
Preclinical Studies of Granulysin-Based Anti-MUC1-Tn Immunotoxins as a New Antitumoral Treatment

 , , , , , , and
, , , , , , and 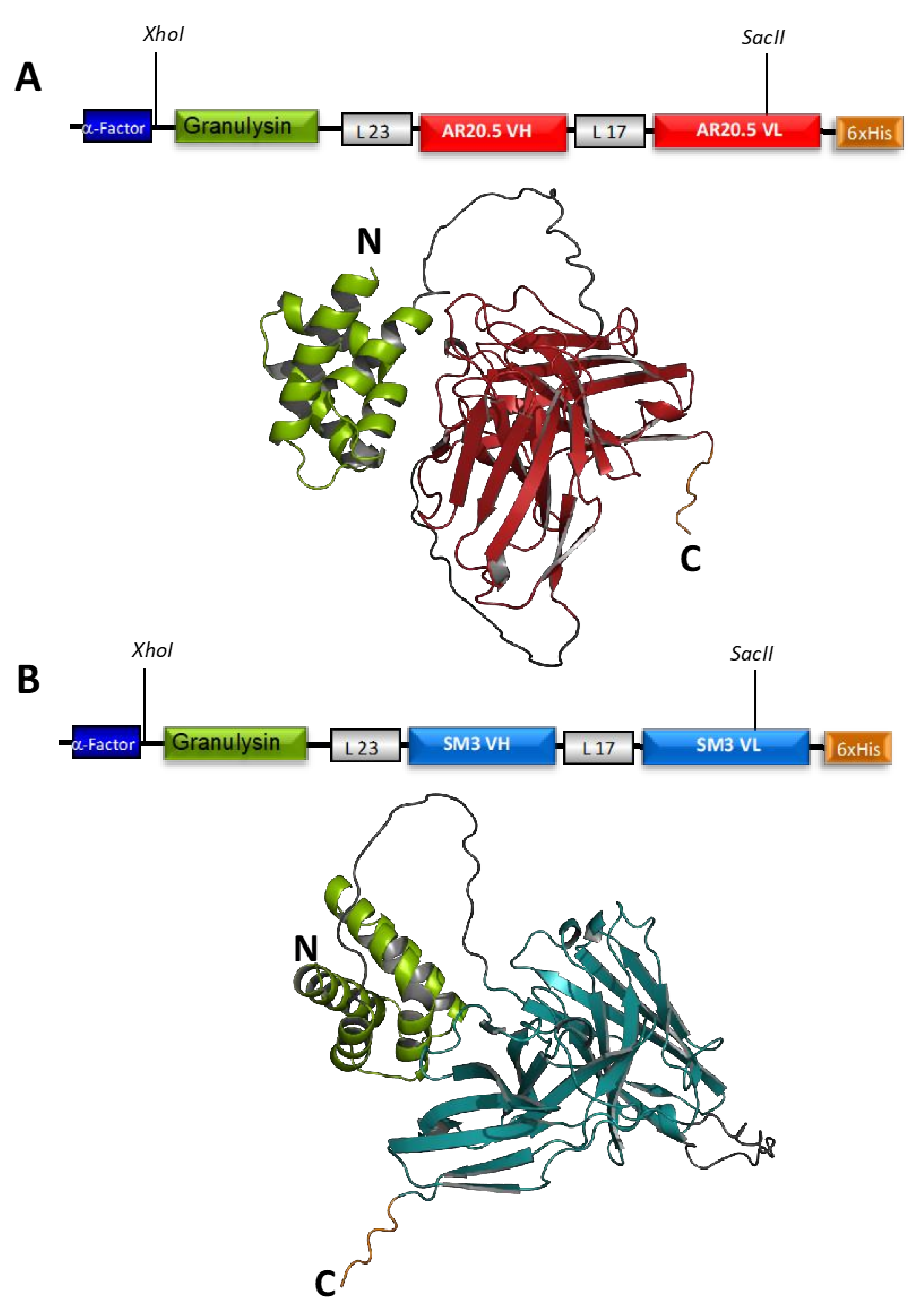{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Culture Conditions
2.2. Expression and Purification of Extracellular Recombinant GRNLY and Immunotoxins in Pichia Pastoris
2.3. Expression and Purification of Intracellular Recombinant Immunotoxins from Pichia Pastoris by Pulsed Electric Field Technology
2.4. Surface Plasmon Resonance
2.5. Cell Culture
2.6. Binding of Immunotoxins to the Tn Antigen Analyzed by Flow Cytometry
2.7. In Vitro Cytotoxicity Assays
2.8. In Vivo Assays
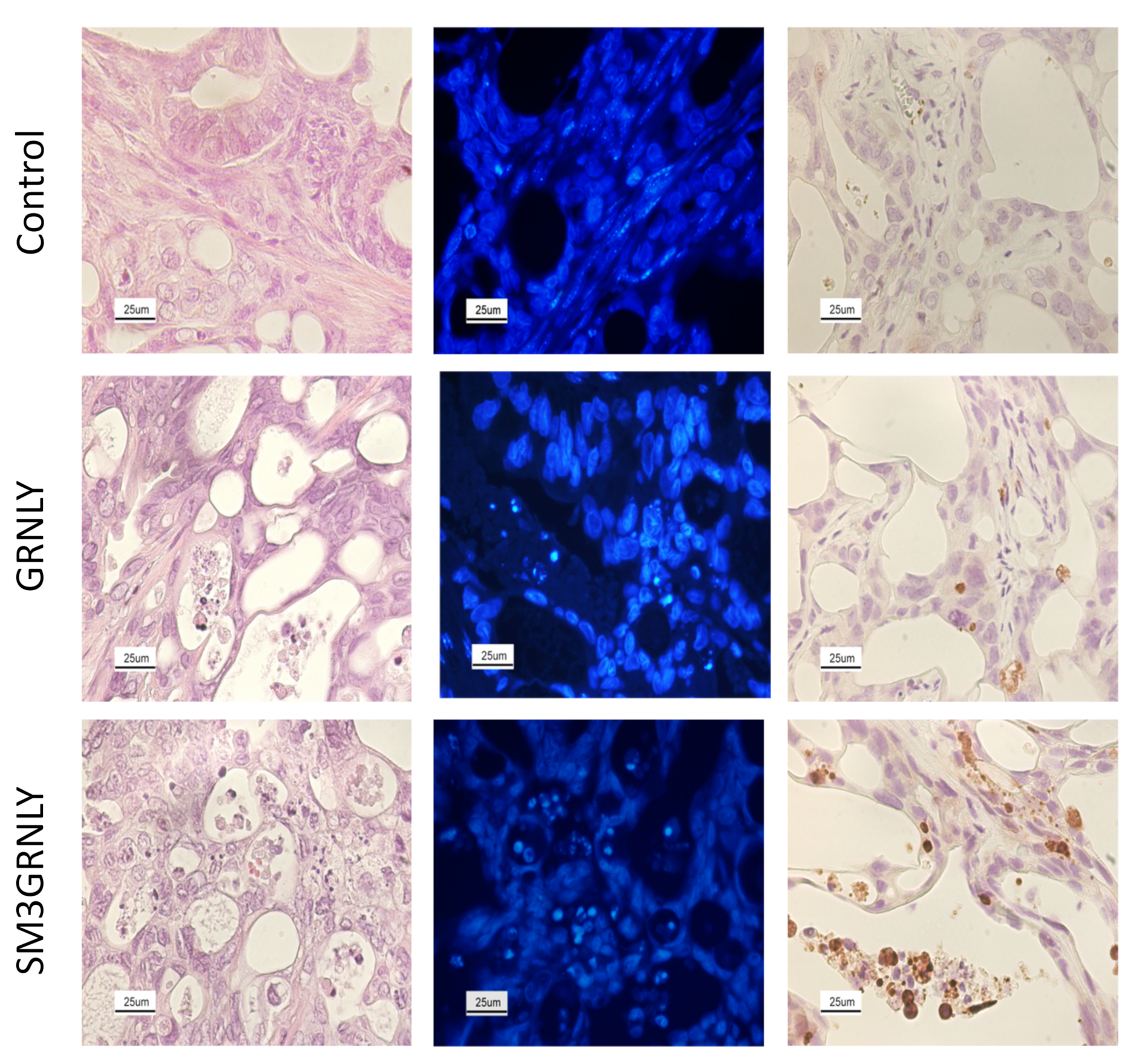
2.9. Histology Analysis
2.10. Statistical Analysis
3. Results
3.1. Design of Recombinant Immunotoxin and Yield of Production
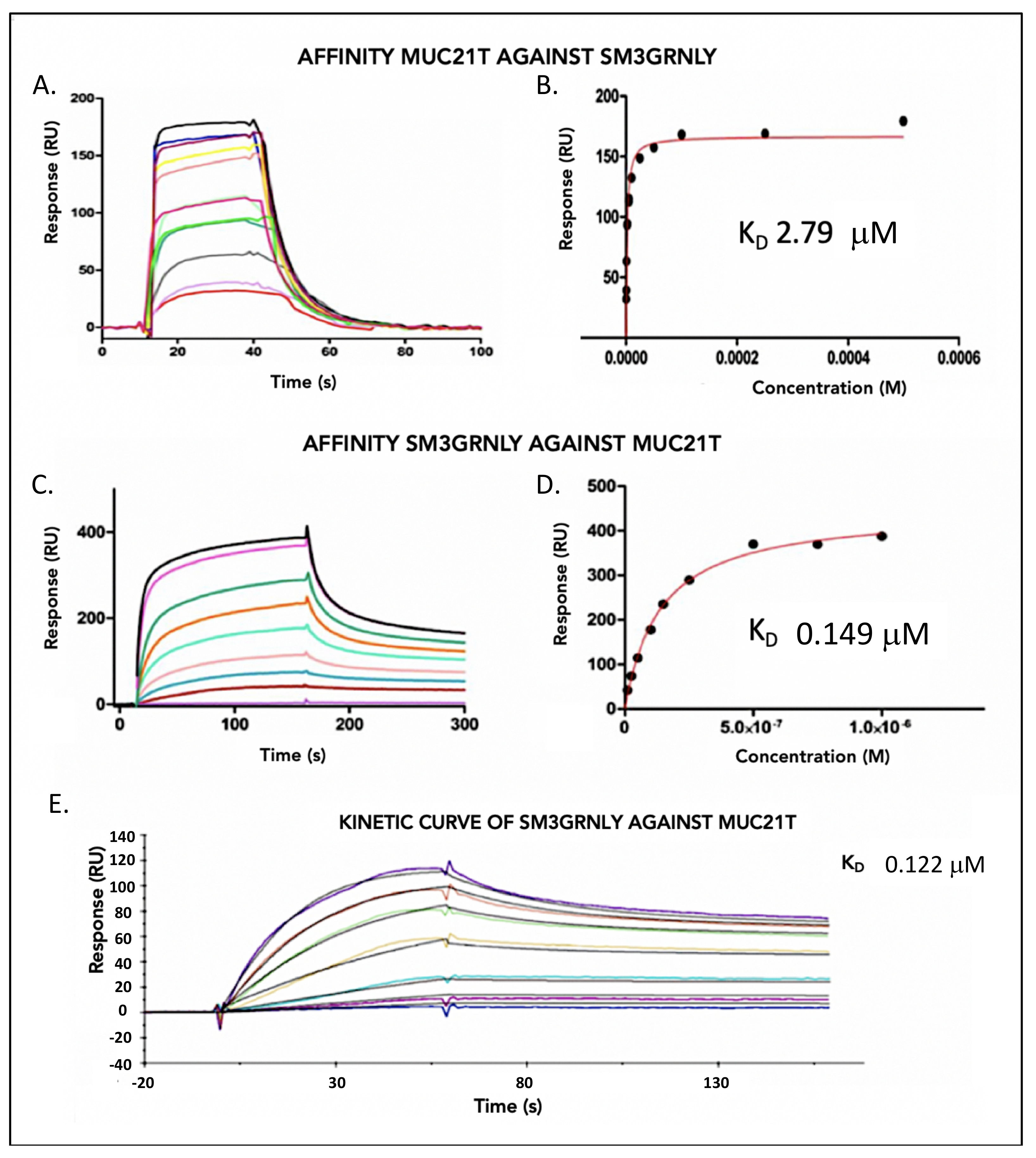
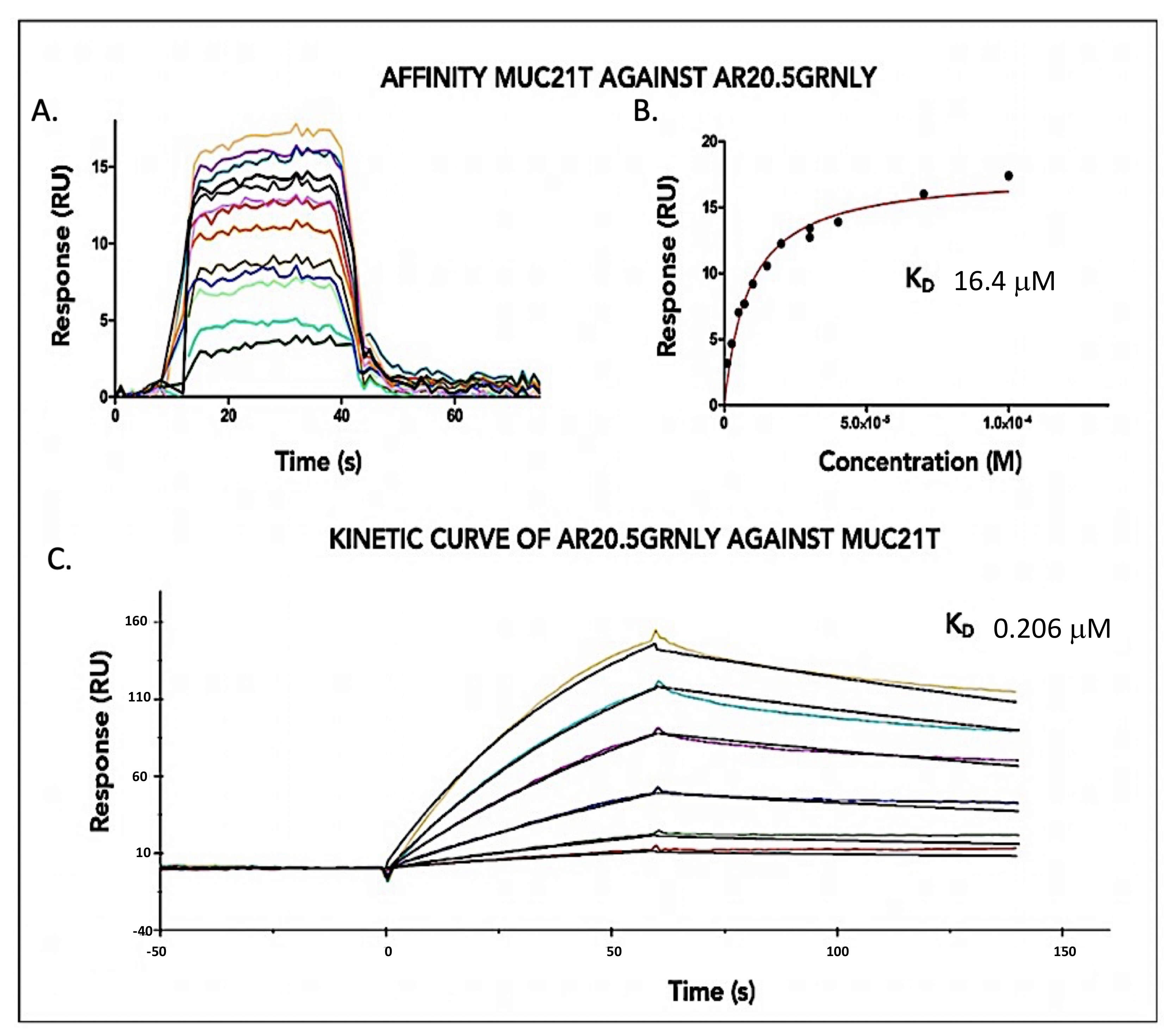
3.2. Immunotoxin Affinity for Its Antigen In Vitro
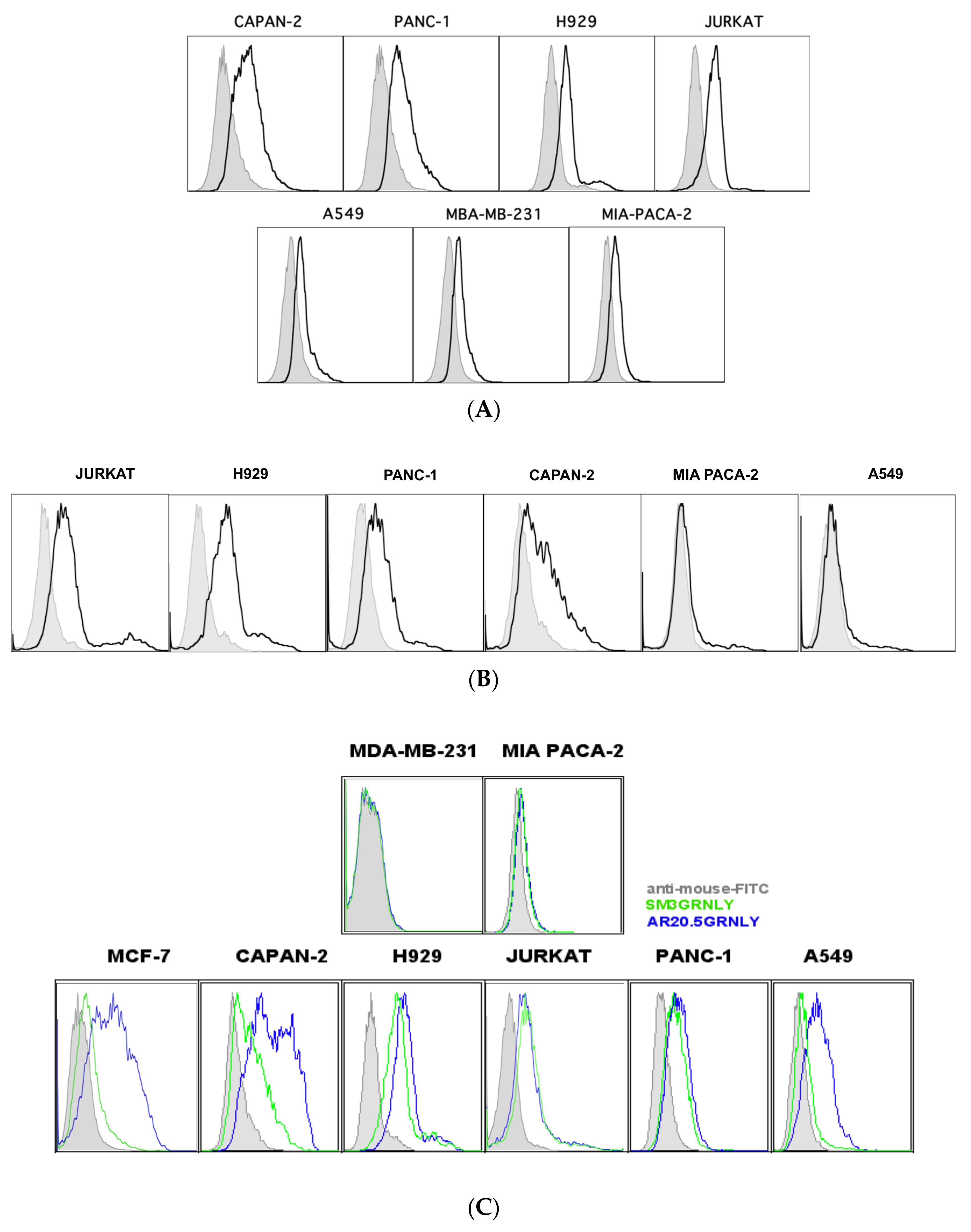
3.3. Immunotoxin Recognition of Its Antigen on the Cell Surface
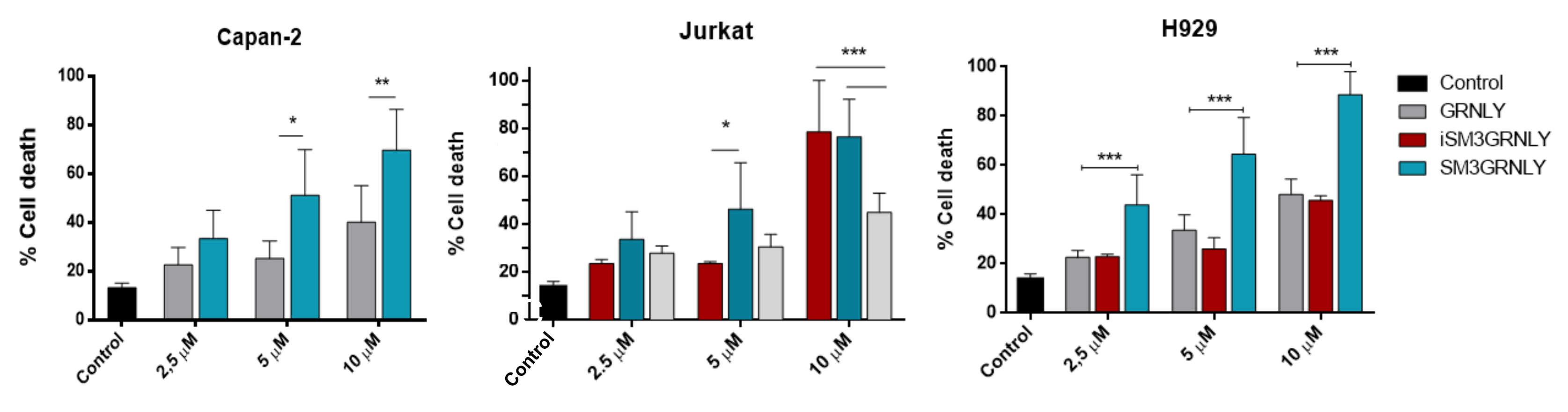
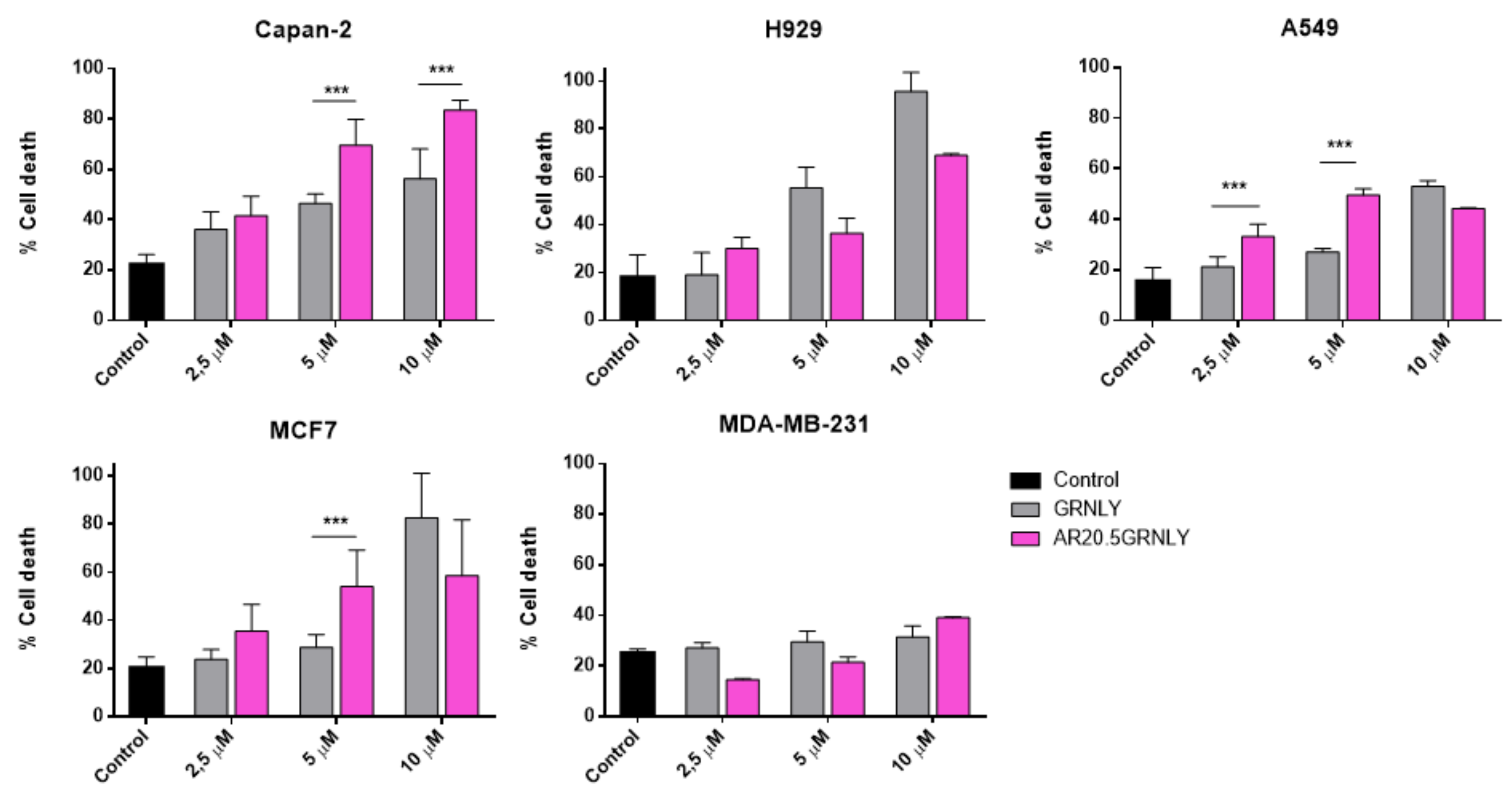
3.4. Cytotoxicity Assays on MUC1-Tn+ Tumor Cells
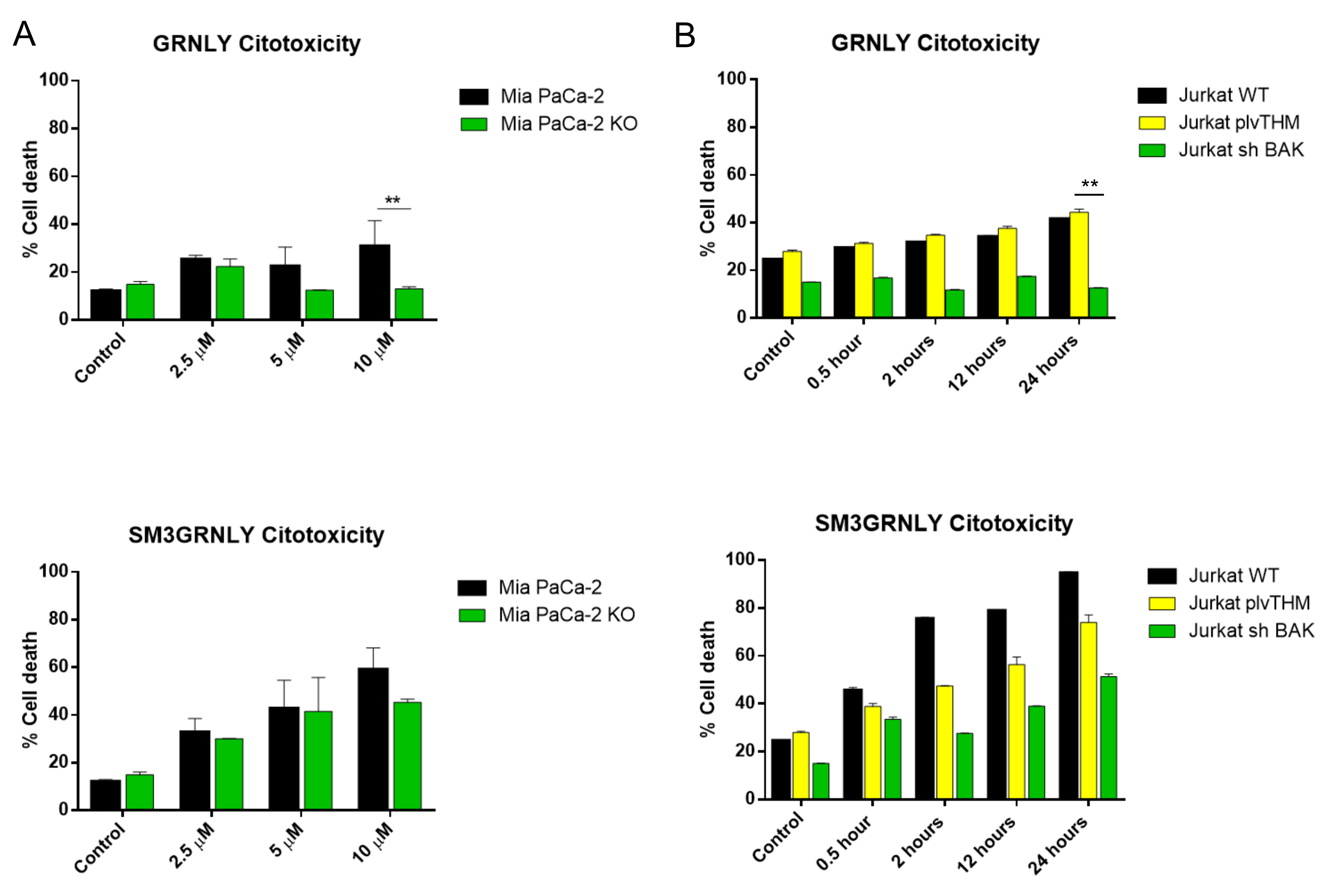
3.5. Mechanism of Cell Death Induced by SM3GRNLY
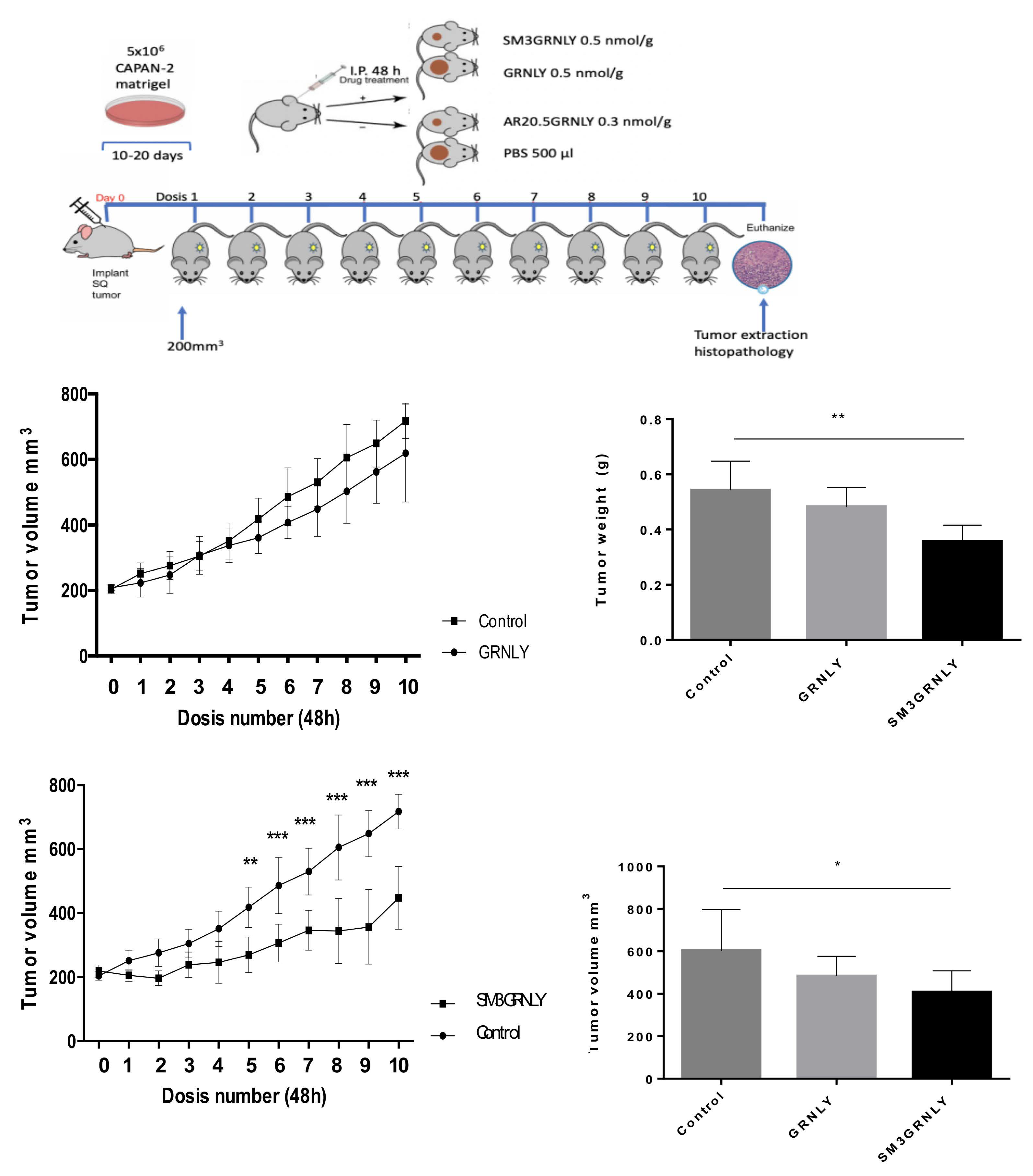
3.6. In Vivo Demonstration of Immunotoxin Targeting after Systemic Injection
- tumor development in a reasonable time after injection in athymic mice
- high immunotoxin binding
- increase in cytotoxicity of the immunotoxins as compared with GRNLY
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Otto, V.; Cummings, R. The Tn antigen-structural simplicity and biological complexity. Angew. Chem. Int. Ed. Engl. 2011, 50, 1770–1791. