protein Kinase Cθ, a Selective Upstream Regulator of Jnk/Sapk and Il-2 Promoter Activation in Jurkat T Cells. Eur. J. Immunol. 1999, 29, 132–142. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Shen, R.R.; Kim, E.; Lock, Y.J.; Xu, M.; Chen, Z.J.; Hahn, W.C. Ikkepsilon-Mediated Tumorigenesis Requires K63-Linked Polyubiquitination by a Ciap1/Ciap2/Traf2 E3 Ubiquitin Ligase Complex. Cell Rep. 2013, 3, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, M.J.; Lippens, S.; Staes, A.; Gilbert, B.; Roelandt, R.; de Medts, J.; Gevaert, K.; Declercq, W.; Vandenabeele, P. Ciap1/2 Are Direct E3 Ligases Conjugating Diverse Types of Ubiquitin Chains to Receptor Interacting Proteins Kinases 1 to 4 Rip1-4. PLoS ONE 2011, 6, e22356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Differential Interactions of Rel-Nf-Kappa B Complexes with I Kappa B Alpha Determine Pools of Constitutive and Inducible Nf-Kappa B Activity. EMBO J. 1994, 13, 4608–4616. [Google Scholar] [CrossRef]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 Proteins P105 and P100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Duan, T.; Du, Y.; Jin, S.; Wang, M.; Cui, J.; Wang, R.F. Lrrc25 Functions as an Inhibitor of Nf-Kappab Signaling Pathway by Promoting P65/Rela for Autophagic Degradation. Sci. Rep. 2017, 7, 13448. [Google Scholar] [CrossRef] [Green Version]
- Kogan, M.; Haine, V.; Ke, Y.; Wigdahl, B.; Fischer-Smith, T.; Rappaport, J. Macrophage Colony Stimulating Factor Regulation by Nuclear Factor Kappa B: A Relevant Pathway in Human Immunodeficiency Virus Type 1 Infected Macrophages. DNA Cell Biol. 2012, 31, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Whelan, J.; Ghersa, P.; van Huijsduijnen, R.H.; Gray, J.; Chandra, G.; Talabot, F.; DeLamarter, J.F. An Nf Kappa B-Like Factor Is Essential but Not Sufficient for Cytokine Induction of Endothelial Leukocyte Adhesion Molecule 1 Elam-1 Gene Transcription. Nucleic Acids Res. 1991, 19, 2645–2653. [Google Scholar] [CrossRef] [Green Version]
- Schindler, U.; Baichwal, V.R. Three Nf-Kappa B Binding Sites in the Human E-Selectin Gene Required for Maximal Tumor Necrosis Factor Alpha-Induced Expression. Mol. Cell. Biol. 1994, 14, 5820–5831. [Google Scholar]
- Libermann, T.A.; Baltimore, D. Activation of Interleukin-6 Gene Expression through the Nf-Kappa B Transcription Factor. Mol Cell. Biol. 1990, 10, 2327–2334. [Google Scholar]
- Son, Y.H.; Jeong, Y.T.; Lee, K.A.; Choi, K.H.; Kim, S.M.; Rhim, B.Y.; Kim, K. Roles of Mapk and Nf-Kappab in Interleukin-6 Induction by Lipopolysaccharide in Vascular Smooth Muscle Cells. J. Cardiovasc. Pharmacol. 2008, 51, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Kao, D.; Blom, H.J.; de Almeida, I.T.; Castro, R.; Loscalzo, J.; Handy, D.E. S-Adenosylhomocysteine Induces Inflammation through Nfkb: A Possible Role for Ezh2 in Endothelial Cell Activation. Biochim. Biophys. Acta 2016, 1862, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astarci, E.; Sade, A.; Cimen, I.; Savas, B.; Banerjee, S. The Nf-Kappab Target Genes Icam-1 and Vcam-1 Are Differentially Regulated During Spontaneous Differentiation of Caco-2 Cells. FEBS J. 2012, 279, 2966–2986. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Linker, R.A.; Deng, J.; Kaltschmidt, C. Cyclooxygenase-2 Is a Neuronal Target Gene of Nf-Kappab. BMC Mol. Biol. 2002, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Matsuoka, A.; Kizuka, F.; Lee, L.; Tamura, I.; Maekawa, R.; Asada, H.; Taketani, T.; Tamura, H.; Sugino, N. Prostaglandin F2alpha Pgf2alpha Stimulates Ptgs2 Expression and Pgf2alpha Synthesis through Nfkb Activation Via Reactive Oxygen Species in the Corpus Luteum of Pseudopregnant Rats. Reproduction 2010, 140, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Kiriakidis, S.; Andreakos, E.; Monaco, C.; Foxwell, B.; Feldmann, M.; Paleolog, E. Vegf Expression in Human Macrophages Is Nf-Kappab-Dependent: Studies Using Adenoviruses Expressing the Endogenous Nf-Kappab Inhibitor Ikappabalpha and a Kinase-Defective Form of the Ikappab Kinase 2. J. Cell Sci. 2003, 116, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. Nf-Κb and Stat1 Control Cxcl1 and Cxcl2 Gene Transcription. Am. J. Physiol-Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [Green Version]
- Bitko, V.; Velazquez, A.; Yang, L.; Yang, Y.C.; Barik, S. Transcriptional Induction of Multiple Cytokines by Human Respiratory Syncytial Virus Requires Activation of Nf-Kappa B and Is Inhibited by Sodium Salicylate and Aspirin. Virology 1997, 232, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Kunsch, C.; Rosen, C.A. Nf-Kappa B Subunit-Specific Regulation of the Interleukin-8 Promoter. Mol. Cell. Biol. 1993, 13, 6137–6146. [Google Scholar]
