
Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs

 ,
,  ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Rutin Preparation
2.3. Circular Dichroism and Fluorescence Spectroscopy
2.4. Proteolytic Activity Assay
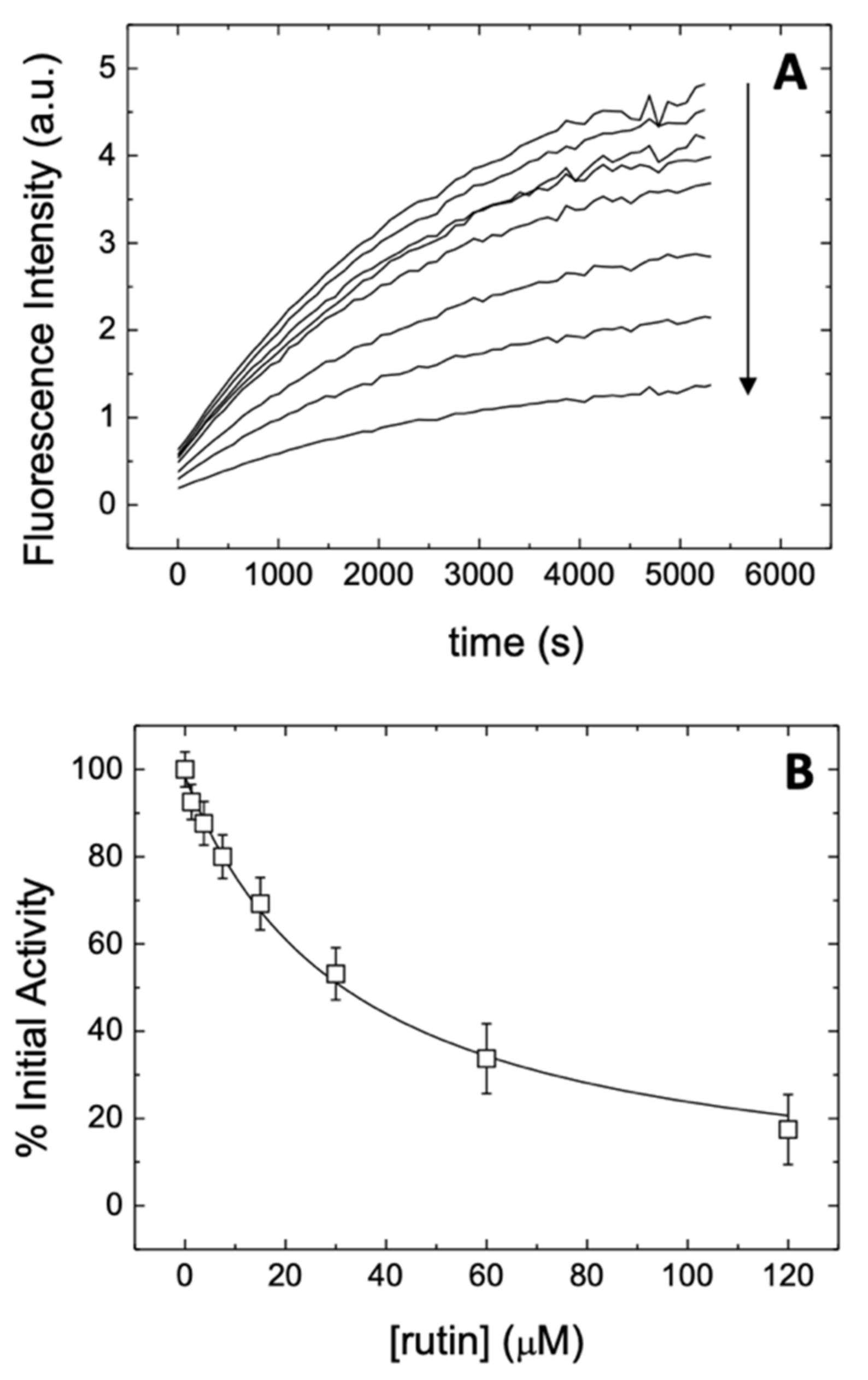
2.5. Inhibition Assay
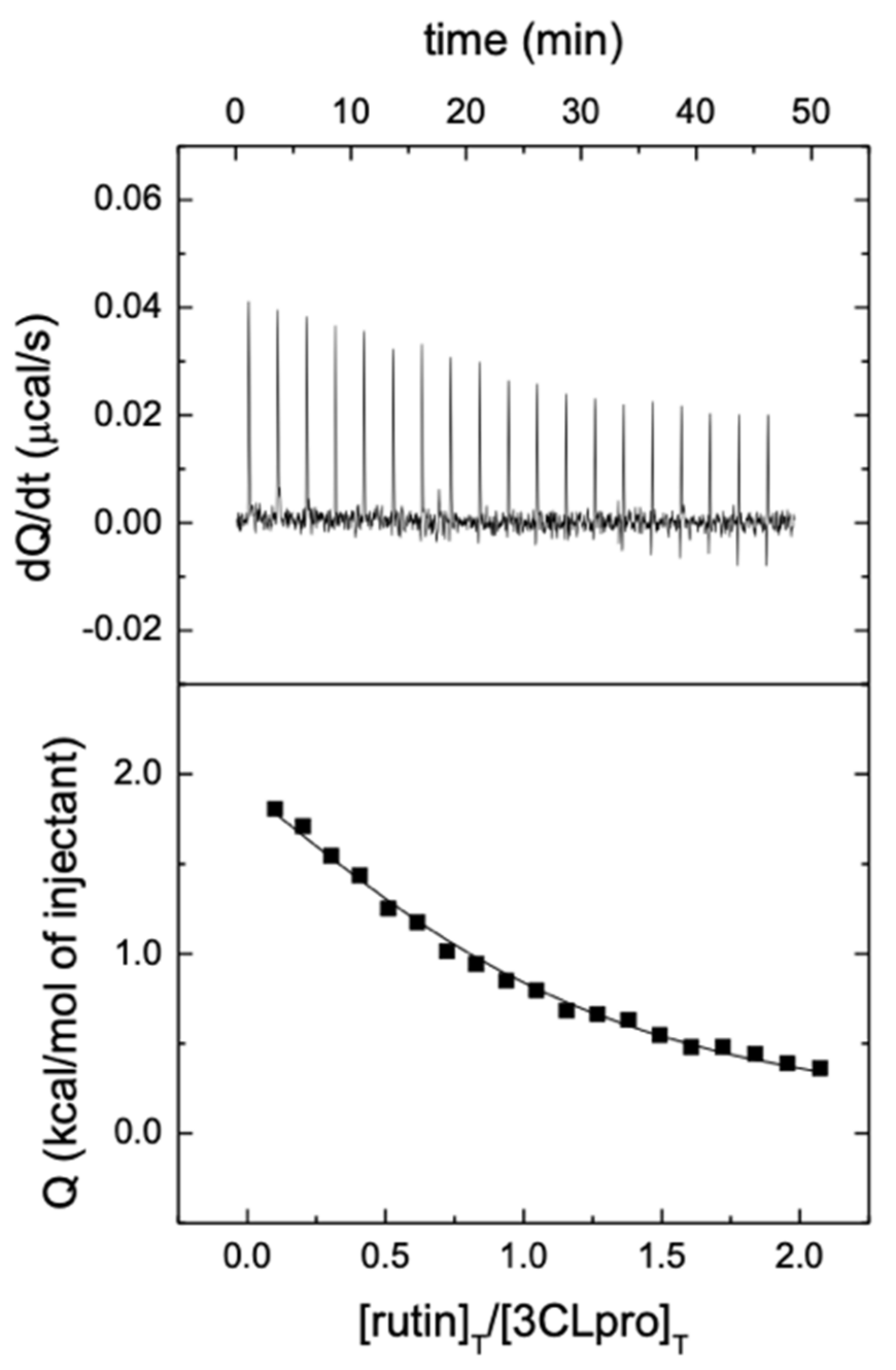
2.6. Isothermal Titration Calorimetry
2.7. Molecular Docking
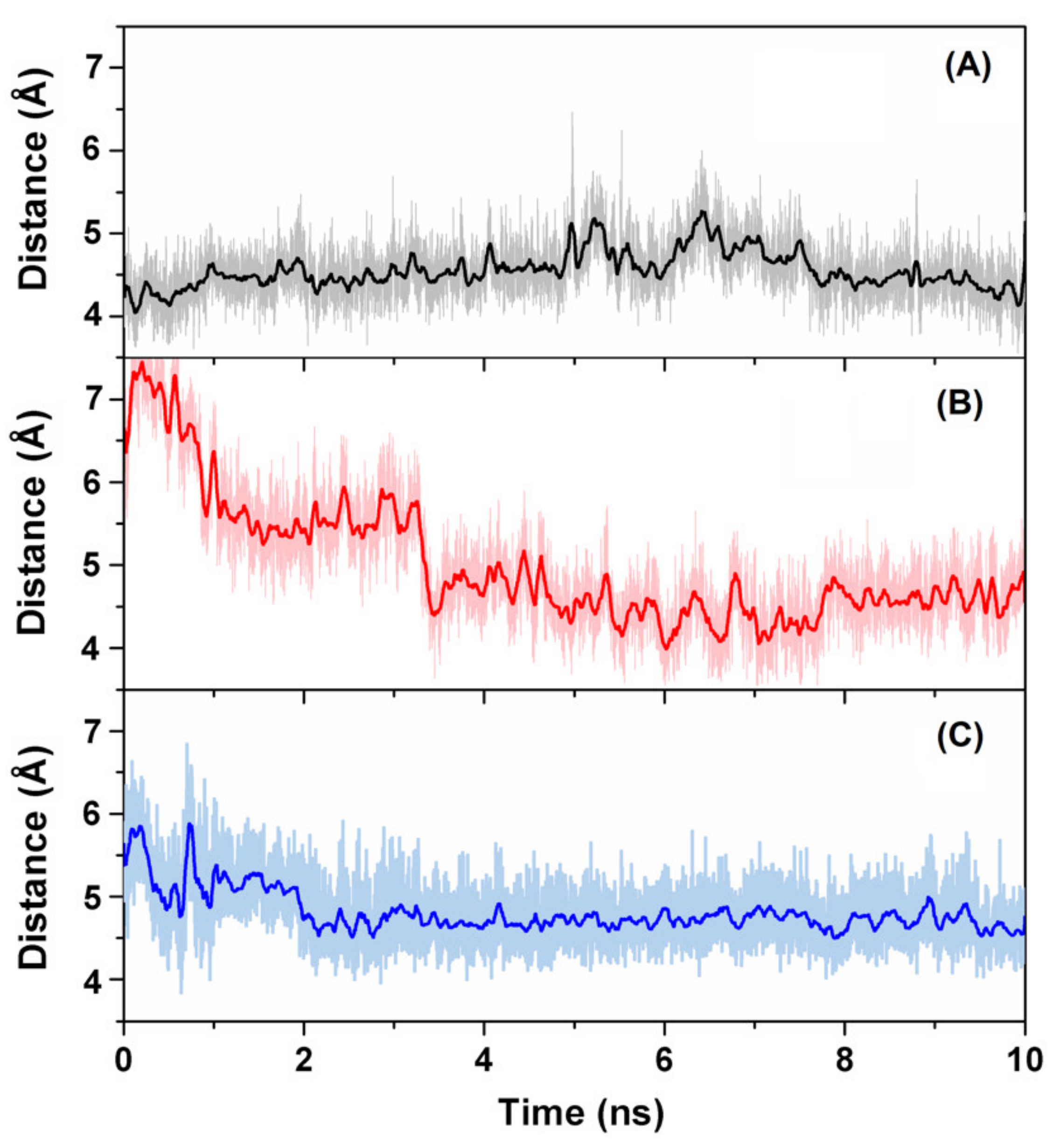
2.8. Molecular Dynamics
3. Results
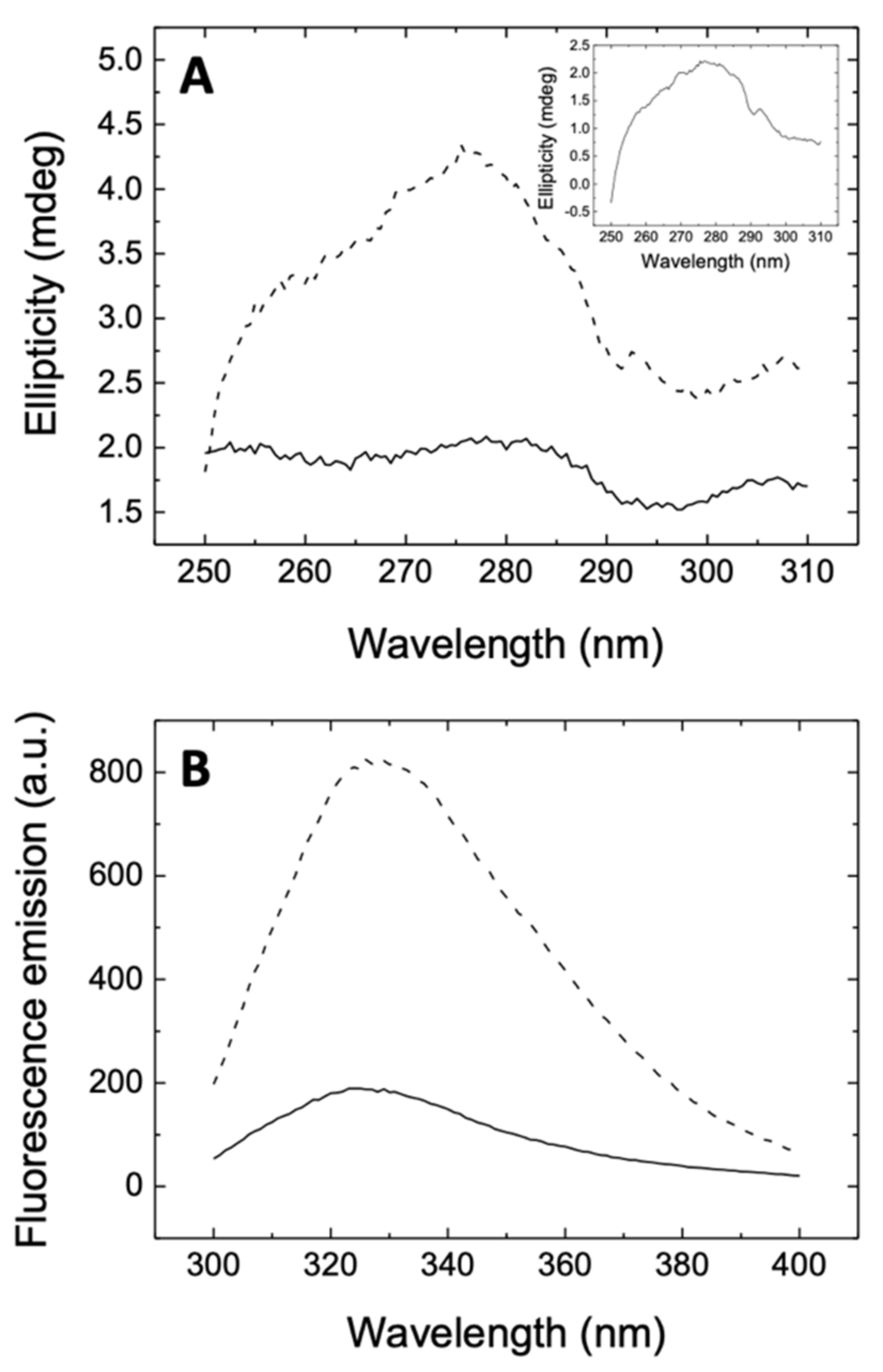
3.1. Rutin Binds and Inhibits the Main Protease 3CLpro
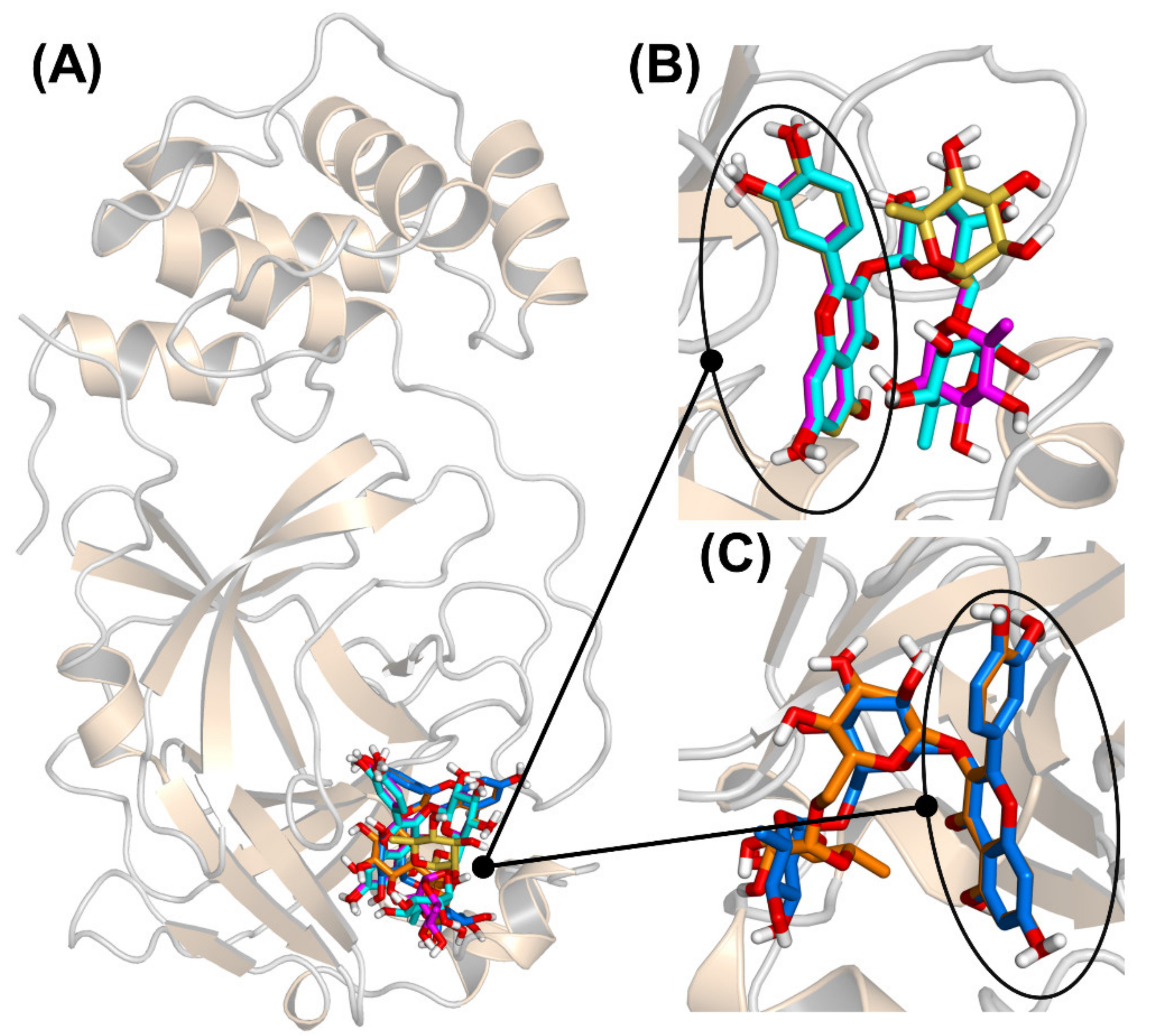
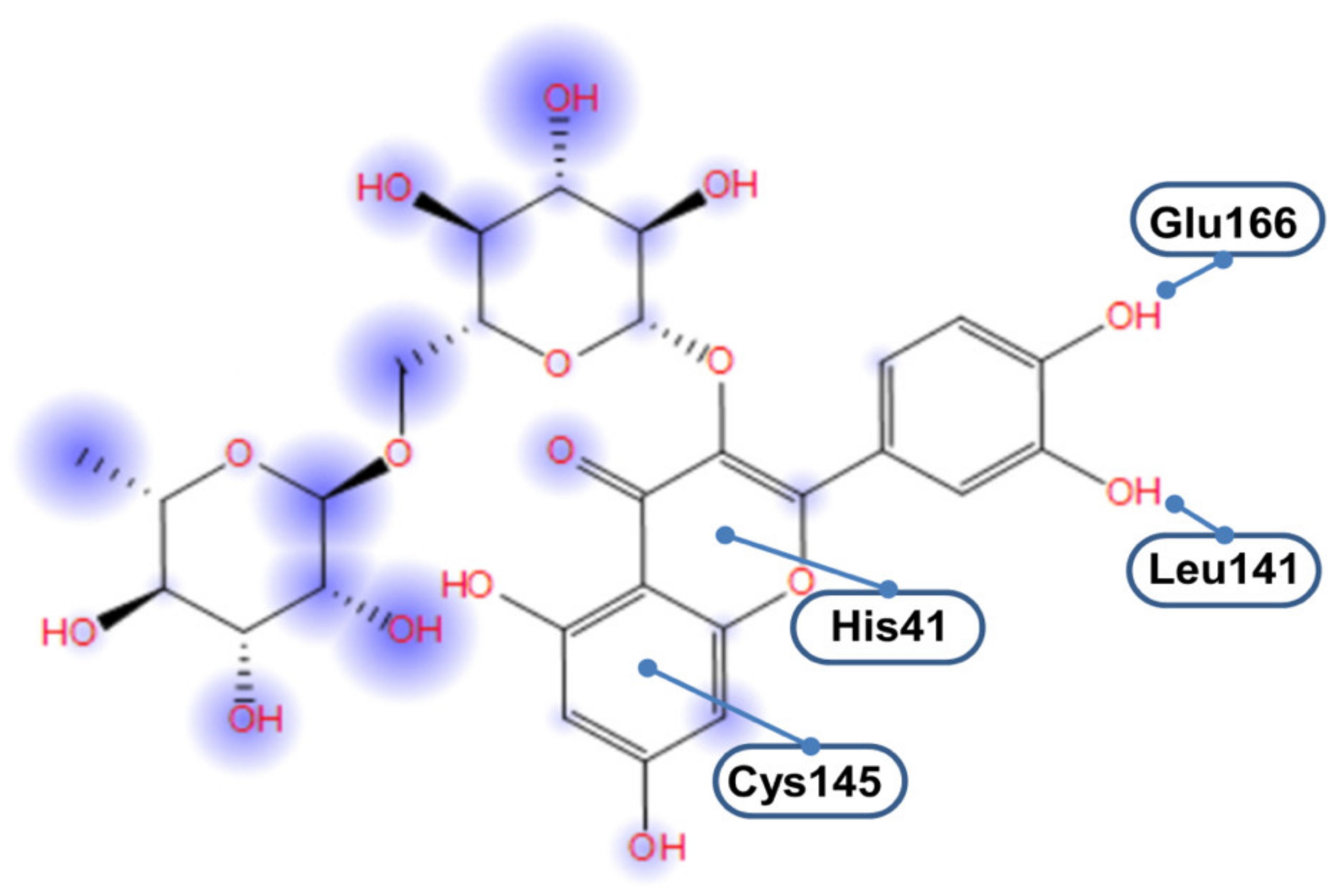
3.2. 3CLpro-Rsutin Interaction Takes Place in the Catalytic Site
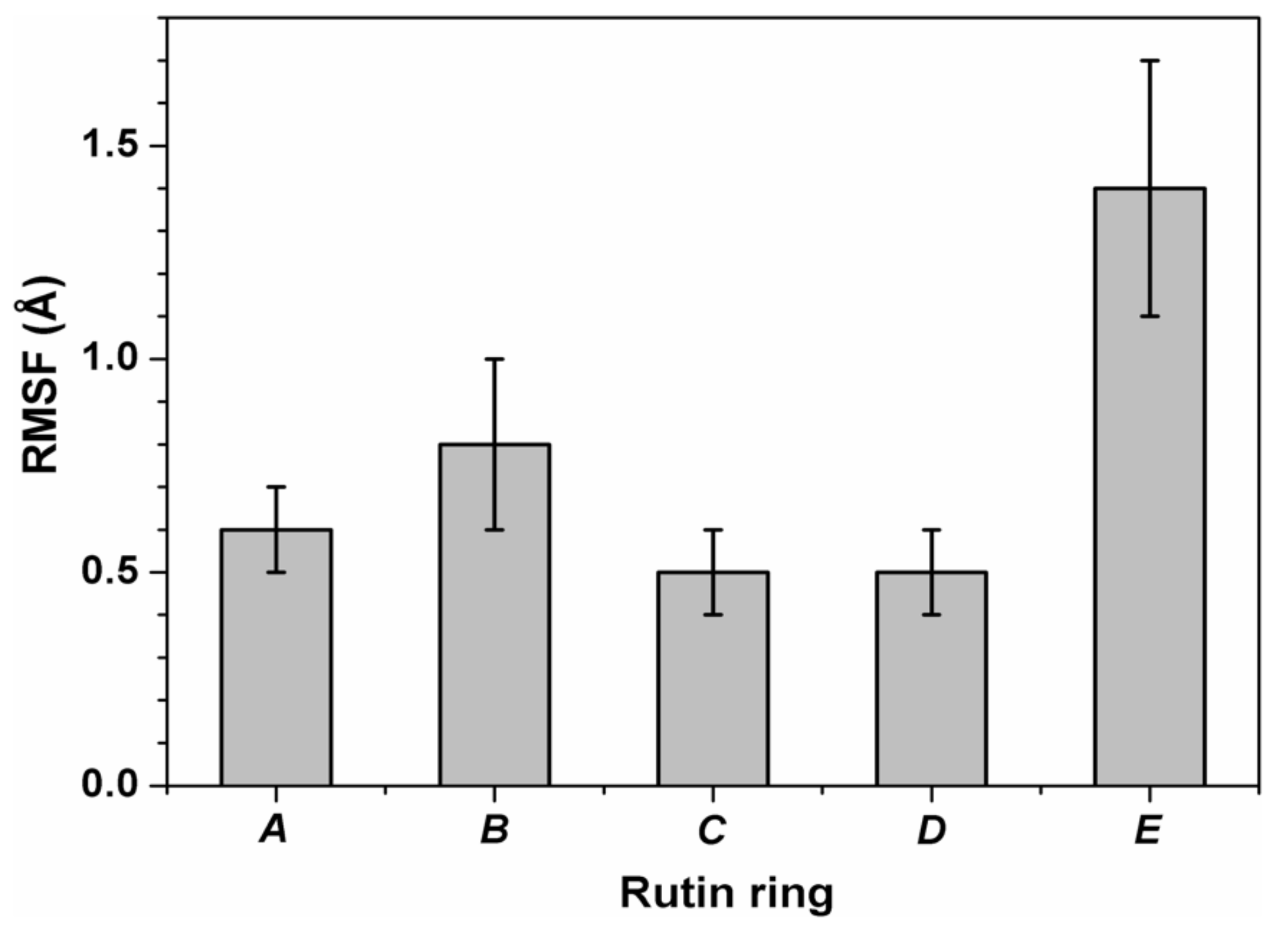
3.3. Binding of Quercetin Scaffold Is Not Hampered by a Bulky Adduct
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta BioMed. 2020, 91, 157–160. [Google Scholar]
- Painter, E.M.; Ussery, E.N.; Patel, A.; Hughes, M.M.; Zell, E.R.; Moulia, D.L.; Scharf, L.G.; Lynch, M.; Ritchey, M.D.; Toblin, R.L.; et al. Demographic Characteristics of Persons Vaccinated During the First Month of the COVID-19 Vaccination Program—United States, December 14, 2020–January 14, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Lee, K.H.; Yoon, S.; Nam, S.W.; Ryu, S.; Seong, D.; Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, J.H.; et al. Treatment of severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS), and coronavirus disease 2019 (COVID-19): A systematic review of in vitro, in vivo, and clinical trials. Theranostics 2021, 11, 1207–1231. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Tao, H.; Satyanarayanan, S.K.; Jin, K.; Su, H. A Comprehensive Summary of the Knowledge on COVID-19 Treatment. Aging Dis. 2021, 12, 155–191. [Google Scholar] [CrossRef] [PubMed]