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, N.; Castro-López, J.; Valero-González, J.; Madariaga, D.; Compañón, I.; Somovilla, V.; Salvadó, M.; Asensio, J.; Jiménez-Barbero, J.; Avenoza, A.; et al. Deciphering the Non-Equivalence of Serine and Threonine O-Glycosylation Points: Implications for Molecular Recognition of the Tn Antigen by an anti-MUC1 Antibody. Angew. Chem. Int. Ed. Engl. 2015, 54, 9830–9834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheever, M.; Allison, J.; Ferris, A.; Finn, O.; Hastings, B.; Hecht, T.; Mellman, I.; Prindiville, S.; Viner, J.; Weiner, L.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, L.; Carrascal, M.; Barbas, A.; Ramalho, J.; Novo, C.; Delannoy, P.; Videira, P. Challenges in Antibody Development against Tn and Sialyl- Tn Antigens. Biomolecules 2015, 5, 1783–1809. [Google Scholar] [PubMed] [Green Version]
- Karsten, U.; Serttas, N.; Paulsen, H.; Danielczyk, A.; Goletz, S. Binding patterns of DTR-specific antibodies reveal a glycosylation-conditioned tumor-specific epitope of the epithelial mucin (MUC1). Glycobiology 2004, 14, 681–692. [Google Scholar]
- Fernández, E.; Navo, C.; Martínez-Sáez, N.; Gonçalves-Pereira, R.; Somovilla, V.; Avenoza, A.; Busto, J.H.; Bernardes, G.J.; Jiménez-Osés, G.; Corzana, F.; et al. Tn Antigen Mimics Based on sp2-Iminosugars with Affinity for an anti-MUC1 Antibody. Org. Lett. 2016, 18, 2796–2799. [Google Scholar]
- Movahedin, M.; Brooks, T.; Supekar, N.; Gokanapudi, N.; Boons, G.; Brooks, C. Glycosylation of MUC1 influences the binding of a therapeutic antibody by altering the conformational equilibrium of the antigen. Glycobiology 2017, 27, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Katayose, Y.; Kudo, T.; Suzuki, M.; Shinoda, M.; Saijyo, S.; Sakurai, N.; Saeki, H.; Fukuhara, K.; Imai, K.; Matsuno, S. MUC1-specific targeting immunotherapy with bispecific antibodies: Inhibition of xenografted human bile duct carcinoma growth. Cancer Res. 1996, 56, 4205–4212. [Google Scholar]
