
Start Me Up: How Can Surrounding Gangliosides Affect Sodium-Potassium ATPase Activity and Steer towards Pathological Ion Imbalance in Neurons?

 , ,
, , 
Abstract
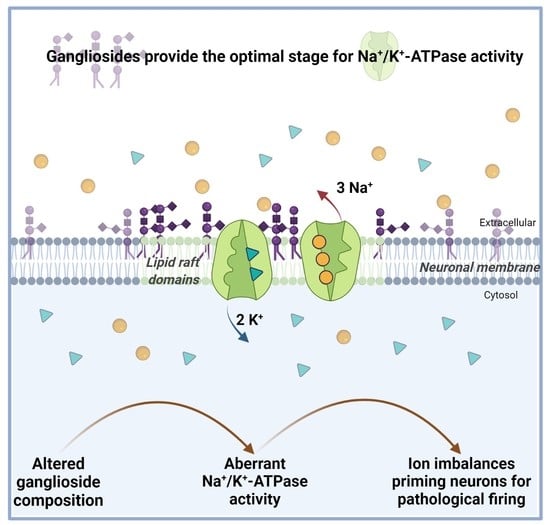
:
1. Membrane Dynamics and Lipid-Protein Interplay
2. Gangliosides
2.1. Short Overwiev of Gangliosides
2.2. Potential Role of Gangliosides in the Molecular Pathogenesis of Seizures
3. Na+/K+-ATPase
Na+/K+-ATPase Involvement in Epileptic Seizures
4. The Effects of Gangliosides on Na+/K+-ATPase
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinigin, K.V.; Kondrashov, O.V.; Jiménez-Munguía, I.; Alexandrova, V.V.; Batishchev, O.; Galimzyanov, T.R.; Akimov, S.A. Elastic Deformations Mediate Interaction of the Raft Boundary with Membrane Inclusions Leading to Their Effective Lateral Sorting. Sci. Rep. 2020, 10, 4087. [Google Scholar] [CrossRef] [PubMed]
- Bodosa, J.; Iyer, S.S.; Srivastava, A. Preferential Protein Partitioning in Biological Membrane with Coexisting Liquid Ordered and Liquid Disordered Phase Behavior: Underlying Design Principles. J. Membr. Biol. 2020, 253, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Ebersberger, L.; Schindler, T.; Kirsch, S.A.; Pluhackova, K.; Schambony, A.; Seydel, T.; Böckmann, R.A.; Unruh, T. Lipid Dynamics in Membranes Slowed Down by Transmembrane Proteins. Front. Cell Dev. Biol. 2020, 8, 1120. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, T.K.M. Lipid-Protein Interplay and Lateral Organization in Biomembranes. Chem. Phys. Lipids 2015, 189, 48–55. [Google Scholar] [CrossRef]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef]
- Laganowsky, A.; Reading, E.; Allison, T.M.; Ulmschneider, M.B.; Degiacomi, M.T.; Baldwin, A.J.; Robinson, C.V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Sarkis, J.; Vié, V. Biomimetic Models to Investigate Membrane Biophysics Affecting Lipid–Protein Interaction. Front. Bioeng. Biotechnol. 2020, 8, 270. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, H.; Chen, Y.; Dong, S.; Wang, F.; Wang, S.; Li, G.L.; Shu, Y.; Xu, F. Structural Insights into the Lipid and Ligand Regulation of a Human Neuronal KCNQ Channel. Neuron 2022, 110, 237–247.e4. [Google Scholar] [CrossRef]
- Corradi, V.; Mendez-Villuendas, E.; Ingólfsson, H.I.; Gu, R.X.; Siuda, I.; Melo, M.N.; Moussatova, A.; Degagné, L.J.; Sejdiu, B.I.; Singh, G.; et al. Lipid-Protein Interactions Are Unique Fingerprints for Membrane Proteins. ACS Cent. Sci. 2018, 4, 709–717. [Google Scholar] [CrossRef]
- Hedger, G.; Sansom, M.S.P. Lipid Interaction Sites on Channels, Transporters and Receptors: Recent Insights from Molecular Dynamics Simulations. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2390–2400. [Google Scholar] [CrossRef]
- Stansfeld, P.J.; Sansom, M.S.P. Molecular Simulation Approaches to Membrane Proteins. Structure 2011, 19, 1562–1572. [Google Scholar] [CrossRef] [Green Version]
- Bostick, D.L.; Berkowitz, M.L. Exterior Site Occupancy Infers Chloride-Induced Proton Gating in a Prokaryotic Homolog of the ClC Chloride Channel. Biophys. J. 2004, 87, 1686–1696. [Google Scholar] [CrossRef] [Green Version]
- Domene, C.; Grottesi, A.; Sansom, M.S.P. Filter Flexibility and Distortion in a Bacterial Inward Rectifier K+ Channel: Simulation Studies of KirBac1.1. Biophys. J. 2004, 87, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.; Tai, K.; Sansom, M.S.P. Molecular Dynamics Simulation of the M2 Helices within the NicotinicAcetylcholine Receptor Transmembrane Domain: Structure and Collective Motions. Biophys. J. 2005, 88, 3233–3321. [Google Scholar] [CrossRef] [Green Version]
- Bond, P.J.; Faraldo-Gömez, J.D.; Sansom, M.S.P. OmpA: A Pore or Not a Pore? Simulation and Modeling Studies. Biophys. J. 2002, 83, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Faraldo-Gómez, J.D.; Smith, G.R.; Sansom, M.S.P. Molecular Dynamics Simulations of the Bacterial Outer Membrane Protein FhuA: A Comparative Study of the Ferrichrome-Free and Bound States. Biophys. J. 2003, 85, 1406–1420. [Google Scholar] [CrossRef] [Green Version]
- Chavent, M.; Duncan, A.L.; Sansom, M.S.P. Molecular Dynamics Simulations of Membrane Proteins and Their Interactions: From Nanoscale to Mesoscale. Curr. Op. Struct. Biol. 2016, 40, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Javanainen, M. Universal Method for Embedding Proteins into Complex Lipid Bilayers for Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2577–2582. [Google Scholar] [CrossRef]