- Kang, H.B.; Kim, Y.E.; Kwon, H.J.; Sok, D.E.; Lee, Y. Enhancement of Nf-Kappab Expression and Activity Upon Differentiation of Human Embryonic Stem Cell Line Snuhes3. Stem Cells Dev. 2007, 16, 615–623. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a Functional Nf-Kappa B Site in the Human Interleukin 1 Beta Promoter: Evidence for a Positive Autoregulatory Loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [PubMed] [Green Version]
- Mori, N.; Prager, D. Transactivation of the Interleukin-1alpha Promoter by Human T-Cell Leukemia Virus Type I and Type Ii Tax Proteins. Blood 1996, 87, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Poleganov, M.A.; Bachmann, M.; Pfeilschifter, J.; Muhl, H. Genome-Wide Analysis Displays Marked Induction of Ebi3/Il-27b in Il-18-Activated Aml-Derived Kg1 Cells: Critical Role of Two Kappab Binding Sites in the Human Ebi3 Promotor. Mol. Immunol. 2008, 45, 2869–2880. [Google Scholar] [CrossRef] [PubMed]
- Pietila, T.E.; Veckman, V.; Lehtonen, A.; Lin, R.; Hiscott, J.; Julkunen, I. Multiple Nf-Kappab and Ifn Regulatory Factor Family Transcription Factors Regulate Ccl19 Gene Expression in Human Monocyte-Derived Dendritic Cells. J. Immunol. 2007, 178, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Lee, J.Y.; Son, J.W.; Oh, J.H.; Shin, D.M.; Yuk, J.M.; Song, C.H.; Paik, T.H.; Jo, E.K. Expression and Regulation of the Cc-Chemokine Ligand 20 During Human Tuberculosis. Scand. J. Immunol. 2008, 67, 77–85. [Google Scholar] [CrossRef]
- Battaglia, F.; Delfino, S.; Merello, E.; Puppo, M.; Piva, R.; Varesio, L.; Bosco, M.C. Hypoxia Transcriptionally Induces Macrophage-Inflammatory Protein-3alpha/Ccl-20 in Primary Human Mononuclear Phagocytes through Nuclear Factor Nf-Kappab. J. Leukoc. Biol. 2008, 83, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Shakhov, A.N.; Collart, M.A.; Vassalli, P.; Nedospasov, S.A.; Jongeneel, C.V. Kappa B-Type Enhancers Are Involved in Lipopolysaccharide-Mediated Transcriptional Activation of the Tumor Necrosis Factor Alpha Gene in Primary Macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef]
- Collart, M.A.; Baeuerle, P.; Vassalli, P. Regulation of Tumor Necrosis Factor Alpha Transcription in Macrophages: Involvement of Four Kappa B-Like Motifs and of Constitutive and Inducible Forms of Nf-Kappa B. Mol. Cell. Biol. 1990, 10, 1498–1506. [Google Scholar]
- Kim, J.-O.; Kim, H.W.; Baek, K.-M.; Kang, C.-Y. Nf-Κb and Ap-1 Regulate Activation-Dependent Cd137 4-1bb Expression in T Cells. FEBS Lett. 2003, 541, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Q.; Xing, L.; Zhang, J.H.; Zhao, M.; Horn, D.; Chan, J.; Boyce, B.F.; Harris, S.E.; Mundy, G.R.; Chen, D. Nf-Kappab Specifically Activates Bmp-2 Gene Expression in Growth Plate Chondrocytes in Vivo and in a Chondrocyte Cell Line in Vitro. J. Biol. Chem. 2003, 278, 29130–29135. [Google Scholar] [CrossRef] [Green Version]
- Fukui, N.; Ikeda, Y.; Ohnuki, T.; Hikita, A.; Tanaka, S.; Yamane, S.; Suzuki, R.; Sandell, L.J.; Ochi, T. Pro-Inflammatory Cytokine Tumor Necrosis Factor-Alpha Induces Bone Morphogenetic Protein-2 in Chondrocytes Via Mrna Stabilization and Transcriptional up-Regulation. J. Biol. Chem. 2006, 281, 27229–27241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.Y.; Wu, S.D.; Tsai, M.H.; Chuang, E.Y.; Chuang, L.L.; Hsu, L.C.; Lai, L.C. Transcription of Tnfaip3 Is Regulated by Nf-Kappab and P38 Via C/Ebpbeta in Activated Macrophages. PLoS ONE 2013, 8, e73153. [Google Scholar]
- Lu, R.; Moore, P.A.; Pitha, P.M. Stimulation of Irf-7 Gene Expression by Tumor Necrosis Factor Alpha: Requirement for Nfkappa B Transcription Factor and Gene Accessibility. J. Biol. Chem. 2002, 277, 16592–16598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the Relb Gene Is Regulated by Nf-Kappab. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [Green Version]
- Sanchavanakit, N.; Saengtong, W.; Manokawinchoke, J.; Pavasant, P. Tnf-Alpha Stimulates Mmp-3 Production Via Pge2 Signalling through the Nf-Kb and P38 Mapk Pathway in a Murine Cementoblast Cell Line. Arch. Oral Biol. 2015, 60, 1066–1074. [Google Scholar] [CrossRef]
- Bond, M. Inhibition of Transcription Factor Nf-Κb Reduces Matrix Metalloproteinase-1, -3 and -9 Production by Vascular Smooth Muscle Cells. Cardiovasc. Res. 2001, 50, 556–565. [Google Scholar] [CrossRef]
- Pach, E.; Kumper, M.; Fromme, J.E.; Zamek, J.; Metzen, F.; Koch, M.; Mauch, C.; Zigrino, P. Extracellular Matrix Remodeling by Fibroblast-Mmp14 Regulates Melanoma Growth. Int. J. Mol. Sci. 2021, 22, 12276. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, G.; Li, X.; Qiu, X.; Ouyang, J.; Dai, J.; Min, S. Matrix Metalloproteinase 3: A Promoting and Destabilizing Factor in the Pathogenesis of Disease and Cell Differentiation. Front. Physiol. 2021, 12, 663978. [Google Scholar] [CrossRef]