- Kodama, H.; Suzuki, M.; Katayose, Y.; Shinoda, M.; Sakurai, N.; Takemura, S.-I.; Yoshida, H.; Saeki, H.; Asano, R.; Ichiyama, M.; et al. Specific and effective targeting cancer immunotherapy with a combination of three bispecific antibodies. Immunol. Lett. 2002, 81, 99–106. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.; Taylor-Papadimitriou, J.; Burchell, J.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [Green Version]
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.; Rha, S.; Stephenson, J.; Schultes, B.; Monroe, P.; Eckhardt, G.; Hammond, L.A.; Whiteside, T.L.; Nicodemus, C.F.; Cermak, J.M.; et al. Phase I trial of a murine antibody to MUC1 in patients with metastatic cancer: Evidence for the activation of humoral and cellular antitumor immunity. Ann. Oncol. 2004, 15, 1825–1833. [Google Scholar] [CrossRef]
- Mehla, K.; Tremayne, J.; Grunkemeyer, J.; O’Connell, K.; Steele, M.; Caffrey, T.; Zhu, X.; Yu, F.; Singh, P.K.; Schultes, B.C.; et al. Combination of mAb-AR20.5, anti-PD-L1 and PolyICLC inhibits tumor progression and prolongs survival of MUC1.Tg mice challenged with pancreatic tumors. Cancer Immunol. Immunother. 2018, 67, 445–457. [Google Scholar] [CrossRef]
- Kreitman, R.; Dearden, C.; Zinzani, P.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.; le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Sanz, L.; Ibáñez-Pérez, R.; Guerrero-Ochoa, P.; Lacadena, J.; Anel, A. Antibody-Based Immunotoxins for Colorectal Cancer Therapy. Biomedicines 2021, 9, 1729. [Google Scholar] [CrossRef]
- Al-Wasaby, S.; de Miguel, D.; Aporta, A.; Naval, J.; Conde, B.; Martínez-Lostao, L.; Anel, A. In vivo potential of recombinant granulysin against human tumors. OncoImmunology 2015, 4, e1036213. [Google Scholar] [CrossRef] [Green Version]