- Kato, K.; Yamaguchi, T.; Yagi-Utsumi, M. Experimental and Computational Characterization of Dynamic Biomolecular Interaction Systems Involving Glycolipid Glycans. Glycoconj. J. 2022, 39, 219–228. [Google Scholar] [CrossRef]
- De Oliveira, E.C.L.; da Costa, K.S.; Taube, P.S.; Lima, A.H.; de Souza de Sales, C., Jr. Biological Membrane-Penetrating Peptides: Computational Prediction and Applications. Front. Cell. Infect. Microbiol. 2022, 12, 276. [Google Scholar] [CrossRef]
- Yang, X.; Lin, C.; Chen, X.; Li, S.; Li, X.; Xiao, B. Structure Deformation and Curvature Sensing of PIEZO1 in Lipid Membranes. Nature 2022, 604, 377–383. [Google Scholar] [CrossRef]
- Lee, I.H.; Imanaka, M.Y.; Modahl, E.H.; Torres-Ocampo, A.P. Lipid Raft Phase Modulation by Membrane-Anchored Proteins with Inherent Phase Separation Properties. ACS Omega 2019, 4, 6551–6559. [Google Scholar] [CrossRef]
- Cornell, C.E.; Mileant, A.; Thakkar, N.; Lee, K.K.; Keller, S.L. Direct Imaging of Liquid Domains in Membranes by Cryo-Electron Tomography. Proc. Natl. Acad. Sci. USA 2020, 117, 19713–19719. [Google Scholar] [CrossRef]
- Leitao, S.M.; Drake, B.; Pinjusic, K.; Pierrat, X.; Navikas, V.; Nievergelt, A.P.; Brillard, C.; Djekic, D.; Radenovic, A.; Persat, A.; et al. Time-Resolved Scanning Ion Conductance Microscopy for Three-Dimensional Tracking of Nanoscale Cell Surface Dynamics. ACS Nano 2021, 15, 17613–17622. [Google Scholar] [CrossRef]
- Flores, A.; Ramirez-Franco, J.; Desplantes, R.; Debreux, K.; Ferracci, G.; Wernert, F.; Blanchard, M.P.; Maulet, Y.; Youssouf, F.; Sangiardi, M.; et al. Gangliosides Interact with Synaptotagmin to Form the High-Affinity Receptor Complex for Botulinum Neurotoxin B. Proc. Natl. Acad. Sci. USA 2019, 116, 18098–18108. [Google Scholar] [CrossRef] [Green Version]
- Gu, R.X.; Ingólfsson, H.I.; De Vries, A.H.; Marrink, S.J.; Tieleman, D.P. Ganglioside-Lipid and Ganglioside-Protein Interactions Revealed by Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [Google Scholar] [CrossRef] [Green Version]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 1004. [Google Scholar] [CrossRef]
- Prinetti, A.; Loberto, N.; Chigorno, V.; Sonnino, S. Glycosphingolipid Behaviour in Complex Membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, T. Thus Started Ganglioside Research. Trends Biochem. Sci. 1988, 13, 452–454. [Google Scholar] [CrossRef]
- Kolter, T. Ganglioside Biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [Green Version]
- Schnaar, R.L. Gangliosides of the Vertebrate Nervous System. J. Mol. Biol. 2016, 428, 3325–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraĉun, I.; Rösner, H.; Ćosović, C.; Stavljenić, A. Topographical Atlas of the Gangliosides of the Adult Human Brain. J. Neurochem. 1984, 43, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Hanada, K. Sphingolipid Metabolism and Interorganellar Transport: Localization of Sphingolipid Enzymes and Lipid Transfer Proteins. Traffic 2015, 16, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Echten-Deckert, G.; Guravi, M. Golgi Localization of Glycosyltransferases Involved in Ganglioside Biosynthesis. Curr. Drug Targets 2008, 9, 282–291. [Google Scholar] [CrossRef]
- Mlinac, K.; Bognar, S.K. Role of Gangliosides in Brain Aging and Neurodegeneration. Transl. Neurosci. 2010, 1, 300–307. [Google Scholar] [CrossRef]
- Schnaar, R.L. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem. 2019, 76, 113–148. [Google Scholar] [CrossRef]
- Li, T.A.; Schnaar, R.L. Congenital Disorders of Ganglioside Biosynthesis. Prog. Mol. Biol. Transl. Sci. 2018, 156, 63–82. [Google Scholar] [CrossRef]
- Sarmento, M.J.; Ricardo, J.C.; Amaro, M.; Sachl, R.; J Sarmento, C.M.; Sachl, R.; Heyrovsk, J. Organization of Gangliosides into Membrane Nanodomains. FEBS Lett. 2020, 594, 3668–3697. [Google Scholar] [CrossRef]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in Membrane Organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar] [CrossRef]
- Julien, S.; Bobowski, M.; Steenackers, A.; Le Bourhis, X.; Delannoy, P. How Do Gangliosides Regulate RTKs Signaling? Cells 2013, 2, 751–767. [Google Scholar] [CrossRef] [Green Version]