- Muhseen, Z.; Hameed, A.; Al-Hasani, H.; Ahmad, S.; Li, G. Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules 2021, 26, 674. [Google Scholar] [CrossRef]
- Romeo, I.; Mesiti, F.; Lupia, A.; Alcaro, S. Current Updates on Naturally Occurring Compounds Recognizing SARS-CoV-2 Druggable Targets. Molecules 2021, 26, 632. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Dev, K.; Sourirajan, A. Antiviral activity of bioactive phytocompounds against coronavirus: An update. J. Virol. Methods 2021, 290, 114070. [Google Scholar] [CrossRef]
- Vougogiannopoulou, K.; Corona, A.; Tramontano, E.; Alexis, M.; Skaltsounis, A.-L. Natural and Nature-Derived Products Targeting Human Coronaviruses. Molecules 2021, 26, 448. [Google Scholar] [CrossRef]
- Latos-Brozio, M.; Masek, A. Structure-Activity Relationships Analysis of Monomeric and Polymeric Polyphenols (Quercetin, Rutin and Catechin) Obtained by Various Polymerization Methods. Chem. Biodivers. 2019, 16, e1900426. [Google Scholar] [CrossRef] [PubMed]
- Magar, R.T.; Sohng, J.K. A Review on Structure, Modifications and Structure-Activity Relation of Quercetin and Its Derivatives. J. Microbiol. Biotechnol. 2020, 30, 11–20. [Google Scholar] [CrossRef]
- Gullón, B.; Lú-Chau, T.A.; Moreira, M.T.; Lema, J.M.; Eibes, G. Rutin: A review on extraction, identification and purification methods, biological activities and approaches to enhance its bioavailability. Trends Food Sci. Technol. 2017, 67, 220–235. [Google Scholar] [CrossRef]