- Al-Wasaby, S.; Guerrero-Ochoa, P.; Ibáñez-Pérez, R.; Soler, R.; Conde, B.; Martínez-Lostao, L.; Anel, A. In vivo potential of recombinant granulysin against human melanoma. Cancer Treat. Res. Commun. 2021, 27, 100355. [Google Scholar] [CrossRef]
- Aporta, A.; Catalán, E.; Galán-Malo, P.; Ramírez-Labrada, A.; Pérez, M.; Azaceta, G.; Palomera, L.; Naval, J.; Marzo, I.; Pardo, J.; et al. Granulysin induces apoptotic cell death and cleavage of the autophagy regulator Atg5 in human hematological tumors. Biochem. Pharmacol. 2014, 87, 410–423. [Google Scholar] [CrossRef]
- Gamen, S.; Hanson, D.A.; Kaspar, A.; Naval, J.; Krensky, A.M.; Anel, A. Granulysin-induced apoptosis. I. Involvement of at least two distinct pathways. J. Immunol. 1998, 161, 1758–1764. [Google Scholar]
- Ibáñez-Pérez, R.; Guerrero-Ochoa, P.; Al-Wasaby, S.; Navarro, R.; Tapia-Galisteo, A.; De Miguel, D.; Gonzalo, O.; Conde, B.; Martínez-Lostao, L.; Hurtado-Guerrero, R.; et al. Anti-tumoral potential of a human granulysinbased, CEA-targeted cytolytic immunotoxin. OncoImmunology 2019, 8, 1641392. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ochoa, P.; Aguilar-Machado, D.; Ibáñez-Pérez, R.; Macías-León, J.; Hurtado-Guerrero, R.; Raso, J.; Anel, A. Production of a Granulysin-Based, Tn-Targeted Cytolytic Immunotoxin Using Pulsed Electric Field Technology. Int. J. Mol. Sci. 2020, 21, 6165. [Google Scholar] [CrossRef]
- Logue, S.; Elgendy, M.; Martin, S. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction With Alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Delano, W. PyMOL: An open-source molecular graphics tool. CCP4 Newslett. Prot. Crystallogr. 2002, 40, 62–92. [Google Scholar]