- Ilic, K.; Auer, B.; Mlinac-Jerkovic, K.; Herrera-Molina, R. Neuronal Signaling by Thy-1 in Nanodomains with Specific Ganglioside Composition: Shall We Open the Door to a New Complexity? Front. Cell Dev. Biol. 2019, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Ilic, K.; Lin, X.; Malci, A.; Stojanović, M.; Puljko, B.; Rožman, M.; Vukelić, Ž.; Heffer, M.; Montag, D.; Schnaar, R.L.; et al. Plasma Membrane Calcium ATPase-Neuroplastin Complexes Are Selectively Stabilized in GM1-Containing Lipid Rafts. Int. J. Mol. Sci. 2021, 22, 3590. [Google Scholar] [CrossRef]
- Yoon, S.J.; Nakayama, K.I.; Hikita, T.; Handa, K.; Hakomori, S.I. Epidermal Growth Factor Receptor Tyrosine Kinase Is Modulated by GM3 Interaction with N-Linked GlcNAc Termini of the Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 18987–18991. [Google Scholar] [CrossRef] [Green Version]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J.I. Dissociation of the Insulin Receptor and Caveolin-1 Complex by Ganglioside GM3 in the State of Insulin Resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef] [Green Version]
- De Laurentiis, A.; Donovan, L.; Arcaro, A. Lipid Rafts and Caveolae in Signaling by Growth Factor Receptors. Open Biochem. J. 2007, 1, 12. [Google Scholar] [CrossRef]
- Prendergast, J.; Umanah, G.K.E.; Yoo, S.W.; Lagerlöf, O.; Motari, M.G.; Cole, R.N.; Huganir, R.L.; Dawson, T.M.; Dawson, V.L.; Schnaar, R.L. Ganglioside Regulation of AMPA Receptor Trafficking. J. Neurosci. 2014, 34, 13246–13258. [Google Scholar] [CrossRef]
- Ichikawa, N.; Iwabuchi, K.; Kurihara, H.; Ishii, K.; Kobayashi, T.; Sasaki, T.; Hattori, N.; Mizuno, Y.; Hozumi, K.; Yamada, Y.; et al. Binding of Laminin-1 to Monosialoganglioside GM1 in Lipid Rafts Is Crucial for Neurite Outgrowth. J. Cell Sci. 2009, 122, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE Classification of the Epilepsies: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Falco-Walter, J.J.; Scheffer, I.E.; Fisher, R.S. The New Definition and Classification of Seizures and Epilepsy. Epilepsy Res. 2018, 139, 73–79. [Google Scholar] [CrossRef]
- McCormick, D.A.; Contreras, D. On the Cellular and Network Bases of Epileptic Seizures. Annu. Rev. Physiol. 2001, 63, 815–846. [Google Scholar] [CrossRef]
- Boccuto, L.; Aoki, K.; Flanagan-Steet, H.; Chen, C.F.; Fan, X.; Bartel, F.; Petukh, M.; Pittman, A.; Saul, R.; Chaubey, A.; et al. A Mutation in a Ganglioside Biosynthetic Enzyme, ST3GAL5, Results in Salt & Pepper Syndrome, a Neurocutaneous Disorder with Altered Glycolipid and Glycoprotein Glycosylation. Hum. Mol. Genet. 2014, 23, 418–433. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced Insulin Sensitivity in Mice Lacking Ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef] [Green Version]
- Kawai, H.; Allende, M.L.; Wada, R.; Kono, M.; Sango, K.; Deng, C.; Miyakawa, T.; Crawley, J.N.; Werth, N.; Bierfreund, U.; et al. Mice Expressing Only Monosialoganglioside GM3 Exhibit Lethal Audiogenic Seizures. J. Biol. Chem. 2001, 276, 6885–6888. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.A.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-Onset Symptomatic Epilepsy Syndrome Caused by a Homozygous Loss-of-Function Mutation of GM3 Synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Fragaki, K.; Ait-El-Mkadem, S.; Chaussenot, A.; Gire, C.; Mengual, R.; Bonesso, L.; Beneteau, M.; Ricci, J.E.; Desquiret-Dumas, V.; Procaccio, V.; et al. Refractory Epilepsy and Mitochondrial Dysfunction Due to GM3 Synthase Deficiency. Eur. J. Hum. Genet. 2013, 21, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.K.; Holley, J.A.; Macala, L.J.; Spencer, D.D. Ganglioside Changes Associated with Temporal Lobe Epilepsy in the Human Hippocampus. Yale J. Biol. Med. 1987, 60, 107. [Google Scholar]
- Abramson, M.B.; Yu, R.K.; Zaby, V. Ionic Properties of Beef Brain Gangliosides. Biochim. Biophys. Acta Lipids Lipid Metab. 1972, 280, 365–372. [Google Scholar] [CrossRef]
- Behr, J.P.; Lehn, J.M. The Binding of Divalent Cations by Purified Gangliosides. FEBS Lett. 1973, 31, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Izumi, T.; Ogawa, T.; Koizumi, H.; Fukuyama, Y. Low Levels of CSF Gangliotetraose-Series Gangliosides in West Syndrome: Implication of Brain Maturation Disturbance. Pediatr. Neurol. 1993, 9, 293–296. [Google Scholar] [CrossRef]
