
SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Aβ: An Early but Transient Response Rescued by Melatonin

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Cell Cultures
2.3. Quantitative Real-Time Polymerase Chain Reaction
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Western Blot
2.6. Immunoprecipitation (IP) & SIRT1 Activity Assay
2.7. Immunocytochemistry
2.8. Statistical Analysis
3. Results
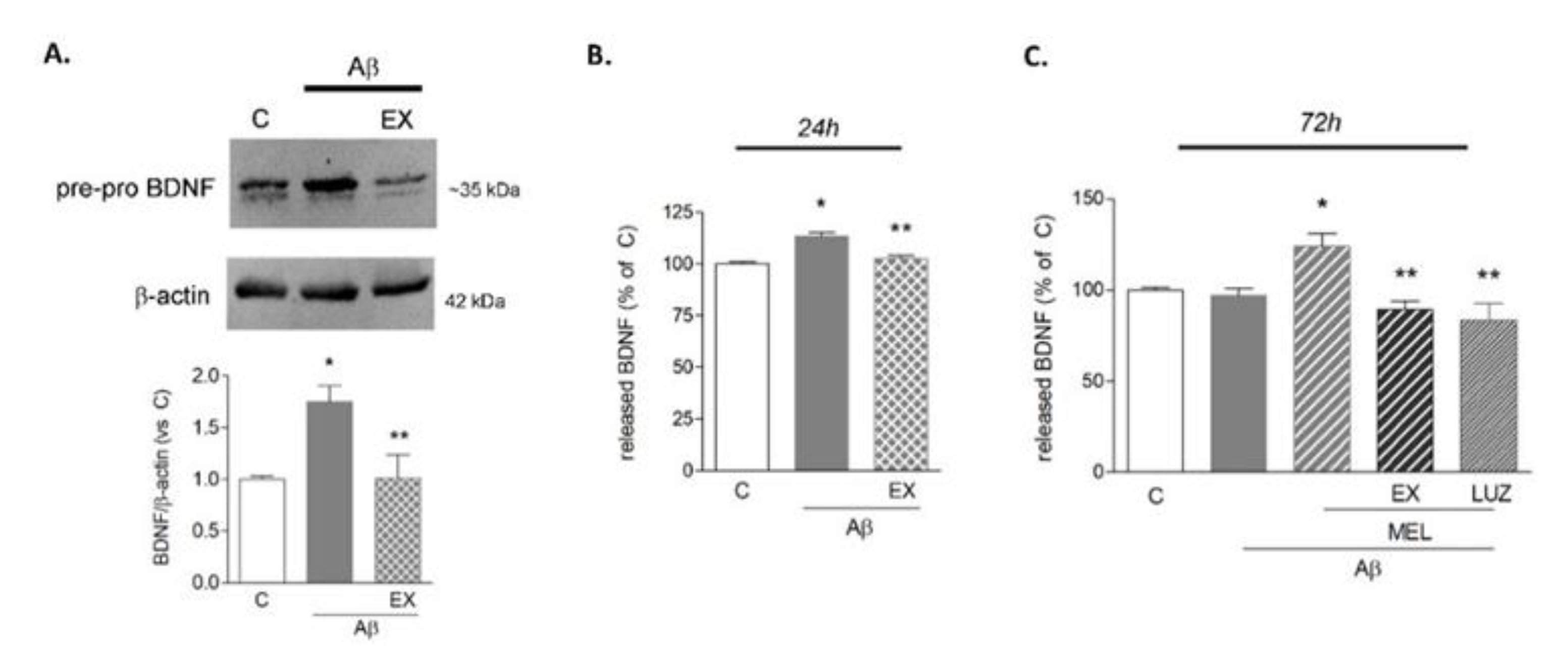
3.1. Microglia Respond to Aβ42 with Transient SIRT1-Mediated BDNF Upregulation That Is Prolonged by Melatonin
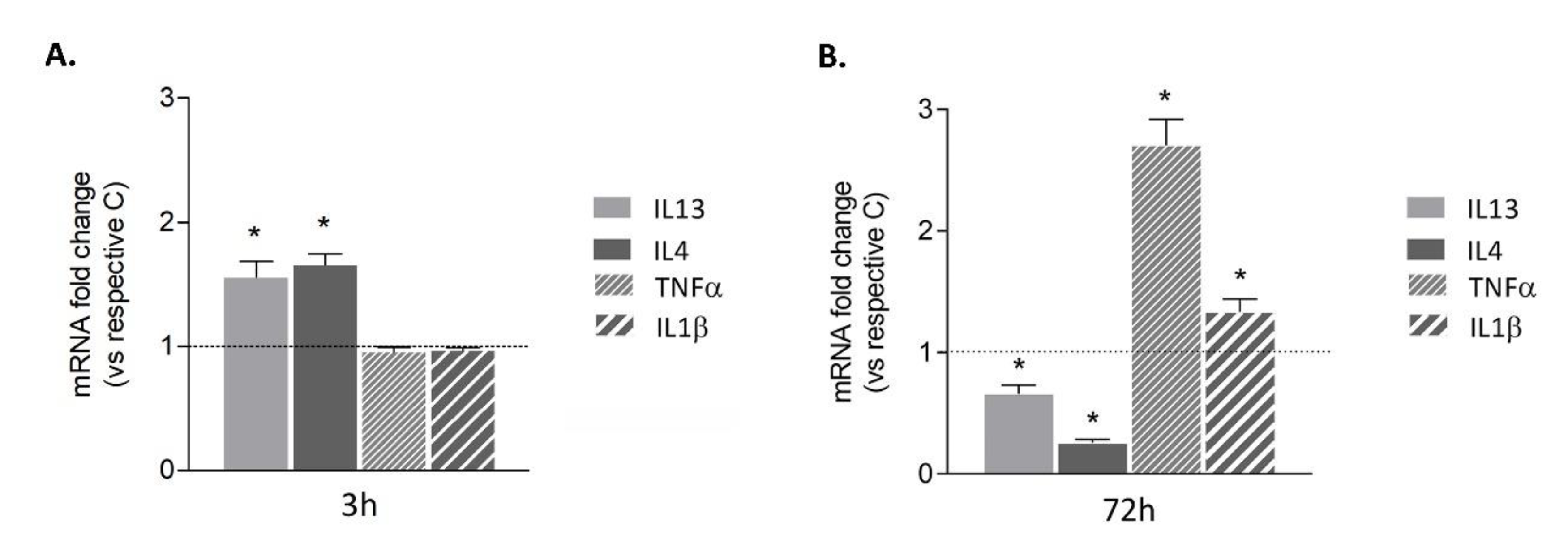
3.2. Microglia Undergo a Pro-Inflammatory Switch Following Prolonged Aβ42 Exposure
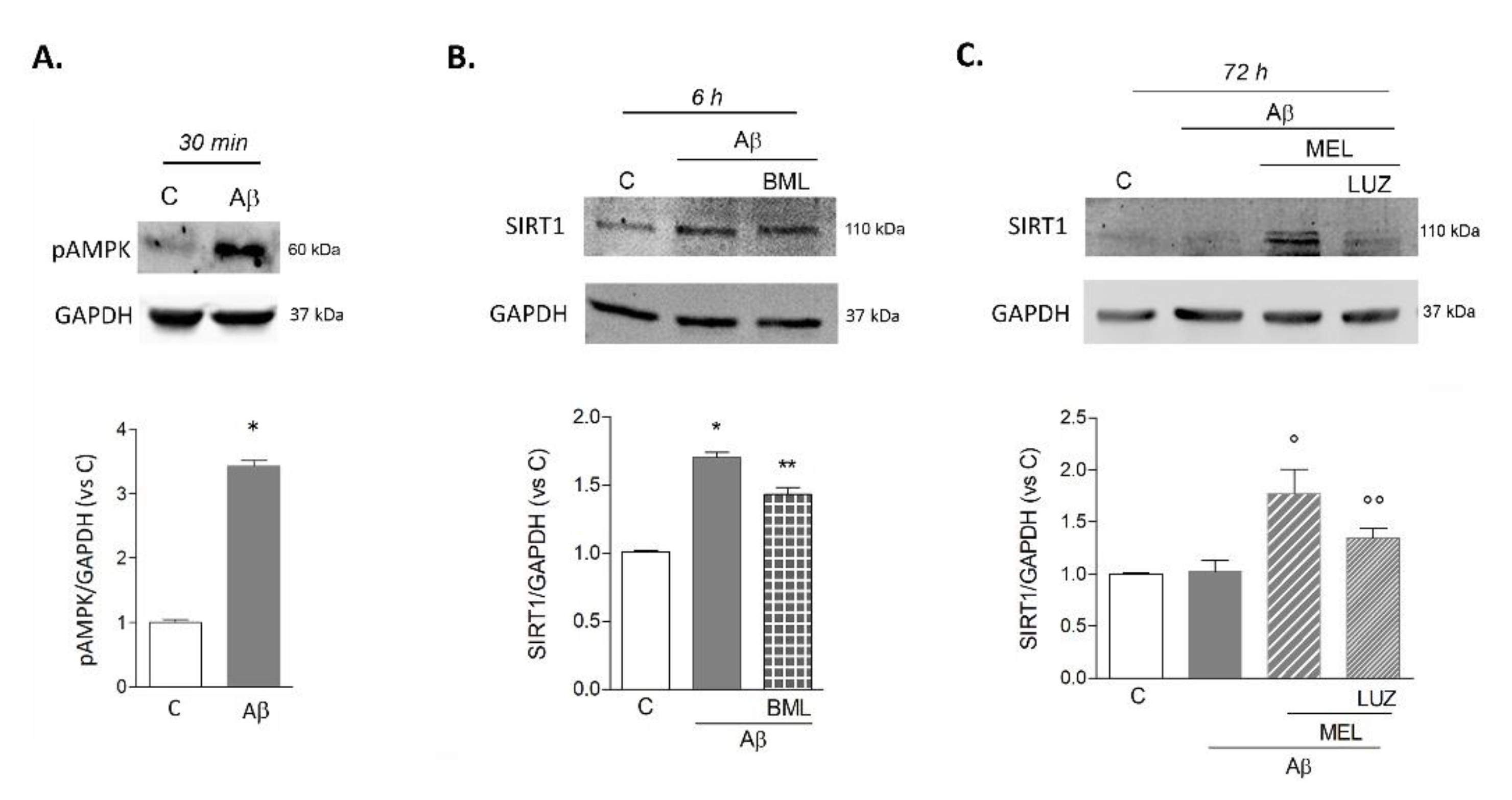
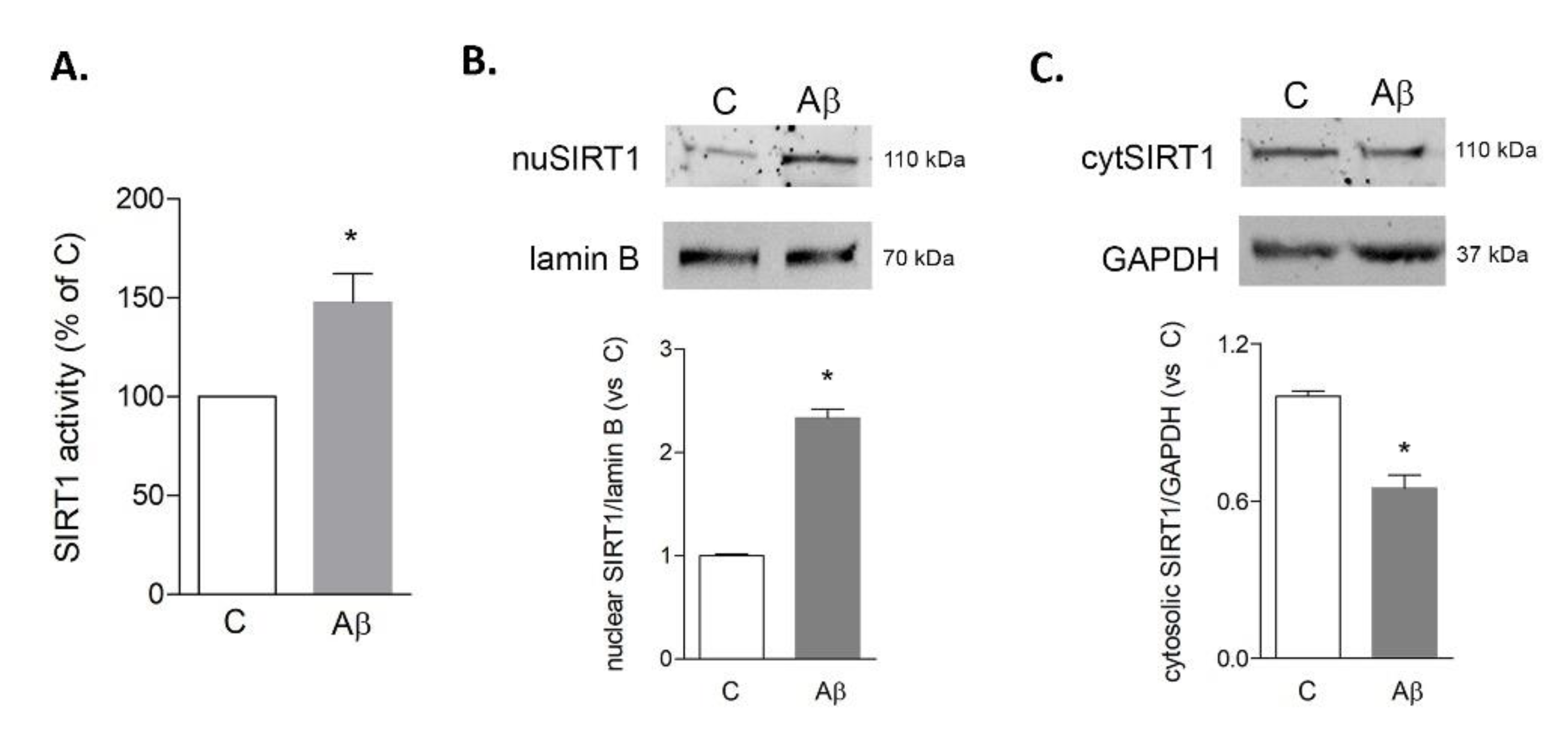
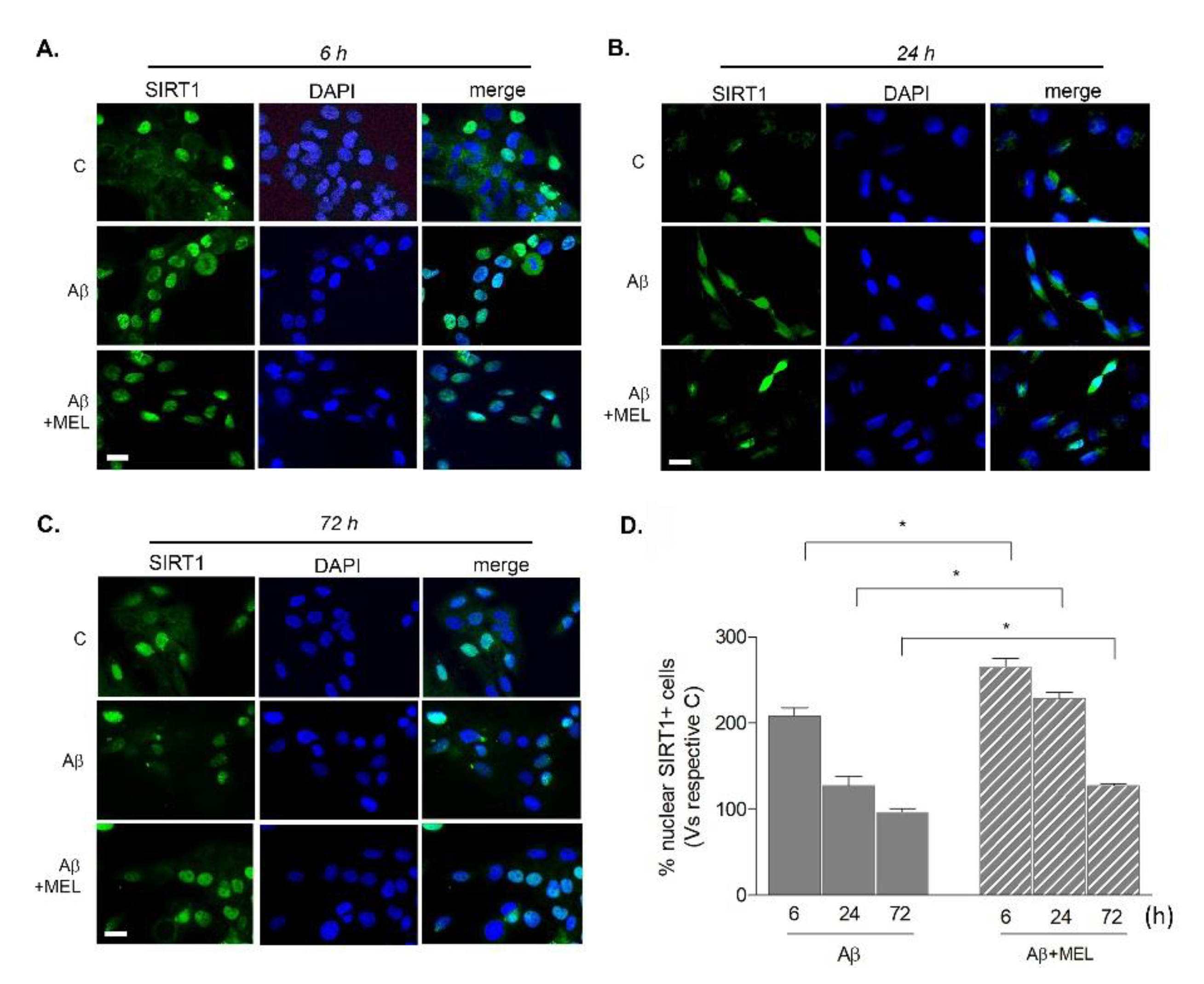
3.3. Melatonin Prolongs Transient Aβ42-Induced Upregulation of SIRT1 Activity and Expression
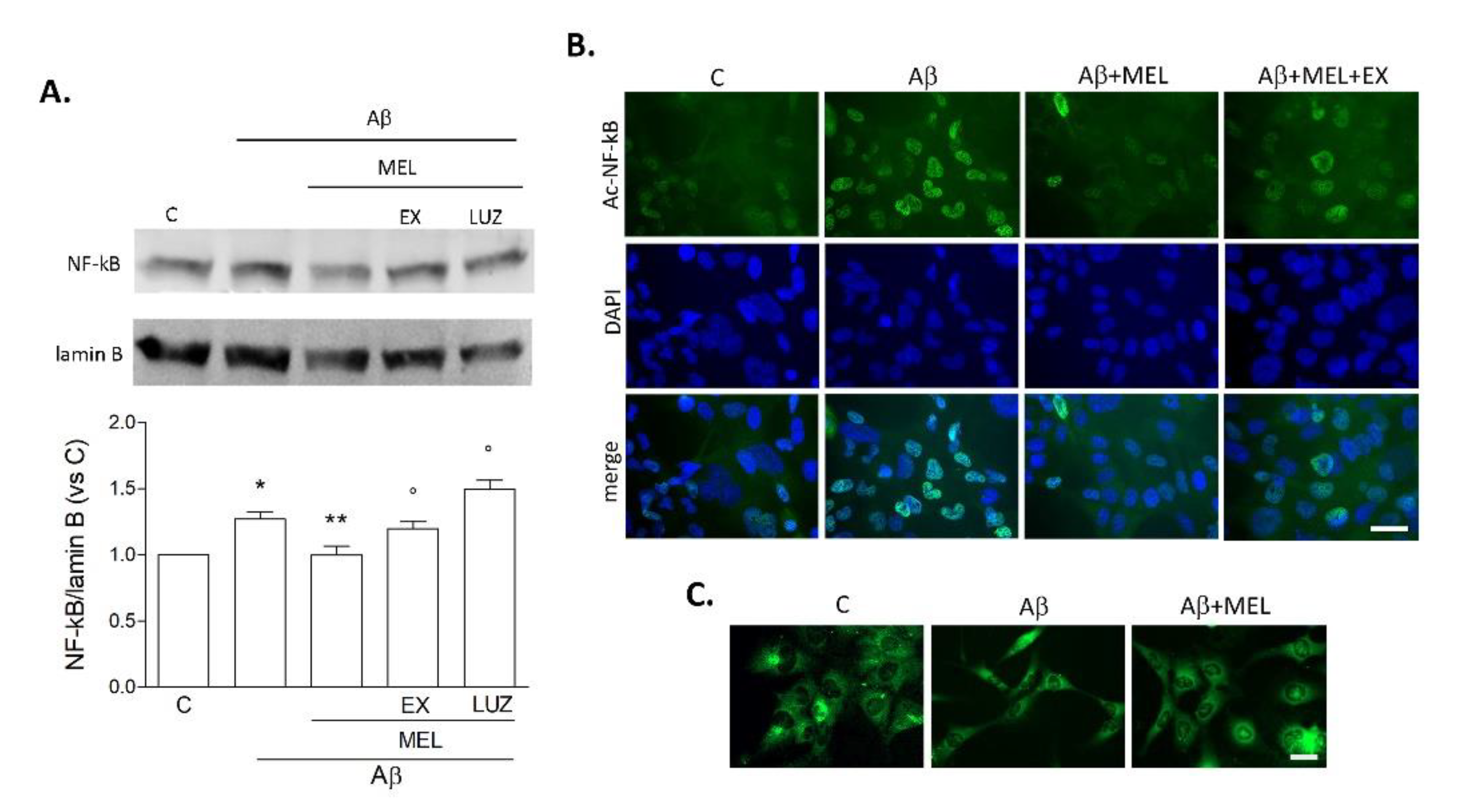
3.4. Melatonin Reduces Microglial NF-kB Expression Induced by Prolonged Aβ42 Exposure
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-beta and tau with synaptic and axonal loss in Alzheimer’s disease. Brain J. Neurol. 2020. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Caruso, G.; Spampinato, S.F.; Cardaci, V.; Caraci, F.; Sortino, M.A.; Merlo, S. Beta-amyloid and oxidative stress: Perspectives in drug development. Curr. Pharm. Des. 2019, 25, 4771–4781. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral Abeta in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell. Mol. Life Sci. CMLS 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, S.; Spampinato, S.F.; Caruso, G.I.; Sortino, M.A. The ambiguous role of microglia in abeta toxicity: Chances for therapeutic intervention. Curr. Neuropharmacol. 2020, 18, 446–455. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Beneventano, M.; Sortino, M.A. The contribution of microglia to early synaptic compensatory responses that precede beta-amyloid-induced neuronal death. Sci. Rep. 2018, 8, 7297. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. TREM2 enables amyloid beta clearance by microglia. Cell Res. 2015, 25, 535–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittington, R.A.; Planel, E.; Terrando, N. Impaired resolution of inflammation in Alzheimer’s disease: A review. Front. Immunol. 2017, 8, 1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.; Bromberg, E.; de Vries, E.F.J. Brain-derived neurotrophic factor in brain disorders: Focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Wong, S.Y.; Tang, B.L. SIRT1 as a therapeutic target for Alzheimer’s disease. Rev. Neurosci. 2016, 27, 813–825. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013, 23, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, S.; Gan, L.; Vosler, P.S.; Gao, Y.; Zigmond, M.J.; Chen, J. Protective effects and mechanisms of sirtuins in the nervous system. Prog. Neurobiol. 2011, 95, 373–395. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.-F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 Protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [Green Version]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromolecular Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef]
- Mahmood, D. Pleiotropic effects of melatonin. Drug Res. 2019, 69, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B. Protective roles of melatonin against the amyloid-dependent development of Alzheimer’s disease: A critical review. Pharmacol. Res. 2018, 134, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.M.; Schamne, M.G.; Sampaio, T.B.; Pertile, R.A.; Fernandes, P.A.; Markus, R.P.; Prediger, R.D. Melatoninergic system in Parkinson’s disease: From neuroprotection to the management of motor and nonmotor symptoms. Oxidative Med. Cell. Longev. 2016, 2016, 3472032. [Google Scholar] [CrossRef] [Green Version]