- Fujita, R.; Hamano, H.; Kameda, Y.; Arai, R.; Shimizu, T.; Ota, M.; Sato, D.; Kobayashi, H.; Iwasaki, N.; Takahata, M. Breast cancer cells expressing cancer-associated sialyl-Tn antigen have less capacity to develop osteolytic lesions in a mouse model of skeletal colonization. Clin. Exp. Metastasis 2019, 36, 539–549. [Google Scholar] [CrossRef]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Brown, T.; Shuford, W.; Wang, W.; Nadler, S.; Bailey, T.; Marquardt, H.; Mittler, R. Characterization of a CD43/leukosialin-mediated pathway for inducing apoptosis in human T-lymphoblastoid cells. J. Biol. Chem. 1996, 271, 27686–27695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, A.; Schwab, R.; Boesteanu, A.; Steentof, C.; Mandel, U.; Engels, B.; Stone, J.; Madsen, T.; Schreiber, K.; Haines, K.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lostao, L.; de Miguel, D.; Al-Wasaby, S.; Gallego-Lleyda, A.; Anel, A. Death ligands and granulysin: Mechanisms of tumor cell death induction and therapeutic opportunities. Immunotherapy 2015, 7, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Reutelingsperger, C.P.M.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.A.A.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatydilserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, D.; Basáñez, G.; Sánchez, D.; Galán, P.; Marzo, I.; Larrad, L.; Naval, J.; Pardo, J.; Anel, A.; Martínez-Lostao, L. Thethering Apo2L/TRAIL to liposomes overcomes chemoresistance of human hematological tumor cells. Mol. Pharm. 2013, 10, 893–904. [Google Scholar] [CrossRef]
- Crespo, Â.; Mulik, S.; Dotiwala, F.; Ansara, J.; Sen Santara, S.; Ingersoll, K.; Ovies, C.; Junqueira, C.; Tilburgs, T.; Strominger, J.; et al. Decidual NK Cells Transfer Granulysin to Selectively Kill Bacteria in Trophoblasts. Cell 2020, 182, 1125–1139. [Google Scholar] [CrossRef]
- Dotiwala, F.; Mulik, S.; Polidoro, R.; Ansara, J.; Burleigh, B.; Walch, M.; Gazzinelli, R.; Lieberman, J. Killer lymphocytes use granulysin, perforin and granzymes to kill intracellular parasites. Nat. Med. 2016, 22, 210–216. [Google Scholar] [CrossRef]