- Van Diepen, L.; Buettner, F.F.R.; Hoffmann, D.; Thiesler, C.T.; von Bohlen und Halbach, O.; von Bohlen und Halbach, V.; Jensen, L.R.; Steinemann, D.; Edvardson, S.; Elpeleg, O.; et al. A Patient-Specific Induced Pluripotent Stem Cell Model for West Syndrome Caused by ST3GAL3 Deficiency. Eur. J. Hum. Genet. 2018, 26, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Indellicato, R.; Domenighini, R.; Malagolini, N.; Cereda, A.; Mamoli, D.; Pezzani, L.; Iascone, M.; Dall’olio, F.; Trinchera, M. A Novel Nonsense and Inactivating Variant of ST3GAL3 in Two Infant Siblings Suffering Severe Epilepsy and Expressing Circulating CA19.9. Glycobiology 2020, 30, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, E.R.; Aoki, K.; Lopez, P.H.H.; Colacurcio, D.; Vajn, K.; Lorenzini, I.; Majić, S.; Yang, W.H.; Heffer, M.; Tiemeyer, M.; et al. Biosynthesis of the Major Brain Gangliosides GD1a and GT1b. Glycobiology 2012, 22, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlalka, G.V.; Lehman, A.; Chioza, B.; Baple, E.L.; Maroofian, R.; Cross, H.; Sreekantan-Nair, A.; Priestman, D.A.; Al-Turki, S.; McEntagart, M.E.; et al. Mutations in B4GALNT1 (GM2 Synthase) Underlie a New Disorder of Ganglioside Biosynthesis. Brain 2013, 136, 3618–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiavegatto, S.; Sun, J.; Nelson, R.J.; Schnaar, R.L. A Functional Role for Complex Gangliosides: Motor Deficits in GM2/GD2 Synthase Knockout Mice. Exp. Neurol. 2000, 166, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Fromholt, S.E.; Hess, E.J.; Crawford, T.O.; Griffin, J.W.; Sheikh, K.A.; Schnaar, R.L. Myelin-Associated Glycoprotein and Complementary Axonal Ligands, Gangliosides, Mediate Axon Stability in the CNS and PNS: Neuropathology and Behavioral Deficits in Single- and Double-Null Mice. Exp. Neurol. 2005, 195, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Takamiya, K.; Yamamoto, A.; Furukawa, K.; Yamashiro, S.; Shin, M.; Okada, M.; Fukumoto, S.; Haraguchi, M.; Takeda, N.; Fujimura, K.; et al. Mice with Disrupted GM2/GD2 Synthase Gene Lack Complex Gangliosides but Exhibit Only Subtle Defects in Their Nervous System. Proc. Natl. Acad. Sci. USA 1996, 93, 10662–10667. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Lu, Z.-H.; Wang, J.; Wang, Y.; Xie, X.; Meyenhofer, M.F.; Ledeen, R.W. Neurobiology of Disease Enhanced Susceptibility to Kainate-Induced Seizures, Neuronal Apoptosis, and Death in Mice Lacking Gangliotetraose Gangliosides: Protection with LIGA 20, a Membrane-Permeant Analog of GM1. J. Neurosci. 2005, 25, 11014–11022. [Google Scholar] [CrossRef] [Green Version]
- Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Oliveira, M.S.; Fiorenza, N.G.; Frussa-Filho, R.; Petry, J.C.; Coelho, R.C.; Mello, C.F. GM1 Ganglioside Prevents Seizures, Na+,K+-ATPase Activity Inhibition and Oxidative Stress Induced by Glutaric Acid and Pentylenetetrazole. Neurobiol. Dis. 2006, 22, 611–623. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tsuji, S.; Nagai, Y. Tetrasialoganglioside GO1b Reactive Monoclonal Antibodies: Their Characterization and Application for Quantification of GQ1b in Some Cell Lines of Neuronal and Adrenal Origin(s). J. Neurochem. 1990, 54, 513–517. [Google Scholar] [CrossRef]
- Kato, K.; Iwamori, M.; Hirabayashi, Y. Increase of GQ1b in the Hippocampus of Mice Following Kindled-Seizures. Neurosci. Lett. 2008, 441, 286–290. [Google Scholar] [CrossRef]
- Nakamura, Y.; Morimoto, K.; Okamoto, M. Modification of Amygdala Kindling by Intracerebroventricularly Administered Gangliosides in Rats. Exp. Neurol. 1989, 106, 61–69. [Google Scholar] [CrossRef]
- De Freitas, R.M.; do Nascimento, K.G.; Ferreira, P.M.P.; Jordán, J. Neurochemical Changes on Oxidative Stress in Rat Hippocampus during Acute Phase of Pilocarpine-Induced Seizures. Pharmacol. Biochem. Behav. 2010, 94, 341–345. [Google Scholar] [CrossRef]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Yoneda, J.S.; Sebinelli, H.G.; Itri, R.; Ciancaglini, P. Overview on Solubilization and Lipid Reconstitution of Na,K-ATPase: Enzyme Kinetic and Biophysical Characterization. Biophys. Rev. 2020, 12, 49–64. [Google Scholar] [CrossRef]