- Lide, D.R.; Milne, G.W.A. Handbook of data on organic compounds, 3rd ed.; CRC Press: Boca Raton, FL, USA, 1994; pp. 1–1570. [Google Scholar]
- Srinivas, K.; King, J.W.; Howard, L.R.; Monrad, J.K. Solubility and solution thermodynamic properties of quercetin and quercetin dihydrate in subcritical water. J. Food Eng. 2010, 100, 208–218. [Google Scholar] [CrossRef]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Reyburn, H.T.; Rizzuti, B.; Velazquez-Campoy, A. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Abian, O.; Vega, S.; Sancho, J.; Velázquez-Campoy, A. Allosteric Inhibitors of the NS3 Protease from the Hepatitis C Virus. PLoS ONE 2013, 8, e69773. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, J.; Latorre, P.; Carrodeguas, J.A.; Velázquez-Campoy, A.; Sancho, J.; Lopez-Buesa, P. Inhibition of Pig Phosphoenolpyruvate Carboxykinase Isoenzymes by 3-Mercaptopicolinic Acid and Novel Inhibitors. PLoS ONE 2016, 11, e0159002. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Villanueva, R.; Romero-Tamayo, S.; Laplaza, R.; Martínez-Olivan, J.; Velázquez-Campoy, A.; Sancho, J.; Ferreira, P.; Medina, M. Redox- and Ligand Binding-Dependent Conformational Ensembles in the Human Apoptosis-Inducing Factor Regulate Its Pro-Life and Cell Death Functions. Antioxid. Redox Signal. 2019, 30, 2013–2029. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Salillas, S.; Velázquez-Campoy, A.; Angarica, V.E.; Fillat, M.F.; Sancho, J.; Lanas, A. Identifying potential novel drugs against Helicobacter pylori by targeting the essential response regulator HsrA. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Savov, V.M.; Galabov, A.S.; Tantcheva, L.P.; Mileva, M.M.; Pavlova, E.L.; Stoeva, E.S.; Braykova, A.A. Effects of rutin and quercetin on monooxygenase activities in experimental influenza virus infection. Exp. Toxicol. Pathol. 2006, 58, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, H.; Kim, S.; Shin, D.H.; Kim, M. Characteristics of flavonoids as potent MERS-CoV 3C-like protease inhibitors. Chem. Biol. Drug Des. 2019, 94, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.K.; Chen, S.Q.; Li, X.; Zeng, S. Quantitative regioselectivity of glucuronidation of quercetin by recombinant UDP-glucuronosyltransferases 1A9 and 1A3 using enzymatic kinetic parameters. Xenobiotica 2005, 35, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Matsukawa, N.; Mineo, H.; Chiji, H.; Hara, H. A Soluble Flavonoid-glycoside, αG-Rutin, Is Absorbed as Glycosides in the Isolated Gastric and Intestinal Mucosa. Biosci. Biotechnol. Biochem. 2004, 68, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Pedriali, C.A.; Fernandes, A.U.; Bernusso, L.D.C.; Polakiewicz, B. The synthesis of a water-soluble derivative of rutin as an antiradical agent. Quim. Nova 2008, 31, 2147–2151. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Choi, J.M.; Cho, E.; Jeong, D.; Shinde, V.V.; Kim, H.; Choi, Y.; Jung, S. Enhancement of Solubility and Bioavailability of Quercetin by Inclusion Complexation with the Cavity of Mono-6-deoxy-6-aminoethylamino-β-cyclodextrin. Bull. Korean Chem. Soc. 2017, 38, 880–889. [Google Scholar] [CrossRef]