- Paprocka, J.; Kijonka, M.; Rzepka, B.; Sokol, M. Melatonin in hypoxic-ischemic brain injury in term and preterm babies. Int. J. Endocrinol. 2019, 2019, 9626715. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhou, Z.; Gao, S.; Guo, Y.; Gao, K.; Wang, H.; Dang, X. melatonin inhibits neural cell apoptosis and promotes locomotor recovery via activation of the Wnt/beta-catenin signaling pathway after spinal cord injury. Neurochem. Res. 2017, 42, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.; Gogenur, I.; Rosenberg, J.; Reiter, R.J. The safety of melatonin in humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Chinchalongporn, V.; Govitrapong, P.; Reiter, R.J. The role of melatonin in targeting cell signaling pathways in neurodegeneration. Ann. N. Y. Acad. Sci. 2019, 1443, 75–96. [Google Scholar] [CrossRef]
- Emet, M.; Ozcan, H.; Ozel, L.; Yayla, M.; Halici, Z.; Hacimuftuoglu, A. A review of melatonin, its receptors and drugs. Eurasian J. Med. 2016, 48, 135–141. [Google Scholar] [CrossRef]
- Ng, K.Y.; Leong, M.K.; Liang, H.; Paxinos, G. Melatonin receptors: Distribution in mammalian brain and their respective putative functions. Brain Struct. Funct. 2017, 222, 2921–2939. [Google Scholar] [CrossRef]
- Alghamdi, B.S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar] [CrossRef]
- Boutin, J.A.; Ferry, G. Is there sufficient evidence that the melatonin binding site MT3 Is quinone reductase 2? J. Pharmacol. Exp. Ther. 2019, 368, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, A.N. Nuclear melatonin receptors. Biochemistry 2001, 66, 19–26. [Google Scholar] [CrossRef]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 175–200. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Abeta production/clearance balance and Abeta neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Shi, Y.; Fang, Y.Y.; Wei, Y.P.; Jiang, Q.; Zeng, P.; Tang, N.; Lu, Y.; Tian, Q. Melatonin in synaptic impairments of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 911–926. [Google Scholar] [CrossRef]
- Mihardja, M.; Roy, J.; Wong, K.Y.; Aquili, L.; Heng, B.C.; Chan, Y.S.; Fung, M.L.; Lim, L.W. Therapeutic potential of neurogenesis and melatonin regulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2020, 1478, 43–62. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: A 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef]
