
Fatty Acid-Binding Proteins Aggravate Cerebral Ischemia-Reperfusion Injury in Mice

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgical Procedures Used for Establishing Transmit Middle Cerebral Artery Occlusion (Tmcao) and Reperfusion
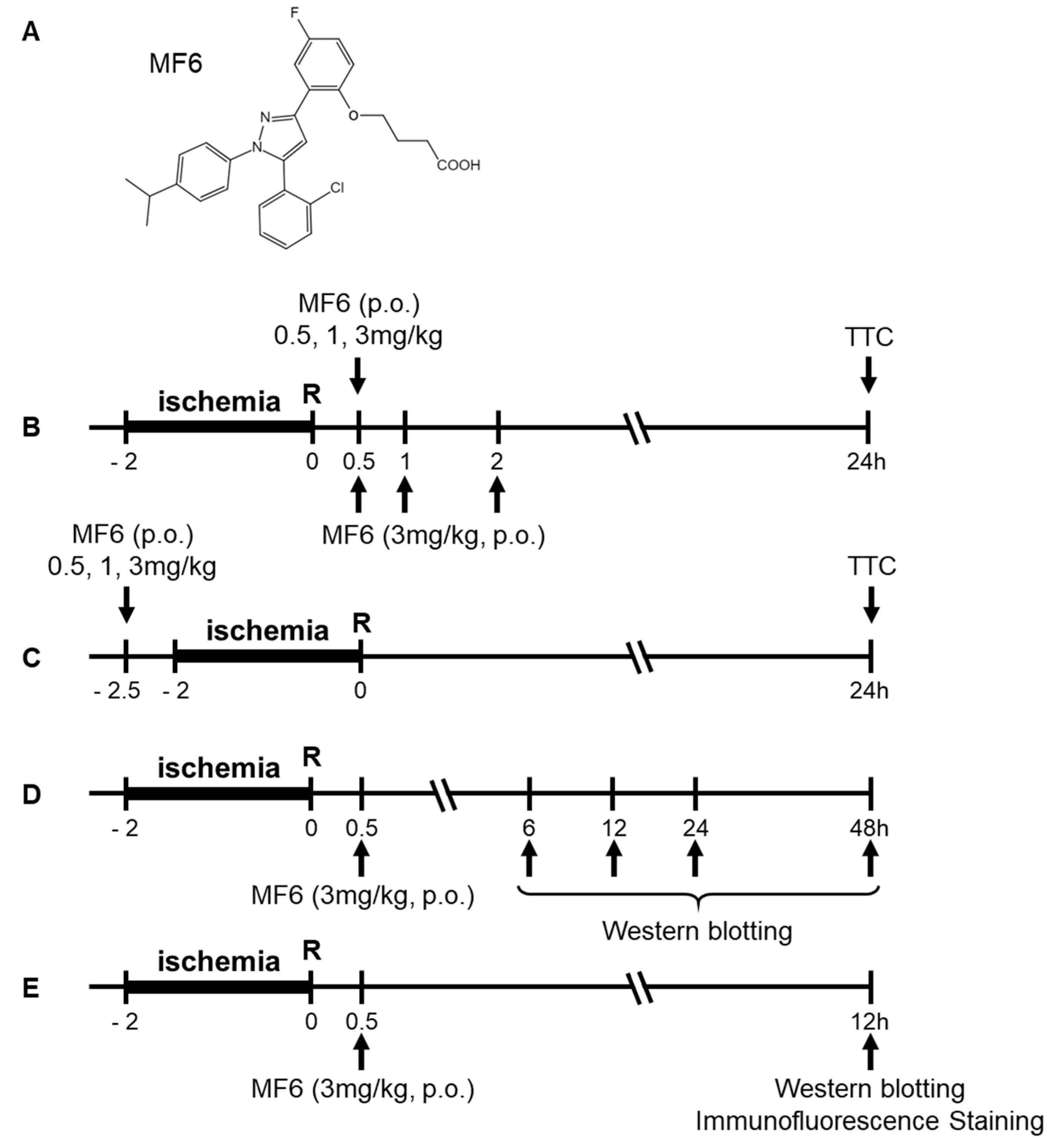
2.3. Drug Treatment
2.4. Infarct Volume Evaluation
2.5. Neurological Score
2.6. Western Blot Analysis
2.7. Immunofluorescence Staining