- Iacopetta, D.; Grande, F.; Caruso, A.; Mordocco, R.A.; Plutino, M.R.; Scrivano, L.; Ceramella, J.; Muià, N.; Saturnino, C.; Puoci, F.; et al. New insights for the use of quercetin analogs in cancer treatment. Future Med. Chem. 2017, 9, 2011–2028. [Google Scholar] [CrossRef] [PubMed]
- Nettore, I.C.; Rocca, C.; Mancino, G.; Albano, L.; Amelio, D.; Grande, F.; Puoci, F.; Pasqua, T.; Desiderio, S.; Mazza, R.; et al. Quercetin and its derivative Q2 modulate chromatin dynamics in adipogenesis and Q2 prevents obesity and metabolic disorders in rats. J. Nutr. Biochem. 2019, 69, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mittal, A.; Patel, K.; Gatuz, J.L.; Truong, L.; Torres, J.; Mulhearn, D.C.; Johnson, M.E. Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorganic Med. Chem. 2014, 22, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a La-marckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Komoto, J.; Yamada, T.; Watanabe, K.; Takusagawa, F. Crystal Structure of Human Prostaglandin F Synthase (AKR1C3). Biochemistry 2004, 43, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castaño, P.; Rizzuti, B.; Pey, Á.L.; Soubeyran, P.; Vidal, M.; Urrutia, R.; Iovanna, J.L.; Neira, J.L. Intrinsically disordered chromatin protein NUPR1 binds to the C-terminal region of Polycomb RING1B. Proc. Natl. Acad. Sci. USA 2017, 114, E6332–E6341. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Guzzi, R.; Rizzuti, B.; Bartucci, R. Dynamics and Binding Affinity of Spin-Labeled Stearic Acids in β-Lactoglobulin: Evidences from EPR Spectroscopy and Molecular Dynamics Simulation. J. Phys. Chem. B 2012, 116, 11608–11615. [Google Scholar] [CrossRef]