- Lee, E.J.; Moon, P.G.; Baek, M.C.; Kim, H.S. Comparison of the Effects of Matrix Metalloproteinase Inhibitors on Tnf-Alpha Release from Activated Microglia and Tnf-Alpha Converting Enzyme Activity. Biomol. Ther. 2014, 22, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.E.; Allen, K.A.P.; Salazar, R.; Maxwell, S.A. Matrix Metalloproteinase-1 and -9 Activation by Plasmin Regulates a Novel Endothelial Cell-Mediated Mechanism of Collagen Gel Contraction and Capillary Tube Regression in Three-Dimensional Collagen Matrices. J. Cell Sci. 2001, 114, 917–930. [Google Scholar] [CrossRef]
- Hahn-Dantona, E.; Ramos-DeSimone, N.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of Prommp-9 by a Plasmin/Mmp-3 Cascade in a Tumor Cell Model. Regulation by Tissue Inhibitors of Metalloproteinases. Ann. N. Y. Acad Sci. 1999, 878, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; Lin, J.X. Cytokine Receptor Signaling Pathways. J. Allergy Clin. Immunol. 2000, 105, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meng, Y.H.; Shang, W.Q.; Liu, L.B.; Chen, X.; Yuan, M.M.; Jin, L.P.; Li, M.Q.; Li, D.J. Chemokine Ccl24 Promotes the Growth and Invasiveness of Trophoblasts through Erk1/2 and Pi3k Signaling Pathways in Human Early Pregnancy. Reproduction 2015, 150, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Ivashkiv, L.B. Costimulation of Chemokine Receptor Signaling by Matrix Metalloproteinase-9 Mediates Enhanced Migration of Ifn-Alpha Dendritic Cells. J. Immunol. 2006, 176, 6022–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the Ccl2/Ccr2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.; Meitei, H.T.; Sonar, S.A.; Sharma, P.K.; Mujeeb, V.R.; Srivastava, S.; Boppana, R.; Lal, G. Ccr6 Signaling Inhibits Suppressor Function of Induced-Treg During Gut Inflammation. J. Autoimmun. 2018, 88, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Sun, S.M.; Lu, X.F.; Chen, P.Y.; Chen, C.F.; Liang, W.Q.; Peng, C.Y. Ccr10 Activation Stimulates the Invasion and Migration of Breast Cancer Cells through the Erk1/2/Mmp-7 Signaling Pathway. Int. Immunopharmacol. 2017, 51, 124–130. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, J.L.; Criado-García, O. The Chemokine Receptor Ccr7 Uses Distinct Signaling Modules with Biased Functionality to Regulate Dendritic Cells. Front. Immunol. 2020, 1, 528. [Google Scholar] [CrossRef]
- Prado, G.N.; Suetomi, K.; Shumate, D.; Maxwell, C.; Ravindran, A.; Rajarathnam, K.; Navarro, J. Chemokine Signaling Specificity: Essential Role for the N-Terminal Domain of Chemokine Receptors. Biochemistry 2007, 46, 8961–8968. [Google Scholar] [CrossRef] [Green Version]
- Legler, D.F.; Thelen, M. New Insights in Chemokine Signaling. F1000Research 2018, 7, 95. [Google Scholar] [CrossRef]
- Lin, L.; Han, M.M.; Wang, F.; Xu, L.L.; Yu, H.X.; Yang, P.Y. Cxcr7 Stimulates Mapk Signaling to Regulate Hepatocellular Carcinoma Progression. Cell Death Dis. 2014, 5, e1488. [Google Scholar] [CrossRef] [PubMed]
- Pozzobon, T.; Goldoni, G.; Viola, A.; Molon, B. Cxcr4 Signaling in Health and Disease. Immunol. Lett. 2016, 177, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Finn, A. Chemokine Receptors and Their Role in Inflammation and Infectious Diseases. Blood 2000, 95, 3032–3043. [Google Scholar] [CrossRef] [PubMed]
- Kuropatwinski, K.K.; de Imus, C.; Gearing, D.; Baumann, H.; Mosley, B. Influence of Subunit Combinations on Signaling by Receptors for Oncostatin M, Leukemia Inhibitory Factor, and Interleukin-6. J. Biol. Chem. 1997, 272, 15135–15144. [Google Scholar] [CrossRef] [Green Version]
- Kitanaka, N.; Nakano, R.; Sugiura, K.; Kitanaka, T.; Namba, S.; Konno, T.; Nakayama, T.; Sugiya, H. Interleukin-1beta Promotes Interleulin-6 Expression Via Erk1/2 Signaling Pathway in Canine Dermal Fibroblasts. PLoS ONE 2019, 14, e0220262. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the Il-6/Jak/Stat3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional Repression of Socs3 Mediated by Il-6/Stat3 Signaling Via Dnmt1 Promotes Pancreatic Cancer Growth and Metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 27. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. Il1-Induced Jak/Stat Signaling Is Antagonized by Tgfbeta to Shape Caf Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Whitley, S.K.; Balasubramani, A.; Zindl, C.L.; Sen, R.; Shibata, Y.; Crawford, G.E.; Weathington, N.M.; Hatton, R.D.; Weaver, C.T. Il-1r Signaling Promotes Stat3 and Nf-Kappab Factor Recruitment to Distal Cis-Regulatory Elements That Regulate Il17a/F Transcription. J. Biol. Chem. 2018, 293, 15790–15800. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Leung, D.Y.; Rafaels, N.M.; Hand, T.; Boguniewicz, M.; Hata, T.R.; Schneider, L.; Hanifin, J.M.; Gallo, R.L.; Gao, L. Genetic Variants in Tslp and Its Receptor, Il7r, Contribute to an Increased Risk for Atopic Dermatitis and Eczema Herpeticum in Two American Populations. J. Allergy Clin. Immunol. 2009, 123, S70. [Google Scholar] [CrossRef]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. Il-27, a Heterodimeric Cytokine Composed of Ebi3 and P28 Protein, Induces Proliferation of Naive Cd4+ T Cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Ghilardi, N.; Li, J.; de Sauvage, F.J. Il-27 Regulates Il-12 Responsiveness of Naive Cd4+ T Cells through Stat1-Dependent and -Independent Mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 15047–15052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibbert, L.; Pflanz, S.; Malefyt, R.d.; Kastelein, R.A. Il-27 and Ifn-Alpha Signal Via Stat1 and Stat3 and Induce T-Bet and Il-12rbeta2 in Naive T Cells. J. Interferon Cytokine Res. 2003, 23, 513–522. [Google Scholar] [CrossRef]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Dual Roles of Il-27 in Cancer Biology and Immunotherapy. Mediat. Inflamm. 2017, 2017, 3958069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owaki, T.; Asakawa, M.; Kamiya, S.; Takeda, K.; Fukai, F.; Mizuguchi, J.; Yoshimoto, T. Il-27 Suppresses Cd28-Mediated [Correction of Medicated] Il-2 Production through Suppressor of Cytokine Signaling 3. J. Immunol. 2006, 176, 2773–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Sullivan, L.; Caligiuri, M.A. Molecular Pathways: Interleukin-15 Signaling in Health and in Cancer. Clin. Cancer Res. 2014, 20, 2044–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, H.P.; Paunovic, V.; Gadina, M. Signalling, Inflammation and Arthritis: Crossed Signals: The Role of Interleukin-15 and -18 in Autoimmunity. Rheumatol. Oxf. 2008, 47, 1269–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, M.M.; Morrison, D.K. Integrating Signals from Rtks to Erk/Mapk. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Kowanetz, M.; Ferrara, N. Vascular Endothelial Growth Factor Signaling Pathways: Therapeutic Perspective. Clin. Cancer Res. 2006, 12, 5018–5022. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.; Gu, X.; Tsai, N.T.; Venema, R.C.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. Vascular Endothelial Growth Factor Activates Stat Proteins in Aortic Endothelial Cells. J. Biol. Chem. 2000, 275, 33189–33192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. Pdgfrs Are Critical for Pi3k/Akt Activation and Negatively Regulated by Mtor. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharibi, B.; Ghuman, M.S.; Hughes, F.J. Akt- and Erk-Mediated Regulation of Proliferation and Differentiation During Pdgfrbeta-Induced Msc Self-Renewal. J. Cell. Mol. Med. 2012, 16, 2789–2801. [Google Scholar] [CrossRef] [PubMed]
- Dell’Albani, P.; Kahn, M.A.; Cole, R.; Condorelli, D.F.; Giuffrida-Stella, A.M.; de Vellis, J. Oligodendroglial Survival Factors, Pdgf-Aa and Cntf, Activate Similar Jak/Stat Signaling Pathways. J. Neurosci. Res. 1998, 54, 191–205. [Google Scholar] [CrossRef]
- Neeli, I.; Liu, Z.; Dronadula, N.; Ma, Z.A.; Rao, G.N. An Essential Role of the Jak-2/Stat-3/Cytosolic Phospholipase a2 Axis in Platelet-Derived Growth Factor Bb-Induced Vascular Smooth Muscle Cell Motility. J. Biol. Chem. 2004, 279, 46122–46128. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, G.; Li, C.; Li, R.; Lu, J.; He, Z.; Wang, Q.; Peng, Z.; Wang, J.; Dong, Y.; et al. Lyn Mediates Fip1l1-Pdgfra Signal Pathway Facilitating Il-5ra Intracellular Signal through Fip1l1-Pdgfra/Jak2/Lyn/Akt Network Complex in Cel. Oncotarget 2017, 8, 64984–64998. [Google Scholar] [CrossRef]
- Tothova, Z.; Tomc, J.; Debeljak, N.; Solar, P. Stat5 as a Key Protein of Erythropoietin Signalization. Int. J. Mol. Sci. 2021, 22, 7109. [Google Scholar] [CrossRef]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a Cytokine Produced by Activated T Cells, Induces Dermatitis in Mice. Nat. Immunol. 2004, 5, 752–760. [Google Scholar] [CrossRef]
- Hintzen, C.; Evers, C.; Lippok, B.E.; Volkmer, R.; Heinrich, P.C.; Radtke, S.; Hermanns, H.M. Box 2 Region of the Oncostatin M Receptor Determines Specificity for Recruitment of Janus Kinases and Stat5 Activation. J. Biol. Chem. 2008, 283, 19465–19477. [Google Scholar] [CrossRef] [Green Version]
- Viswanadhapalli, S.; Dileep, K.V.; Zhang, K.Y.J.; Nair, H.B.; Vadlamudi, R.K. Targeting Lif/Lifr Signaling in Cancer. Genes Dis. 2021, in press. [CrossRef]