- Stenger, S.; Hanson, D.A.; Teitelbaum, R.; Dewan, P.; Niazi, K.R.; Froelich, C.J.; Ganz, T.; Thoma-Uszynski, S.; Melián, A.; Bogdan, C.; et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 1998, 282, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Kishi, A.; Takamori, Y.; Ogawa, K.; Takano, S.; Tomita, S.; Tanigawa, M.; Niman, M.; Kishida, T.; Fujita, S. Differential expression of granulysin and perforin by NK cells in cancer patients and correlation of impaired granulysin expression with progression of cancer. Cancer Immunol. Immunother. 2002, 50, 604–614. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector Memory T Cells, Early Metastasis, and Survival in Colorectal Cancer. N. Eng. J. Med. 2005, 353, 2654–2666. [Google Scholar]
- Sparrow, E.; Bodman-Smith, M. Granulysin: The attractive side of a natural born killer. Immunol. Lett. 2020, 217, 126–132. [Google Scholar] [CrossRef]
- Tong, X.; Qu, X.; Wang, M. A Four-Gene-Based Prognostic Model Predicts Overall Survival in Patients with Cutaneous Melanoma. Front. Oncol. 2021, 11, 639874. [Google Scholar] [CrossRef]
- Huang, L.P.; Lyu, S.C.; Clayberger, C.; Krensky, A.M. Granulysin-Mediated Tumor Rejection in Transgenic Mice. J. Immunol. 2007, 178, 77–84. [Google Scholar] [CrossRef]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Li, Y.; Song, Y.; Rizvi, S.; Raja, C.; Zhang, D.; Samra, J.; Smith, R.; Perkins, A.; Apostolidis, C.; et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br. J. Cancer 2004, 91, 2086–2093. [Google Scholar] [CrossRef] [Green Version]
- Mazal, D.; Lo-Man, R.; Bay, S.; Pritsch, O.; Dériaud, E.; Ganneau, C.; Medeiros, A.; Ubillos, L.; Obal, G.; Berois, N.; et al. Monoclonal antibodies toward different Tn-amino acid backbones display distinct recognition patterns on human cancer cells. Implications for effective immuno-targeting of cancer. Cancer Immunol. Immunother. 2013, 62, 1107–1122. [Google Scholar] [CrossRef]