- Neira, J.L.; Rizzuti, B.; Iovanna, J.L. Determinants of the pKa values of ionizable residues in an intrinsically disordered protein. Arch. Biochem. Biophys. 2016, 598, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, A.; Rizzuti, B.; Guzzi, R. Stereoselective and domain-specific effects of ibuprofen on the thermal stability of human serum albumin. Eur. J. Pharm. Sci. 2018, 112, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CLpro. Biochemistry 2004, 43, 4906–4912. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Rizzuti, B.; Grande, F. Virtual screening in drug discovery: A precious tool for a still-demanding challenge. In Protein Homeostasis Diseases; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 309–327. [Google Scholar]
- Komatsu, T.S.; Okimoto, N.; Koyama, Y.M.; Hirano, Y.; Morimoto, G.; Ohno, Y.; Taiji, M. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Avasthi, K.; Shukla, L.; Kant, R.; Ravikumar, K. Folded conformations due to arene interactions in dissymmetric and symmetric butylidene-linker models based on pyrazolo[3,4-d]pyrimidine, purine and 7-deazapurine. Acta Crystallogr. Sect. C Struct. Chem. 2014, 70, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.-H.; De Vaca, I.C.; Liosi, M.-E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533. [Google Scholar] [CrossRef]
- Chodera, J.; Lee, A.A.; London, N.; Von Delft, F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020, 12, 581. [Google Scholar] [CrossRef]
- Chua, L.S. A review on plant-based rutin extraction methods and its pharmacological activities. J. Ethnopharmacol. 2013, 150, 805–817. [Google Scholar] [CrossRef]