- Suman, P.; Malhotra, S.S.; Gupta, S.K. Lif-Stat Signaling and Trophoblast Biology. JAKSTAT 2013, 2, e25155. [Google Scholar] [CrossRef] [Green Version]
- Malaval, L.; Liu, F.; Vernallis, A.B.; Aubin, J.E. Gp130/Osmr Is the Only Lif/Il-6 Family Receptor Complex to Promote Osteoblast Differentiation of Calvaria Progenitors. J. Cell. Physiol. 2005, 204, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Soman, S.; Patil, Y.B.; Advani, J.; Thomas, J.K.; Desai, D.V.; Kulkarni-Kale, U.; Harsha, H.C.; Prasad, T.S.; Raju, R.; et al. Il-11/Il11ra Receptor Mediated Signaling: A Web Accessible Knowledgebase. Cell Commun. Adhes. 2013, 20, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, J.V.; Kristensen, A.M.; Pallesen, L.T.; Bauer, J.; Vaegter, C.B.; Nielsen, M.S.; Madsen, P.; Petersen, C.M. Cytokine-Like Factor 1, an Essential Facilitator of Cardiotrophin-Like Cytokine:Ciliary Neurotrophic Factor Receptor Alpha Signaling and Sorla-Mediated Turnover. Mol. Cell. Biol. 2016, 36, 1272–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin Inhibits Neuronal Cell Death by Interacting with a Cytokine Receptor Complex or Complexes Involving Cntf Receptor Alpha/Wsx-1/Gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef] [Green Version]
- Lelievre, E.; Plun-Favreau, H.; Chevalier, S.; Froger, J.; Guillet, C.; Elson, G.C.; Gauchat, J.F.; Gascan, H. Signaling Pathways Recruited by the Cardiotrophin-Like Cytokine/Cytokine-Like Factor-1 Composite Cytokine: Specific Requirement of the Membrane-Bound Form of Ciliary Neurotrophic Factor Receptor Alpha Component. J. Biol. Chem. 2001, 276, 22476–22484. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Marquez, C.P.; Sperberg, R.A.P.; Wu, J.; Bae, W.G.; Huang, P.S.; Sweet-Cordero, E.A.; Cochran, J.R. Engineering a Potent Receptor Superagonist or Antagonist from a Novel Il-6 Family Cytokine Ligand. Proc. Natl. Acad. Sci. USA 2020, 117, 14110–14118. [Google Scholar] [CrossRef]
- Sun, Z.J.; Chen, G.; Hu, X.; Zhang, W.; Liu, Y.; Zhu, L.X.; Zhou, Q.; Zhao, Y.F. Activation of Pi3k/Akt/Ikk-Alpha/Nf-Kappab Signaling Pathway Is Required for the Apoptosis-Evasion in Human Salivary Adenoid Cystic Carcinoma: Its Inhibition by Quercetin. Apoptosis 2010, 15, 850–863. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-Dependent Regulation of Nf-{Kappa}B Is Controlled by Mtor and Raptor in Association with Ikk. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A Positive Feedback Loop Involving Egfr/Akt/Mtorc1 and Ikk/Nf-Kb Regulates Head and Neck Squamous Cell Carcinoma Proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. Nf-Kappab Is a Target of Akt in Anti-Apoptotic Pdgf Signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef]
- Legendre, F.; Dudhia, J.; Pujol, J.P.; Bogdanowicz, P. Jak/Stat but Not Erk1/Erk2 Pathway Mediates Interleukin Il-6/Soluble Il-6r Down-Regulation of Type Ii Collagen, Aggrecan Core, and Link Protein Transcription in Articular Chondrocytes. Association with a Down-Regulation of Sox9 Expression. J. Biol. Chem. 2003, 278, 2903–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, S.J.; Doecke, J.D.; Ghazawi, I.; Yang, J.; Griffiths, L.R.; Spring, K.J.; Ralph, S.J.; Mellick, A.S. Novel Stat Binding Elements Mediate Il-6 Regulation of Mmp-1 and Mmp-3. Sci. Rep. 2017, 7, 8526. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small Molecule Inhibitors Targeting Activator Protein 1 Ap-1. J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 Cooperate with C-Jun/C-Fos to Mediate Tgf-Beta-Induced Transcription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef]
- Washio, A.; Kitamura, C.; Morotomi, T.; Terashita, M.; Nishihara, T. Possible Involvement of Smad Signaling Pathways in Induction of Odontoblastic Properties in Kn-3 Cells by Bone Morphogenetic Protein-2: A Growth Factor to Induce Dentin Regeneration. Int. J. Dent. 2012, 2012, 258469. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lee, C.E.; Denard, B.; Sustained, J.Y. Induction of Collagen Synthesis by Tgf-Beta Requires Regulated Intramembrane Proteolysis of Creb3l1. PLoS ONE 2014, 9, e108528. [Google Scholar]