- Merlo, S.; Sortino, M.A. Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell. Neurosci. 2012, 49, 423–429. [Google Scholar] [CrossRef]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia mediated neuroinflammation: Focus on PI3K modulation. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef]
- Gupta, N.; Shyamasundar, S.; Patnala, R.; Karthikeyan, A.; Arumugam, T.V.; Ling, E.A.; Dheen, S.T. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin. Ther. Targets 2018, 22, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain J. Neurol. 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain J. Neurol. 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET imaging of microglial activation: Implications for microglial therapeutics in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Gyengesi, E.; Munch, G. In search of an anti-inflammatory drug for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 131–132. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Capani, F.; Sortino, M.A. Early beta-Amyloid-induced synaptic dysfunction is counteracted by estrogen in organotypic hippocampal cultures. Curr. Alzheimer Res. 2016, 13, 631–640. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflammation 2018, 15, 259. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): A systematic review and meta-analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Laske, C.; Stellos, K.; Hoffmann, N.; Stransky, E.; Straten, G.; Eschweiler, G.W.; Leyhe, T. Higher BDNF serum levels predict slower cognitive decline in Alzheimer’s disease patients. Int. J. Neuropsychopharmacol. 2011, 14, 399–404. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Wittorf, A.; Richartz, E.; Bartels, M.; Buchkremer, G.; Schott, K. Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1217–1224. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Negishi, T.; Inoue, M.; Tashiro, T.; Tabira, T.; Kimura, N. Sendai virus vector-mediated brain-derived neurotrophic factor expression ameliorates memory deficits and synaptic degeneration in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. Res. 2012, 90, 981–989. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagai, N.; Tanaka, A.; Sato, A.; Uchiumi, F.; Tanuma, S.I. low levels of brain-derived neurotrophic factor trigger self-aggregated amyloid beta-induced neuronal cell death in an alzheimer’s cell model. Biol. Pharm. Bull. 2020, 43, 1073–1080. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Yarkov, R.S.; Mishchenko, T.A.; Krut, V.G.; Gavrish, M.S.; Epifanova, E.A.; Babaev, A.A.; Vedunova, M.V. Brain-Derived Neurotrophic Factor (BDNF) preserves the functional integrity of neural networks in the beta-amyloidopathy model in vitro. Front. Cell Dev. Biol. 2020, 8, 582. [Google Scholar] [CrossRef]
- Arancibia, S.; Silhol, M.; Mouliere, F.; Meffre, J.; Hollinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Pan, B.S.; Tsai, S.F.; Chiang, Y.T.; Huang, B.M.; Mo, F.E.; Kuo, Y.M. BDNF reverses aging-related microglial activation. J. Neuroinflammation 2020, 17, 210. [Google Scholar] [CrossRef]
- Hisahara, S.; Chiba, S.; Matsumoto, H.; Tanno, M.; Yagi, H.; Shimohama, S.; Sato, M.; Horio, Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 15599–15604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- Hardeland, R. Aging, melatonin, and the pro- and anti-inflammatory networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Michan, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Graff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Lee, H.G.; Camins, A.; Pallas, M.; Casadesus, G.; Smith, M.A.; Zhu, X. The sirtuin pathway in ageing and Alzheimer disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2011, 10, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Elibol, B.; Kilic, U. High Levels of SIRT1 expression as a protective mechanism against disease-related conditions. Front. Endocrinol. 2018, 9, 614. [Google Scholar] [CrossRef]