- Forrest, M.D. The Sodium-Potassium Pump Is an Information Processing Element in Brain Computation. Front. Physiol. 2014, 5, 472. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.; Bulygina, E.; Gerassimova, O.; Lyapina, L.; Schoner, W. Functional Relationship between Na/K-ATPase and NMDA-Receptors in Rat Cerebellum Granule Cells. Biochem. Mosc. 2004, 69, 429–434. [Google Scholar] [CrossRef]
- Fuchs, R.; Schmid, S.; Mellman, I. A Possible Role for Na+,K+-ATPase in Regulating ATP-Dependent Endosome Acidification. Proc. Natl. Acad. Sci. USA 1989, 86, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Jørgensen, P.L.; Andersen, J.P. Structural Basis for E1-E2 Conformational Transitions in Na,K-Pump and Ca-Pump Proteins. J. Membr. Biol. 1988, 103, 95–120. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Rael, E. The FXYD Gene Family of Small Ion Transport Regulators or Channels: CDNA Sequence, Protein Signature Sequence, and Expression. Genomics 2000, 68, 41–56. [Google Scholar] [CrossRef]
- Zhang, L.N.; Sun, Y.J.; Pan, S.; Li, J.X.; Qu, Y.E.; Li, Y.; Wang, Y.L.; Gao, Z. Bin Na+-K+-ATPase, a Potent Neuroprotective Modulator against Alzheimer Disease. Fundam. Clin. Pharmacol. 2013, 27, 96–103. [Google Scholar] [CrossRef]
- Chauhan, N.B.; Lee, J.M.; Siegel, G.J. Na,K-ATPase MRNA Levels and Plaque Load in Alzheimer’s Disease. J. Mol. Neurosci. 1997, 9, 151–166. [Google Scholar] [CrossRef]
- Fu, Y.J.; Xiong, S.; Lovell, M.A.; Lynn, B.C. Quantitative Proteomic Analysis of Mitochondria in Aging PS-1 Transgenic Mice. Cell. Mol. Neurobiol. 2009, 29, 649–694. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na,K-ATPase A3 Is a Death Target of Alzheimer Patient Amyloid-β Assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474. [Google Scholar] [CrossRef] [Green Version]
- Holm, T.H.; Isaksen, T.J.; Glerup, S.; Heuck, A.; Bøttger, P.; Füchtbauer, E.M.; Nedergaard, S.; Nyengaard, J.R.; Andreasen, M.; Nissen, P.; et al. Cognitive Deficits Caused by a Disease-Mutation in the A3 Na(+)/K(+)-ATPase Isoform. Sci. Rep. 2016, 6, 31972. [Google Scholar] [CrossRef] [Green Version]
- Sweney, M.T.; Newcomb, T.M.; Swoboda, K.J. The Expanding Spectrum of Neurological Phenotypes in Children with ATP1A3 Mutations, Alternating Hemiplegia of Childhood, Rapid-Onset Dystonia-Parkinsonism, CAPOS and Beyond. Pediatr. Neurol. 2015, 52, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Funck, V.R.; Ribeiro, L.R.; Pereira, L.M.; de Oliveira, C.V.; Grigoletto, J.; Della-Pace, I.D.; Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Larrick, J.W.; et al. Contrasting Effects of Na+,K+-ATPase Activation on Seizure Activity in Acute versus Chronic Models. Neuroscience 2015, 298, 171–179. [Google Scholar] [CrossRef]
- Krishnan, G.P.; Filatov, G.; Shilnikov, A.; Bazhenov, M. Electrogenic Properties of the Na+/K+ ATPase Control Transitions between Normal and Pathological Brain States. J. Neurophysiol. 2015, 113, 3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, D.L.W.; Tsukada, Y.; Barbeau, A. Ouabain Induced Seizures: Site of Production and Response to Anticonvulsants. Can. J. Neurol. Sci. 1978, 5, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarren, M.; Alger, B.E. Sodium-Potassium Pump Inhibitors Increase Neuronal Excitability in the Rat Hippocampal Slice: Role of a Ca2+-Dependent Conductance. J. Neurophysiol. 1987, 57, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.R.; Janicot, R.; Stafstrom, C.E. Na+-K+-ATPase Functions in the Developing Hippocampus: Regional Differences in CA1 and CA3 Neuronal Excitability and Role in Epileptiform Network Bursting. J. Neurophysiol. 2021, 125, 1–11. [Google Scholar] [CrossRef]
- Hosseinzadeh, Z.; Hauser, S.; Singh, Y.; Pelzl, L.; Schuster, S.; Sharma, Y.; Höflinger, P.; Zacharopoulou, N.; Stournaras, C.; Rathbun, D.L.; et al. Decreased Na+/K+ ATPase Expression and Depolarized Cell Membrane in Neurons Differentiated from Chorea-Acanthocytosis Patients. Sci. Rep. 2020, 10, 8391. [Google Scholar] [CrossRef]
- Rapport, R.L.; Harris, A.B.; Friel, P.N.; Ojemann, G.A. Human Epileptic Brain Na, K ATPase Activity and Phenytoin Concentrations. Arch. Neurol. 1975, 32, 549–554. [Google Scholar] [CrossRef]
- Ygberg, S.; Akkuratov, E.E.; Howard, R.J.; Taylan, F.; Jans, D.C.; Mahato, D.R.; Katz, A.; Kinoshita, P.F.; Portal, B.; Nennesmo, I.; et al. A Missense Mutation Converts the Na+,K+-ATPase into an Ion Channel and Causes Therapy-Resistant Epilepsy. J. Biol. Chem. 2021, 297, 101355. [Google Scholar] [CrossRef]