- Greenwood, M.P.; Greenwood, M.; Gillard, B.T.; Devi, R.C.; Murphy, D. Regulation of Camp Responsive Element Binding Protein 3-Like 1 Creb3l1 Expression by Orphan Nuclear Receptor Nr4a1. Front. Mol. Neurosci. 2017, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A Distinct Role of Riplet-Mediated K63-Linked Polyubiquitination of the Rig-I Repressor Domain in Human Antiviral Innate Immune Responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. Trim25 Ring-Finger E3 Ubiquitin Ligase Is Essential for Rig-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Shi, Y.; Yuan, B.; Zhu, W.; Zhang, R.; Li, L.; Hao, X.; Chen, S.; Hou, F. Ube2d3 and Ube2n Are Essential for Rig-I-Mediated Mavs Aggregation in Antiviral Innate Immunity. Nat. Commun. 2017, 8, 15138. [Google Scholar] [CrossRef] [Green Version]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase Riplet in Innate Immunity. Cell 2019, 177, 1187–1200.e16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Thomas, K.; Blanco, J.C.; Salkowski, C.A.; Vogel, S.N. The Role of the Interferon Regulatory Factors, Irf-1 and Irf-2, in Lps-Induced Cyclooxygenase-2 Cox-2 Expression in Vivo and in Vitro. J. Endotoxin Res. 2002, 8, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Oshima, S.; Nakamura, T.; Namiki, S.; Okada, E.; Tsuchiya, K.; Okamoto, R.; Yamazaki, M.; Yokota, T.; Aida, M.; Yamaguchi, Y.; et al. Interferon Regulatory Factor 1 Irf-1 and Irf-2 Distinctively up-Regulate Gene Expression and Production of Interleukin-7 in Human Intestinal Epithelial Cells. Mol. Cell. Biol. 2004, 24, 6298–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Fleming, J.A.; Kim, J.; Spencer, T.E.; Bazer, F.W. Pregnancy and Interferon Tau Regulate Ddx58 and Plscr1 in the Ovine Uterus During the Peri-Implantation Period. Reproduction 2011, 141, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Maratheftis, C.I.; Giannouli, S.; Spachidou, M.P.; Panayotou, G.; Voulgarelis, M. Rna Interference of Interferon Regulatory Factor-1 Gene Expression in Thp-1 Cell Line Leads to Toll-Like Receptor-4 Overexpression/Activation as Well as up-Modulation of Annexin-Ii. Neoplasia 2007, 9, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Pigino, G.; Ishikawa, T. Axonemal Radial Spokes: 3d Structure, Function and Assembly. Bioarchitecture 2012, 2, 50–58. [Google Scholar] [CrossRef]
- Lobry, C.; Lopez, T.; Israel, A.; Weil, R. Negative Feedback Loop in T Cell Activation through Ikappab Kinase-Induced Phosphorylation and Degradation of Bcl10. Proc. Natl. Acad. Sci. USA 2007, 104, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Pelaez, M.; Lamont, D.J.; Peggie, M.; Shpiro, N.; Gray, N.S.; Cohen, P. Protein Kinase Ikkbeta-Catalyzed Phosphorylation of Irf5 at Ser462 Induces Its Dimerization and Nuclear Translocation in Myeloid Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 17432–17437. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Peggie, M.; Plater, L.; Sorcek, R.J.; Young, E.R.; Madwed, J.B.; Hough, J.; McIver, E.G.; Cohen, P. Novel Cross-Talk within the Ikk Family Controls Innate Immunity. Biochem. J. 2011, 434, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Han, K.J.; Su, X.; Xu, L.G.; Bin, L.H.; Zhang, J.; Shu, H.B. Mechanisms of the Trif-Induced Interferon-Stimulated Response Element and Nf-Kappab Activation and Apoptosis Pathways. J. Biol. Chem. 2004, 279, 15652–15661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.K.; Yu, J.; Shimoda, M.; Takada, Y. Integrin Binding to the Trimeric Interface of Cd40l Plays a Critical Role in Cd40/Cd40l Signaling. J. Immunol. 2019, 203, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, S.; Nakano, H.; Ishida, T.; Horie, R.; Nagai, M.; Ito, K.; Yagita, H.; Okumura, K.; Inoue, J.; Watanabe, T. Tumor Necrosis Factor Receptor-Associated Factor Traf 5 and Traf2 Are Involved in Cd30-Mediated Nfkappab Activation. J. Biol. Chem. 1997, 272, 2042–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, C.; Patskovsky, Y.; Yan, Q.; Li, Z.; Ramagopal, U.; Cheng, H.; Brenowitz, M.; Hui, X.; Nathenson, S.G.; Almo, S.C. Decoy Strategies: The Structure of Tl1a:Dcr3 Complex. Structure 2011, 19, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.D.; Kaltschmidt, C.; Kaltschmidt, B.; Offenhauser, N.; Bohm-Matthaei, R.; Baeuerle, P.A.; Barde, Y.A. Selective Activation of Nf-Kappa B by Nerve Growth Factor through the Neurotrophin Receptor P75. Science 1996, 272, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Baeuerle, P.A. Nf-Kappa B as Inducible Transcriptional Activator of the Granulocyte-Macrophage Colony-Stimulating Factor Gene. Mol. Cell. Biol. 1990, 10, 1281–1286. [Google Scholar]
- Azimi, N.; Brown, K.; Bamford, R.N.; Tagaya, Y.; Siebenlist, U.; Waldmann, T.A. Human T Cell Lymphotropic Virus Type I Tax Protein Trans-Activates Interleukin 15 Gene Transcription through an Nf-Kappab Site. Proc. Natl. Acad. Sci. USA 1998, 95, 2452–2457. [Google Scholar] [CrossRef] [Green Version]
- Lenardo, M.J.; Fan, C.-M.; Maniatis, T.; Baltimore, D. The Involvement of Nf-Κb in Β-Interferon Gene Regulation Reveals Its Role as Widely Inducible Mediator of Signal Transduction. Cell 1989, 57, 287–294. [Google Scholar] [CrossRef]