- Ng, F.; Wijaya, L.; Tang, B.L. SIRT1 in the brain—Connections with aging-associated disorders and lifespan. Front. Cell. Neurosci. 2015, 9, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Gimenez-Llort, L.; Sarkis, C.; Pallas, M.; Sanfeliu, C. SIRT1 Overexpression in mouse hippocampus induces cognitive enhancement through proteostatic and neurotrophic mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Kumar, R.; Chaterjee, P.; Sharma, P.K.; Singh, A.K.; Gupta, A.; Gill, K.; Tripathi, M.; Dey, A.B.; Dey, S. Sirtuin1: A promising serum protein marker for early detection of Alzheimer’s disease. PLoS ONE 2013, 8, e61560. [Google Scholar] [CrossRef] [Green Version]
- Pukhalskaia, A.E.; Dyatlova, A.S.; Linkova, N.S.; Kozlov, K.L.; Kvetnaia, T.V.; Koroleva, M.V.; Kvetnoy, I.M. Sirtuins as possible predictors of aging and Alzheimer’s disease development: Verification in the hippocampus and saliva. Bull. Exp. Biol. Med. 2020, 169, 821–824. [Google Scholar] [CrossRef]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-beta genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.Z.; Zheng, L.J.; Shen, J.; Li, X.Y.; Zhang, Q.; Bai, X.; Wang, Q.S.; Ji, J.G. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural. Regen. Res. 2018, 13, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, N.; Andolina, G.; Cannata, A.; Costanzo, M.G.; Rizzo, V.; Curro, M.; Ientile, R.; Caccamo, D. Is melatonin the cornucopia of the 21st century? Antioxidants 2020, 9, 1088. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Cappoli, N.; Mezzogori, D.; Tabolacci, E.; Coletta, I.; Navarra, P.; Pani, G.; Dello Russo, C. The mTOR kinase inhibitor rapamycin enhances the expression and release of pro-inflammatory cytokine interleukin 6 modulating the activation of human microglial cells. EXCLI J. 2019, 18, 779–798. [Google Scholar] [CrossRef]
- Akhter, R.; Shao, Y.; Formica, S.; Khrestian, M.; Bekris, L.M. TREM2 alters the phagocytic, apoptotic and inflammatory response to Abeta42 in HMC3 cells. Mol. Immunol. 2021, 131, 171–179. [Google Scholar] [CrossRef]
- Tan, H.Y.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Pharmacological effects of melatonin as neuroprotectant in rodent model: A review on the current biological evidence. Cell. Mol. Neurobiol. 2020, 40, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.K.; Srivastava, G.; Agrawal, S.; Jain, S.K.; Singh, M.P. Melatonin as a neuroprotective agent in the rodent models of Parkinson’s disease: Is it all set to irrefutable clinical translation? Mol. Neurobiol. 2012, 45, 186–199. [Google Scholar] [CrossRef]