- Vetro, A.; Nielsen, H.N.; Holm, R.; Hevner, R.F.; Parrini, E.; Powis, Z.; Møller, R.S.; Bellan, C.; Simonati, A.; Lesca, G.; et al. ATP1A2- and ATP1A3-Associated Early Profound Epileptic Encephalopathy and Polymicrogyria. Brain 2021, 144, 1435–1450. [Google Scholar] [CrossRef]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 Encoding the Na+/K+ Pump Alpha2 Subunit Associated with Familial Hemiplegic Migraine Type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef]
- Deprez, L.; Weckhuysen, S.; Peeters, K.; Deconinck, T.; Claeys, K.G.; Claes, L.R.F.; Suls, A.; Van Dyck, T.; Palmini, A.; Matthijs, G.; et al. Epilepsy as Part of the Phenotype Associated with ATP1A2 Mutations. Epilepsia 2008, 49, 500–508. [Google Scholar] [CrossRef]
- Costa, C.; Prontera, P.; Sarchielli, P.; Tonelli, A.; Bassi, M.T.; Cupini, L.M.; Caproni, S.; Siliquini, S.; Donti, E.; Calabresi, P. A Novel ATP1A2 Gene Mutation in Familial Hemiplegic Migraine and Epilepsy. Cephalalgia 2014, 34, 68–72. [Google Scholar] [CrossRef]
- Segall, L.; Scanzano, R.; Kaunisto, M.A.; Wessman, M.; Palotie, A.; Gargus, J.J.; Blostein, R. Kinetic Alterations Due to a Missense Mutation in the Na,K-ATPase Alpha2 Subunit Cause Familial Hemiplegic Migraine Type 2. J. Biol. Chem. 2004, 279, 43692–43696. [Google Scholar] [CrossRef] [Green Version]
- Segall, L.; Mezzetti, A.; Scanzano, R.; Gargus, J.J.; Purisima, E.; Blostein, R. Alterations in the Alpha2 Isoform of Na,K-ATPase Associated with Familial Hemiplegic Migraine Type 2. Proc. Natl. Acad. Sci. USA 2005, 102, 11106–11111. [Google Scholar] [CrossRef] [Green Version]
- Vanmolkot, K.R.J.; Stroink, H.; Koenderink, J.B.; Kors, E.E.; Van Den Heuvel, J.J.M.W.; Van Den Boogerd, E.H.; Stam, A.H.; Haan, J.; De Vries, B.B.A.; Terwindt, G.M.; et al. Severe Episodic Neurological Deficits and Permanent Mental Retardation in a Child with a Novel FHM2 ATP1A2 Mutation. Ann. Neurol. 2006, 59, 310–314. [Google Scholar] [CrossRef]
- Li, Y.; Tang, W.; Kang, L.; Kong, S.; Dong, Z.; Zhao, D.; Liu, R.; Yu, S. Functional Correlation of ATP1A2 Mutations with Phenotypic Spectrum: From Pure Hemiplegic Migraine to Its Variant Forms. J. Headache Pain 2021, 22, 92. [Google Scholar] [CrossRef]
- Moya-Mendez, M.E.; Mueller, D.M.; Pratt, M.; Bonner, M.; Elliott, C.; Hunanyan, A.; Kucera, G.; Bock, C.; Prange, L.; Jasien, J.; et al. Early Onset Severe ATP1A2 Epileptic Encephalopathy: Clinical Characteristics and Underlying Mutations. Epilepsy Behav. 2021, 116, 107732. [Google Scholar] [CrossRef]
- Calame, D.G.; Houck, K.; Lotze, T.; Emrick, L.; Parnes, M. A Novel ATP1A2 Variant Associated with Severe Stepwise Regression, Hemiplegia, Epilepsy and Movement Disorders in Two Unrelated Patients. Eur. J. Paediatr. Neurol. 2021, 31, 21–26. [Google Scholar] [CrossRef]
- Ishii, A.; Saito, Y.; Mitsui, J.; Ishiura, H.; Yoshimura, J.; Arai, H.; Yamashita, S.; Kimura, S.; Oguni, H.; Morishita, S.; et al. Identification of ATP1A3 Mutations by Exome Sequencing as the Cause of Alternating Hemiplegia of Childhood in Japanese Patients. PLoS ONE 2013, 8, e56120. [Google Scholar] [CrossRef] [Green Version]
- Paciorkowski, A.R.; McDaniel, S.S.; Jansen, L.A.; Tully, H.; Tuttle, E.; Ghoneim, D.H.; Tupal, S.; Gunter, S.A.; Vasta, V.; Zhang, Q.; et al. Novel Mutations in ATP1A3 Associated with Catastrophic Early Life Epilepsy, Episodic Prolonged Apnea, and Postnatal Microcephaly. Epilepsia 2015, 56, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Kirshenbaum, G.S.; Dawson, N.; Mullins, J.G.L.; Johnston, T.H.; Drinkhill, M.J.; Edwards, I.J.; Fox, S.H.; Pratt, J.A.; Brotchie, J.M.; Roder, J.C.; et al. Alternating Hemiplegia of Childhood-Related Neural and Behavioural Phenotypes in Na+,K+-ATPase A3 Missense Mutant Mice. PLoS ONE 2013, 8, e60141. [Google Scholar] [CrossRef] [Green Version]
- Clapcote, S.J.; Duffy, S.; Xie, G.; Kirshenbaum, G.; Bechard, A.R.; Schack, V.R.; Petersen, J.; Sinai, L.; Saab, B.J.; Lerch, J.P.; et al. Mutation I810N in the 3 Isoform of Na,K-ATPase Causes Impairments in the Sodium Pump and Hyperexcitability in the CNS. Proc. Natl. Acad. Sci. USA 2009, 106, 14085–14090. [Google Scholar] [CrossRef] [Green Version]