- Hiscott, J.; Alper, D.; Cohen, L.; Leblanc, J.F.; Sportza, L.; Wong, A.; Xanthoudakis, S. Induction of Human Interferon Gene Expression Is Associated with a Nuclear Factor That Interacts with the Nf-Kappa B Site of the Human Immunodeficiency Virus Enhancer. J. Virol. 1989, 63, 2557–2566. [Google Scholar] [CrossRef] [Green Version]
- Bash, J.; Zong, W.X.; Banga, S.; Rivera, A.; Ballard, D.W.; Ron, Y.; Gelinas, C. Rel/Nf-Kappab Can Trigger the Notch Signaling Pathway by Inducing the Expression of Jagged1, a Ligand for Notch Receptors. EMBO J. 1999, 18, 2803–2811. [Google Scholar] [CrossRef] [Green Version]
- Wickremasinghe, M.I.; Thomas, L.H.; O’Kane, C.M.; Uddin, J.; Friedland, J.S. Transcriptional Mechanisms Regulating Alveolar Epithelial Cell-Specific Ccl5 Secretion in Pulmonary Tuberculosis. J. Biol. Chem. 2004, 279, 27199–27210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix Metalloproteinase-9: Many Shades of Function in Cardiovascular Disease. Physiol. 2013, 28, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlet, A.; Kappenstein, M.; Keye, P.; Klasener, K.; Endres, C.; Poggio, T.; Gorantla, S.P.; Kreutmair, S.; Sanger, J.; Illert, A.L.; et al. The Il-3, Il-5, and Gm-Csf Common Receptor Beta Chain Mediates Oncogenic Activity of Flt3-Itd-Positive Aml. Leukemia 2022, 36, 701–711. [Google Scholar] [CrossRef]
- Ratthe, C.; Girard, D. Interleukin-15 Enhances Human Neutrophil Phagocytosis by a Syk-Dependent Mechanism: Importance of the Il-15ralpha Chain. J. Leukoc. Biol. 2004, 76, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.D.; Schwartz, O.M.; Brown, M.R.; Leto, T.L.; Holland, S.M. Characterization of a Dipeptide Motif Regulating Ifn-Gamma Receptor 2 Plasma Membrane Accumulation and Ifn-Gamma Responsiveness. J. Immunol. 2004, 173, 3991–3999. [Google Scholar] [CrossRef] [Green Version]
- Kotenko, S.V.; Izotova, L.S.; Pollack, B.P.; Mariano, T.M.; Donnelly, R.J.; Muthukumaran, G.; Cook, J.R.; Garotta, G.; Silvennoinen, O.; Ihle, J.N.; et al. Interaction between the Components of the Interferon Gamma Receptor Complex. J. Biol. Chem. 1995, 270, 20915–20921. [Google Scholar] [CrossRef] [Green Version]
- Sakatsume, M.; Igarashi, K.; Winestock, K.D.; Garotta, G.; Larner, A.C.; Finbloom, D.S. The Jak Kinases Differentially Associate with the Alpha and Beta Accessory Factor Chains of the Interferon Gamma Receptor to Form a Functional Receptor Unit Capable of Activating Stat Transcription Factors. J. Biol. Chem. 1995, 270, 17528–17534. [Google Scholar] [CrossRef] [Green Version]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An Overview on the Role of Flt3-Tyrosine Kinase Receptor in Acute Myeloid Leukemia: Biology and Treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskas, A. Pdgfs and Their Receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef]
- Kamalakar, A.; McKinney, J.M.; Duron, D.S.; Amanso, A.M.; Ballestas, S.A.; Drissi, H.; Willett, N.J.; Bhattaram, P.; Garcia, A.J.; Wood, L.B.; et al. Jagged1 Stimulates Cranial Neural Crest Cell Osteoblast Commitment Pathways and Bone Regeneration Independent of Canonical Notch Signaling. Bone 2021, 143, 115657. [Google Scholar] [CrossRef]
- Ip, N.Y.; McClain, J.; Barrezueta, N.X.; Aldrich, T.H.; Pan, L.; Li, Y.; Wiegand, S.J.; Friedman, B.; Davis, S.; Yancopoulos, G.D. The A Component of the Cntf Receptor Is Required for Signaling and Defines Potential Cntf Targets in the Adult and During Development. Neuron 1993, 10, 89–102. [Google Scholar] [CrossRef]
- Kim, J.W.; Marquez, C.P.; Kostyrko, K.; Koehne, A.L.; Marini, K.; Simpson, D.R.; Lee, A.G.; Leung, S.G.; Sayles, L.C.; Shrager, J.; et al. Antitumor Activity of an Engineered Decoy Receptor Targeting Clcf1-Cntfr Signaling in Lung Adenocarcinoma. Nat. Med. 2019, 25, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, R.; Chen, J.; Liu, L.; Cui, X.; Shen, S.; Cui, G.; Ren, Z.; Yu, Z. Crosstalk Mechanisms between Hgf/C-Met Axis and Ncrnas in Malignancy. Front. Cell Dev. Biol. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Derijard, B.; Davis, R.J. Mkk3- and Mkk6-Regulated Gene Expression Is Mediated by the P38 Mitogen-Activated Protein Kinase Signal Transduction Pathway. Mol. Cell. Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieser, R.; Wrana, J.L.; Massagué, J. Gs Domain Mutations That Constitutively Activate T Beta R-I, the Downstream Signaling Component in the Tgf-Beta Receptor Complex. EMBO J. 1995, 1410, 2199–2208. [Google Scholar] [CrossRef]