- Medeiros, A.; Berois, N.; Incerti, M.; Bay, S.; Franco Fraguas, L.; Osinaga, E. A Tn antigen binding lectin from Myrsine coriacea displays toxicity in human cancer cell lines. J. Nat. Med. 2013, 67, 247–254. [Google Scholar] [CrossRef]
- Xu, F.; Liu, F.; Zhao, H.; An, G.; Feng, G. Prognostic Significance of Mucin Antigen MUC1 in Various Human Epithelial Cancers. A Meta-Analysis. Medicine 2015, 94, e2286. [Google Scholar] [CrossRef]
- Anderson, D.H.; Sawaya, M.R.; Cascio, D.; Ernst, W.; Modlin, R.L.; Krensky, A.M.; Eisenberg, D. Granulysin crystal structure and a structure-derived lytic mechanism. J. Mol. Biol. 2003, 325, 355–365. [Google Scholar] [CrossRef]
- Barman, H.; Walch, M.; Latinovic-Golic, S.; Dumrese, C.; Dolder, M.; Groscurth, P.; Ziegler, U. Cholesterol in Negatively Charged Lipid Bilayers Modulates the Effect of the Antimicrobial Protein Granulysin. J. Membr. Biol. 2006, 212, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Anel, A.; Naval, J.; Desportes, P.; Gonzalez, B.; Uriel, J.; Piñeiro, A. Increased cytotoxicity of polyunsaturated fatty acids on human tumoral B and T-cell lines compared with normal lymphocytes. Leukemia 1992, 6, 680–688. [Google Scholar]
- Anel, A.; Naval, J.; González, B.; Torres, J.M.; Mishal, Z.; Uriel, J.; Piñeiro, A. Fatty acid metabolism in human lymphocytes. I. Time-course changes in fatty acid composition and membrane fluidity during blastic transformation of peripheral blood lymphocytes. Biochim. Biophys. Acta 1990, 1044, 323–331. [Google Scholar] [CrossRef]
- Aporta, A. La Granulisina Como Péptido Anti-Tumoral y el Papel de la Apoptosis en la Inmunidad Frente a M. Tuberculosis. Ph.D. Thesis, University of Zaragoza, Zaragoza, Spain, 2014. [Google Scholar]