- Azzaz, F.; Yahi, N.; di Scala, C.; Chahinian, H.; Fantini, J. Ganglioside Binding Domains in Proteins: Physiological and Pathological Mechanisms. Adv. Protein Chem. Struct. Biol. 2022, 128, 289–324. [Google Scholar] [CrossRef]
- Cantu’, L.; Corti, M.; Brocca, P.; del Favero, E. Structural Aspects of Ganglioside-Containing Membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Gangliosides in Health and Disease. In Progress in Molecular Biology and Translational Science, 1st ed.; Schnaar, R.L., Lopez, P.H.H., Eds.; Academic Press Inc.: Dordrecht, The Netherlands, 2018; Volume 156, pp. 1–462. [Google Scholar]
- Rodriguez, P.E.A.; Maggio, B.; Cumar, F.A. Acid and Enzymatic Hydrolysis of the Internal Sialic Acid Residue in Native and Chemically Modified Ganglioside GM1. J. Lipid Res. 1996, 37, 382–390. [Google Scholar] [CrossRef]
- Cornelius, F. Cholesterol Modulation of Molecular Activity of Reconstituted Shark Na+, K(+)-ATPase. Biochim. Biophys. Acta Biomembr. 1995, 1235, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Habeck, M.; Haviv, H.; Katz, A.; Kapri-Pardes, E.; Ayciriex, S.; Shevchenko, A.; Ogawa, H.; Toyoshima, C.; Karlish, S.J.D. Stimulation, Inhibition, or Stabilization of Na,K-ATPase Caused by Specific Lipid Interactions at Distinct Sites. J. Biol. Chem. 2015, 290, 4829–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, R.; Cornelius, F.; Ogawa, H.; Motoyama, K.; Vilsen, B.; Toyoshima, C. Binding of Cardiotonic Steroids to Na+,K+-ATPase in the E2P State. Proc. Natl. Acad. Sci. USA 2020, 118, e2020438118. [Google Scholar] [CrossRef]
- Puljko, B.; Stojanović, M.; Ilic, K.; Maček Hrvat, N.; Zovko, A.; Damjanović, V.; Mlinac-Jerkovic, K.; Kalanj-Bognar, S. Redistribution of Gangliosides Accompanies Thermally Induced Na+,K+-ATPase Activity Alternation and Submembrane Localisation in Mouse Brain. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183475. [Google Scholar] [CrossRef]
- Vyas, K.A.; Patel, H.V.; Vyas, A.A.; Schnaar, R.L. Segregation of Gangliosides GM1 and GD3 on Cell Membranes, Isolated Membrane Rafts, and Defined Supported Lipid Monolayers. Biol. Chem. 2001, 382, 241–250. [Google Scholar] [CrossRef]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a Pool of Non-Pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- Liebmann, T.; Fritz, N.; Kruusmägi, M.; Westin, L.; Bernhem, K.; Bondar, A.; Aperia, A.; Brismar, H. Regulation of Neuronal Na,K-ATPase by Extracellular Scaffolding Proteins. Int. J. Mol. Sci. 2018, 19, 2214. [Google Scholar] [CrossRef] [Green Version]
- Esmann, M.; Marsh, D.; Schwarzmann, G.; Sandhoff, K. Ganglioside-Protein Interactions: Spin-Label Electron Spin Resonance Studies with (Na+,K+)-ATPase Membranes. Biochemistry 1988, 27, 2398–2403. [Google Scholar] [CrossRef]
- Jeserich, G.; Breer, H.; Düvel, M. Effect of Exogenous Gangliosides on Synaptosomal Membrane ATPase Activity. Neurochem. Res. 1981, 6, 465–474. [Google Scholar] [CrossRef]
- Wood, P.A.; McBride, M.R.; Baker, H.J.; Christian, S.T. Fluorescence Polarization Analysis, Lipid Composition, and Na+,K+-ATPase Kinetics of Synaptosomal Membranes in Feline GM1 and GM2 Gangliosidosis. J. Neurochem. 1985, 44, 947–956. [Google Scholar] [CrossRef]
- Mirzoyan, S.A.; Mkheyan, E.E.; Sekoyan, E.S.; Sotskii, O.P.; Akopov, S.E. Effect of Gangliosides on Na,K-ATPase Activity and Conformation of Microsomal Membranes. Bull. Exp. Biol. Med. 1978, 86, 1607–1610. [Google Scholar] [CrossRef]
- Caputto, R.; Maccioni, A.H.R.; Caputto, B.L. Activation of Deoxycholate Solubilized Adenosine Triphosphatase by Ganglioside and Asialoganglioside Preparations. Biochem. Biophys. Res. Commun. 1977, 74, 1046–1052. [Google Scholar] [CrossRef]
- Leon, A.; Facci, L.; Toffano, G.; Sonnino, S.; Tettamanti, G. Activation of (Na+,K+)-ATPase by Nanomolar Concentrations of GM1 Ganglioside. J. Neurochem. 1981, 37, 350–357. [Google Scholar] [CrossRef]
- Nagata, Y.; Ando, M.; Hori, S. Stimulative Effect of Nerve Growth Factor on Alpha-Aminoisobutyric Acid Uptake and Na,K-ATPase Activity in Superior Cervical Sympathetic Ganglia Excised from Adult Rats. Neurochem. Res. 1985, 10, 1173–1185. [Google Scholar] [CrossRef]
- Ando, M.; Nakashima, Y.; Nagata, Y. Effects of GM1-Ganglioside and α-Sialyl Cholesterol on Amino Acid Uptake, Protein Synthesis, and Na+,K+-ATPase Activity in Superior Cervical and Nodose Ganglia Excised from Adult Rats. Mol. Chem. Neuropathol. 1990, 13, 33–46. [Google Scholar] [CrossRef]