- Merlo, S.; Luaces, J.P.; Spampinato, S.F.; Toro-Urrego, N.; Caruso, G.I.; D’Amico, F.; Capani, F.; Sortino, M.A. SIRT1 mediates melatonin’s effects on microglial activation in hypoxia: In vitro and in vivo evidence. Biomolecules 2020, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bald, E.M.; Nance, C.S.; Schultz, J.L. Melatonin may slow disease progression in amyotrophic lateral sclerosis: Findings from the pooled resource open-access ALS clinic trials database. Muscle Nerve 2021. [Google Scholar] [CrossRef]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef]
- Lee, J.G.; Woo, Y.S.; Park, S.W.; Seog, D.H.; Seo, M.K.; Bahk, W.M. The neuroprotective effects of melatonin: Possible role in the pathophysiology of neuropsychiatric disease. Brain Sci. 2019, 9, 285. [Google Scholar] [CrossRef] [Green Version]
- Nous, A.; Engelborghs, S.; Smolders, I. Melatonin levels in the Alzheimer’s disease continuum: A systematic review. Alzheimer’s Res. Ther. 2021, 13, 52. [Google Scholar] [CrossRef]
- Labban, S.; Alghamdi, B.S.; Alshehri, F.S.; Kurdi, M. Effects of melatonin and resveratrol on recognition memory and passive avoidance performance in a mouse model of Alzheimer’s disease. Behav. Brain. Res. 2021, 402, 113100. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Hsu, W.L.; Ma, Y.L.; Lee, E.H.Y. Melatonin induction of APP intracellular domain 50 SUMOylation alleviates AD through enhanced transcriptional activation and abeta degradation. Mol. Ther. 2021, 29, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Qiu, X.; Wang, Y.; Liu, J.; Li, Q.; Jiang, H.; Li, S.; Song, C. Long-term oral melatonin alleviates memory deficits, reduces amyloid-beta deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice. Neurosci. Lett. 2020, 735, 135192. [Google Scholar] [CrossRef]