- Matsuo, M.; Sasaki, N.; Saga, K.; Kaneko, T. Cytotoxicity of Flavonoids toward Cultured Normal Human Cells. Biol. Pharm. Bull. 2005, 28, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Iriti, M.; Kubina, R.; Cochis, A.; Sorrentino, R.; Varoni, E.M.; Kabała-Dzik, A.; Azzimonti, B.; Dziedzic, A.; Rimondini, L.; Wojtyczka, R.D. Rutin, a Quercetin Glycoside, Restores Chemosensitivity in Human Breast Cancer Cells. Phytother. Res. 2017, 31, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Al-Zahrani, A.A. Rutin as a Promising Inhibitor of Main Protease and Other Protein Targets of COVID-19: In Silico Study. Nat. Prod. Commun. 2020, 15. [Google Scholar] [CrossRef]
- Puttaswamy, H.; Gowtham, H.G.; Ojha, M.D.; Yadav, A.; Choudhir, G.; Raguraman, V.; Kongkham, B.; Selvaraju, K.; Shareef, S.; Gehlot, P.; et al. In silico studies evidenced the role of structurally diverse plant secondary metabolites in reducing SARS-CoV-2 pathogenesis. Sci. Rep. 2020, 10, 1–24. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, L.; Zhang, X.; Zhang, Q.; Yang, Z.; Liu, Y.; Wei, S.; Liu, W. Discovery of Potential Flavonoid Inhibitors Against COVID-19 3CL Proteinase Based on Virtual Screening Strategy. Front. Mol. Biosci. 2020, 7, 556481. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Rajput, V.S.; Nagpal, P.; Kukrety, H.; Grover, S.; Grover, A. Dual inhibition of SARS-CoV-2 spike and main protease through a repurposed drug, rutin. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Huynh, T.; Wang, H.; Luan, B. Structure-based lead optimization of herbal medicine rutin for inhibiting SARS-CoV-2’s main protease. Phys. Chem. Chem. Phys. 2020, 22, 25335–25343. [Google Scholar] [CrossRef] [PubMed]