- Nagata, Y.; Ando, M.; Iwata, M.; Hara, A.; Taketomi, T. Effect of Exogenous Gangliosides on Amino Acid Uptake and Na+,K+-ATPase Activity in Superior Cervical and Nodose Ganglia of Rats. J. Neurochem. 1987, 49, 201–207. [Google Scholar] [CrossRef]
- Karpiak, S.E.; Li, Y.S.; Mahadik, S.P. Gangliosides (GM1 and AGF2) Reduce Mortality Due to Ischemia: Protection of Membrane Function. Stroke 1987, 18, 184–187. [Google Scholar] [CrossRef] [Green Version]
- Mahadik, S.P.; Hawver, D.B.; Hungund, B.L.; Li, Y.S.; Karpiak, S.E. GM1 Ganglioside Treatment after Global Ischemia Protects Changes in Membrane Fatty Acids and Properties of Na+,K+-ATPase and Mg2+-ATPase. J. Neurosci. Res. 1989, 24, 402–412. [Google Scholar] [CrossRef]
- Lodovici, M.; Dolara, P.; Amerini, S.; Mantelli, L.; Ledda, F.; Bennardini, F.; Fazi, M.; Montereggi, A.; Dini, G. Effects of GM1 Ganglioside on Cardiac Function Following Experimental Hypoxia-Reoxygenation. Eur. J. Pharmacol. 1993, 243, 255–263. [Google Scholar] [CrossRef]
- Bianchi, R.; Zhu, X.; Fiori, M.G.; Eichberg, J. Effect of Gangliosides on Diacylglycerol Content and Molecular Species in Nerve from Diabetic Rats. Eur. J. Pharmacol. 1993, 239, 55–61. [Google Scholar] [CrossRef]
- Kreutz, F.; Scherer, E.B.; Ferreira, A.G.K.; Petry, F.D.S.; Pereira, C.L.; Santana, F.; De Souza Wyse, A.T.; Salbego, C.G.; Trindade, V.M.T. Alterations on Na+,K+-ATPase and Acetylcholinesterase Activities Induced by Amyloid-β Peptide in Rat Brain and GM1 Ganglioside Neuroprotective Action. Neurochem. Res. 2013, 38, 2342–2350. [Google Scholar] [CrossRef]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sørensen, T.L.M.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal Structure of the Sodium–Potassium Pump. Nature 2007, 450, 1043–1049. [Google Scholar] [CrossRef]
- Fazzari, M.; Lunghi, G.; Chiricozzi, E.; Mauri, L.; Sonnino, S. Gangliosides and the Treatment of Neurodegenerative Diseases: A Long Italian Tradition. Biomedicines 2022, 10, 363. [Google Scholar] [CrossRef]
- Mancini, G.; Loberto, N.; Olioso, D.; Dechecchi, M.C.; Cabrini, G.; Mauri, L.; Bassi, R.; Schiumarini, D.; Chiricozzi, E.; Lippi, G.; et al. GM1 as Adjuvant of Innovative Therapies for Cystic Fibrosis Disease. Int. J. Mol. Sci. 2020, 21, 4486. [Google Scholar] [CrossRef]


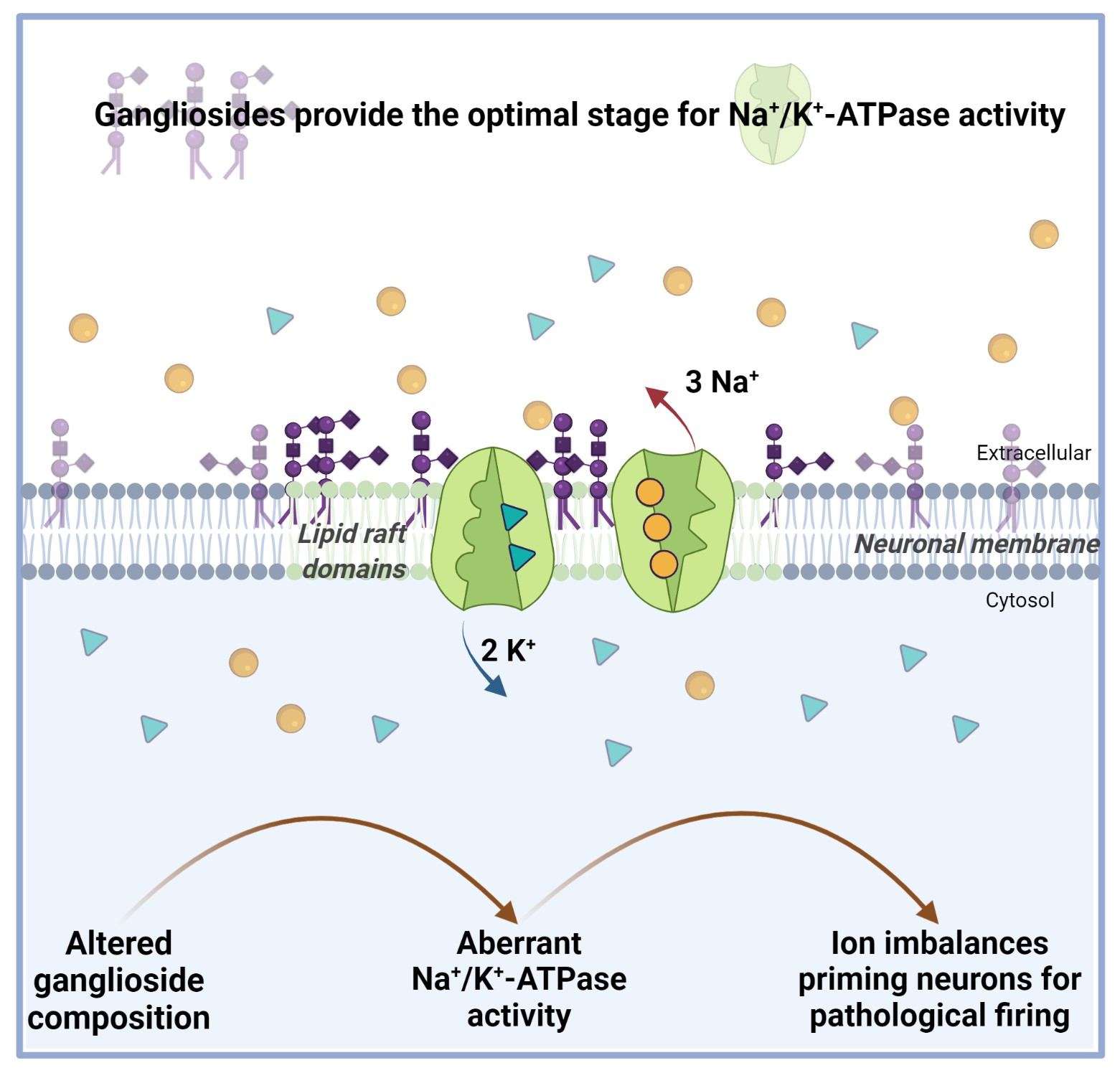{kind=link}
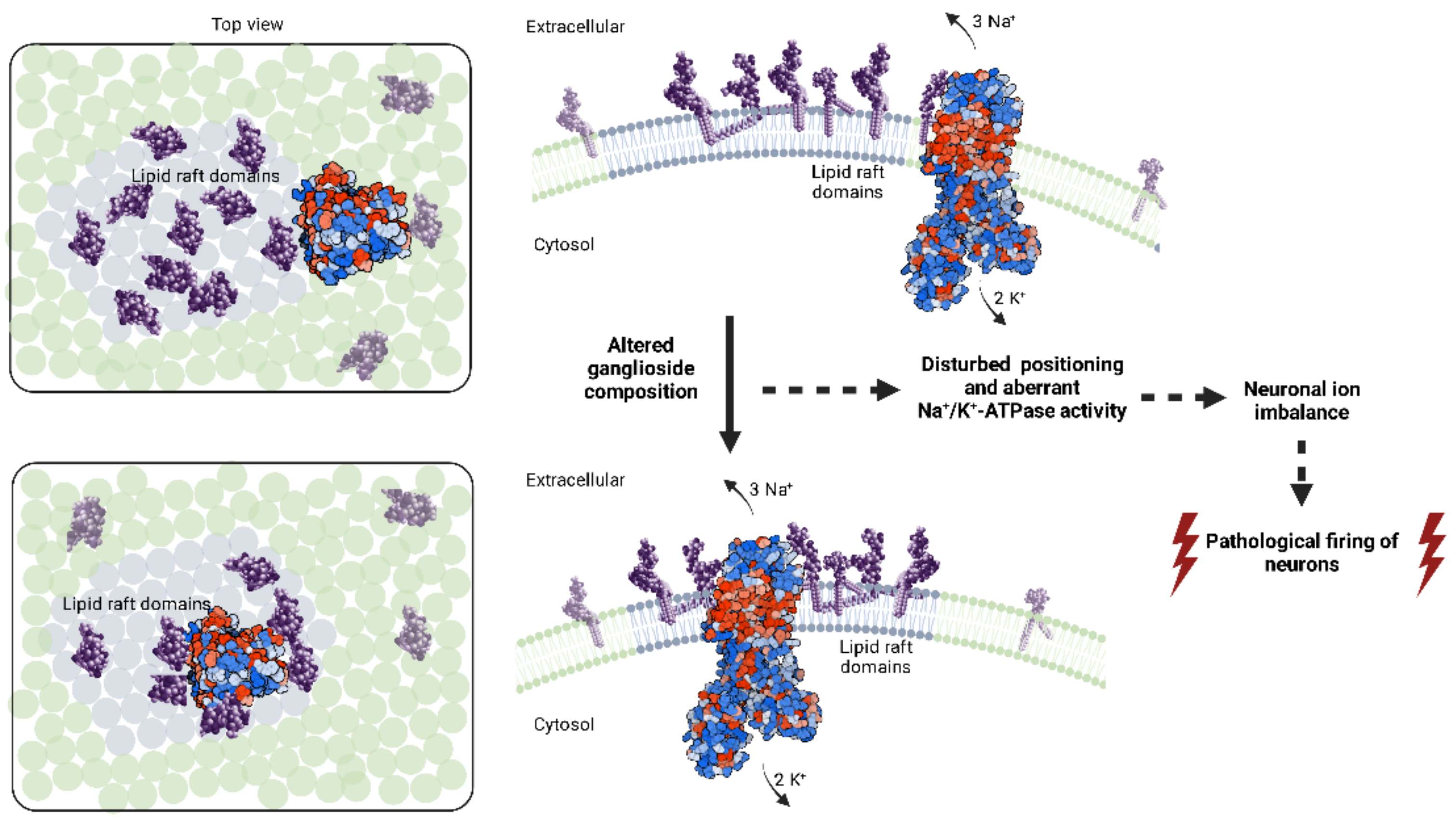{kind=link}
| Animal Model | Sample Type | Gangliosides Studied | NKA Activity | Publication |
|---|---|---|---|---|
| Chicken | Brain synaptosomes | Total porcine brain gangliosides | ↓ | [121] |
| Cat | Brain synaptosomes | GM1 | No change | [122] |
| Rat | Brain microsomes | Total human brain gangliosides | ↑↓ * | [123] |
| Rat | Brain microsomes | Total human brain gangliosides | ↑ | [124] |
| Rat | Brain mitochondrial fractions | GM1, GD1a, GD1b, GT1b | ↑ | [125] |
| Rat | SCG homogenates | GM1 | ↓ | [126,127] |
| Rat | NG homogenates | GM1 | ↑ | [127,128] |
| Mongolian gerbil | Brain homogenates | GM1 | No change | [129,130] |
| Rat | Striatal homogenates | GM1 | ↑ | [68] |
| Rat | Heart | GM1 | ↑ | [131] |
| Rat | Nerve homogenates | Mixed bovine gangliosides | ↑ | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puljko, B.; Stojanović, M.; Ilic, K.; Kalanj-Bognar, S.; Mlinac-Jerkovic, K. Start Me Up: How Can Surrounding Gangliosides Affect Sodium-Potassium ATPase Activity and Steer towards Pathological Ion Imbalance in Neurons? Biomedicines 2022, 10, 1518. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071518
Puljko B, Stojanović M, Ilic K, Kalanj-Bognar S, Mlinac-Jerkovic K. Start Me Up: How Can Surrounding Gangliosides Affect Sodium-Potassium ATPase Activity and Steer towards Pathological Ion Imbalance in Neurons? Biomedicines. 2022; 10(7):1518. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071518
Chicago/Turabian StylePuljko, Borna, Mario Stojanović, Katarina Ilic, Svjetlana Kalanj-Bognar, and Kristina Mlinac-Jerkovic. 2022. "Start Me Up: How Can Surrounding Gangliosides Affect Sodium-Potassium ATPase Activity and Steer towards Pathological Ion Imbalance in Neurons?" Biomedicines 10, no. 7: 1518. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071518