- Jurgenson, M.; Zharkovskaja, T.; Noortoots, A.; Morozova, M.; Beniashvili, A.; Zapolski, M.; Zharkovsky, A. Effects of the drug combination memantine and melatonin on impaired memory and brain neuronal deficits in an amyloid-predominant mouse model of Alzheimer’s disease. J. Pharm. Pharmacol. 2019, 71, 1695–1705. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, G.; Liu, L.; Xu, S. Suppressive effects of melatonin on amyloid-beta-induced glial activation in rat hippocampus. Arch. Med Res. 2007, 38, 284–290. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer’s Disease? CNS Neurosci. Ther. 2019, 25, 5–13. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Cat. No. |
|---|---|---|
| BDNF | Hs_BDNF_1_SG QuantiTect Primer Assay | QT00235368 |
| IL-13 | Hs_IL13_1_SG QuantiTect Primer Assay | QT00000511 |
| IL-4 | Hs_IL4_1_SG QuantiTect Primer Assay | QT00012565 |
| TNFα | Hs_TNF_1_SG QuantiTect Primer Assay | QT00029162 |
| IL-1β | Hs_IL1B_1_SG QuantiTect Primer Assay | QT00021385 |
| RPLP0 | Hs_RPLP0_1_SG QuantiTect Primer Assay | QT00075012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations All authors have read and agreed to the published version of the manuscript.. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, G.I.; Spampinato, S.F.; Costantino, G.; Merlo, S.; Sortino, M.A. SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Aβ: An Early but Transient Response Rescued by Melatonin. Biomedicines 2021, 9, 466. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050466
Caruso GI, Spampinato SF, Costantino G, Merlo S, Sortino MA. SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Aβ: An Early but Transient Response Rescued by Melatonin. Biomedicines. 2021; 9(5):466. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050466
Chicago/Turabian StyleCaruso, Grazia Ilaria, Simona Federica Spampinato, Giuseppe Costantino, Sara Merlo, and Maria Angela Sortino. 2021. "SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Aβ: An Early but Transient Response Rescued by Melatonin" Biomedicines 9, no. 5: 466. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050466