- Shivanika, C.; Kumar, S.D.; Ragunathan, V.; Tiwari, P.; Sumitha, A.; Devi, P.B. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020, 83, 1–27. [Google Scholar] [CrossRef]
- Verma, S.; Pandey, A.K. Factual insights of the allosteric inhibition mechanism of SARS-CoV-2 main protease by quercetin: An in silico analysis. 3 Biotech 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Woo, H.-J.; Kang, H.-K.; Nguyen, V.D.; Kim, Y.-M.; Kim, D.-W.; Ahn, S.-A.; Xia, Y.; Kim, D. Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol. Lett. 2012, 34, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, J.; Luo, C.; Liu, H.; Xu, W.; Chen, G.; Liew, O.W.; Zhu, W.; Puah, C.M.; Shen, X.; et al. Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: Structure–activity relationship studies reveal salient pharmacophore features. Bioorganic Med. Chem. 2006, 14, 8295–8306. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040375
Rizzuti B, Grande F, Conforti F, Jimenez-Alesanco A, Ceballos-Laita L, Ortega-Alarcon D, Vega S, Reyburn HT, Abian O, Velazquez-Campoy A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines. 2021; 9(4):375. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040375
Chicago/Turabian StyleRizzuti, Bruno, Fedora Grande, Filomena Conforti, Ana Jimenez-Alesanco, Laura Ceballos-Laita, David Ortega-Alarcon, Sonia Vega, Hugh T. Reyburn, Olga Abian, and Adrian Velazquez-Campoy. 2021. "Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs" Biomedicines 9, no. 4: 375. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040375