- Kaspar, A.A.; Okada, S.; Kumar, J.; Poulain, F.R.; Drouvalakis, K.A.; Kelekar, A.; Hanson, D.A.; Kluck, R.M.; Hitoshi, Y.; Johnson, D.E.; et al. A distinct pathway of cell-mediated apoptosis initiated by granulysin. J. Immunol. 2001, 167, 350–356. [Google Scholar] [CrossRef]
- Saini, R.V.; Wilson, C.; Finn, M.W.; Wang, T.; Krensky, A.M.; Clayberger, C. Granulysin delivered by cytotoxic cells damages endoplasmic reticulum and activates caspase-7 in target cells. J. Immunol. 2011, 186, 3497–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef] [Green Version]
- Hlongwane, P.; Mungra, N.; Madheswaran, S.; Akinrinmade, O.; Chetty, S.; Barth, S. Human Granzyme B Based Targeted Cytolytic Fusion Proteins. Biomedicines 2018, 6, 72. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Ochoa, P.; Ibáñez-Pérez, R.; Berbegal-Pinilla, G.; Aguilar, D.; Marzo, I.; Corzana, F.; Minjárez-Sáenz, M.; Macías-León, J.; Conde, B.; Raso, J.; et al. Preclinical Studies of Granulysin-Based Anti-MUC1-Tn Immunotoxins as a New Antitumoral Treatment. Biomedicines 2022, 10, 1223. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061223
Guerrero-Ochoa P, Ibáñez-Pérez R, Berbegal-Pinilla G, Aguilar D, Marzo I, Corzana F, Minjárez-Sáenz M, Macías-León J, Conde B, Raso J, et al. Preclinical Studies of Granulysin-Based Anti-MUC1-Tn Immunotoxins as a New Antitumoral Treatment. Biomedicines. 2022; 10(6):1223. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061223
Chicago/Turabian StyleGuerrero-Ochoa, Patricia, Raquel Ibáñez-Pérez, Germán Berbegal-Pinilla, Diederich Aguilar, Isabel Marzo, Francisco Corzana, Martha Minjárez-Sáenz, Javier Macías-León, Blanca Conde, Javier Raso, and et al. 2022. "Preclinical Studies of Granulysin-Based Anti-MUC1-Tn Immunotoxins as a New Antitumoral Treatment" Biomedicines 10, no. 6: 1223. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061223