- Macias-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific Activation of Smad1 Signaling Pathways by the Bmp7 Type I Receptor, Alk2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting Edge: A Novel Toll/Il-1 Receptor Domain-Containing Adapter That Preferentially Activates the Ifn-Beta Promoter in the Toll-Like Receptor Signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. Ticam-1, an Adaptor Molecule That Participates in Toll-Like Receptor 3-Mediated Interferon-Beta Induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-Fadd Death Domain Complex Structure Unravels Signalling by Receptor Clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Chandler, J.M.; Cohen, G.M.; MacFarlane, M. Different Subcellular Distribution of Caspase-3 and Caspase-7 Following Fas-Induced Apoptosis in Mouse Liver. J. Biol. Chem. 1998, 273, 10815–10818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Pappalardo, Z.; Chopra, D.G.; Hennings, T.G.; Vaughn, I.; Lan, C.; Choe, J.J.; Ang, K.; Chen, S.; Arkin, M.; et al. A Genetic Interaction Map of Insulin Production Identifies Mfi as an Inhibitor of Mitochondrial Fission. Endocrinology 2018, 159, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Pozo, M.; Nieto, E.; Fernandez, M.; Alemany, S. Traf6 and Src Kinase Activity Regulates Cot Activation by Il-1. Cell. Signal. 2006, 18, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Lang, V.; Symons, A.; Watton, S.J.; Janzen, J.; Soneji, Y.; Beinke, S.; Howell, S.; Ley, S.C. Abin-2 Forms a Ternary Complex with Tpl-2 and Nf-Kappa B1 P105 and Is Essential for Tpl-2 Protein Stability. Mol. Cell. Biol. 2004, 24, 5235–5248. [Google Scholar] [CrossRef] [Green Version]
- Stack, J.; Doyle, S.L.; Connolly, D.J.; Reinert, L.S.; O’Keeffe, K.M.; McLoughlin, R.M.; Paludan, S.R.; Bowie, A.G. Tram Is Required for Tlr2 Endosomal Signaling to Type I Ifn Induction. J. Immunol. 2014, 193, 6090–6102. [Google Scholar] [CrossRef] [Green Version]
- McGettrick, A.F.; Brint, E.K.; Palsson-McDermott, E.M.; Rowe, D.C.; Golenbock, D.T.; Gay, N.J.; Fitzgerald, K.A.; O’Neill, L.A. Trif-Related Adapter Molecule Is Phosphorylated by Pkc{Epsilon} During Toll-Like Receptor 4 Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9196–9201. [Google Scholar] [CrossRef] [Green Version]
- Waterfield, M.R.; Zhang, M.; Norman, L.P.; Sun, S.-C. Nf-Κb1/P105 Regulates Lipopolysaccharide-Stimulated Map Kinase Signaling by Governing the Stability and Function of the Tpl2 Kinase. Mol. Cell 2003, 11, 685–694. [Google Scholar] [CrossRef]
- Jager, J.; Gremeaux, T.; Gonzalez, T.; Bonnafous, S.; Debard, C.; Laville, M.; Vidal, H.; Tran, A.; Gual, P.; le Marchand-Brustel, Y.; et al. Tpl2 Kinase Is Upregulated in Adipose Tissue in Obesity and May Mediate Interleukin-1beta and Tumor Necrosis Factor-{Alpha} Effects on Extracellular Signal-Regulated Kinase Activation and Lipolysis. Diabetes 2010, 59, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, M.; Nagata, S. Regulatory Elements Responsible for Inducible Expression of the Granulocyte Colony-Stimulating Factor Gene in Macrophages. Mol. Cell. Biol. 1990, 10, 2002–2011. [Google Scholar]
- Quehenberger, P.; Bierhaus, A.; Fasching, P.; Muellner, C.; Klevesath, M.; Hong, M.; Stier, G.; Sattler, M.; Schleicher, E.; Speiser, W.; et al. Endothelin 1 Transcription Is Controlled by Nuclear Factor-Kappab in Age-Stimulated Cultured Endothelial Cells. Diabetes 2000, 49, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M. Bcl2a1: The Underdog in the Bcl2 Family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, W.X.; Edelstein, L.C.; Chen, C.; Bash, J.; Gelinas, C. The Prosurvival Bcl-2 Homolog Bfl-1/A1 Is a Direct Transcriptional Target of Nf-Kappab That Blocks Tnfalpha-Induced Apoptosis. Genes Dev. 1999, 13, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Stange, K.; Thieme, T.; Hertel, K.; Kuhfahl, S.; Janecke, A.R.; Piza-Katzer, H.; Penttinen, M.; Hietala, M.; Dathe, K.; Mundlos, S.; et al. Molecular Analysis of Two Novel Missense Mutations in the Gdf5 Proregion That Reduce Protein Activity and Are Associated with Brachydactyly Type C. J. Mol. Biol. 2014, 426, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Kleijn, K.M.A.; Straasheijm, K.R.; Zuure, W.A.; Martens, G.J.M. Molecular Signature of Neuroinflammation Induced in Cytokine-Stimulated Human Cortical Spheroids. Biomedicines 2022, 10, 1025. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051025
De Kleijn KMA, Straasheijm KR, Zuure WA, Martens GJM. Molecular Signature of Neuroinflammation Induced in Cytokine-Stimulated Human Cortical Spheroids. Biomedicines. 2022; 10(5):1025. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051025
Chicago/Turabian StyleDe Kleijn, Kim M. A., Kirsten R. Straasheijm, Wieteke A. Zuure, and Gerard J. M. Martens. 2022. "Molecular Signature of Neuroinflammation Induced in Cytokine-Stimulated Human Cortical Spheroids" Biomedicines 10, no. 5: 1025. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051025