2.8. Measuring MF6 Concentrations in the Blood and Brain
2.9. Measuring Brain PGE2 Concentrations by Performing Enzyme-Linked Immunosorbent Assays (ELISAs)
2.10. Cell Culture and Luciferase Reporter Assay
2.11. Statistical Analysis
3. Results
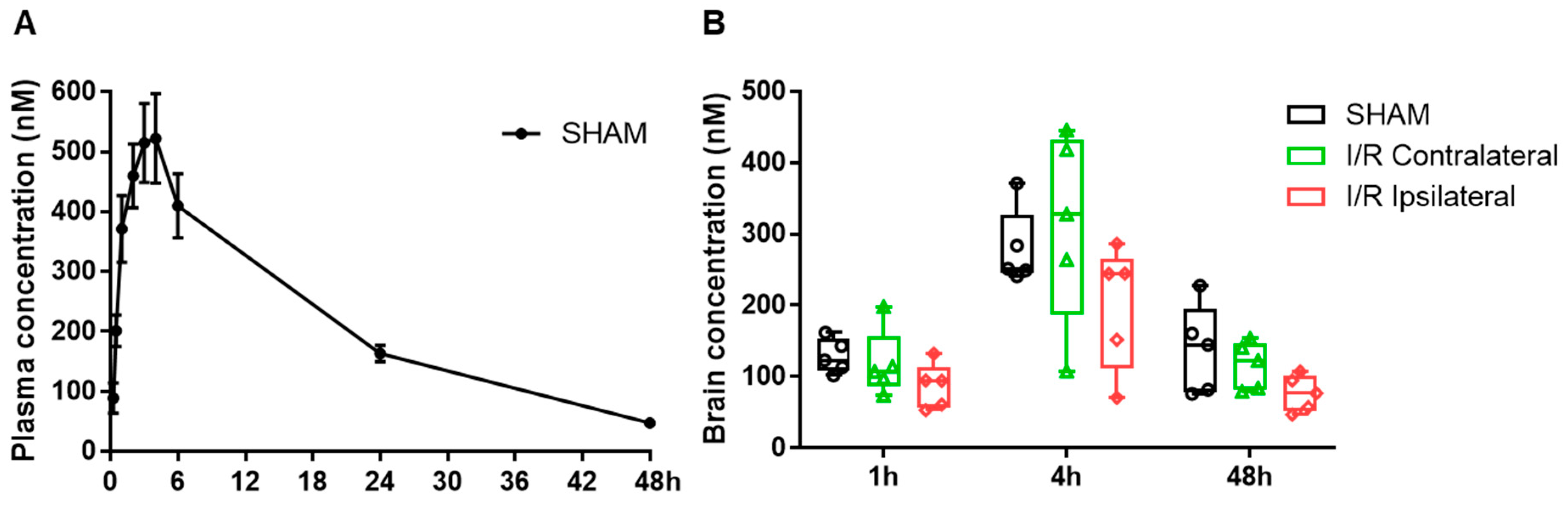
3.1. Pharmacokinetics of MF6 in the Blood and Brain after I/R
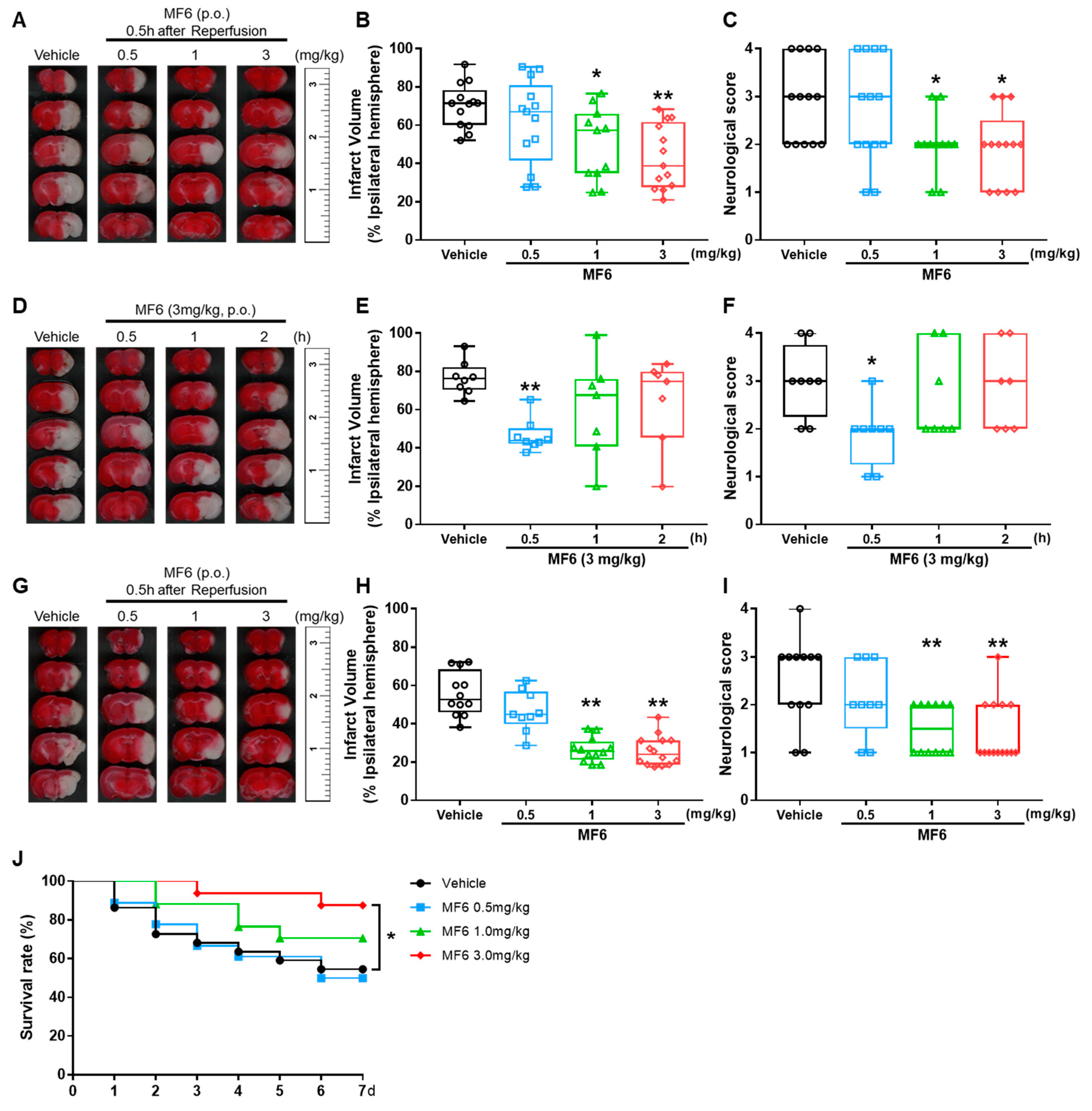
3.2. MF6 Reduced Infarct Volumes and Ameliorated Neurological Deficits in I/R Mice
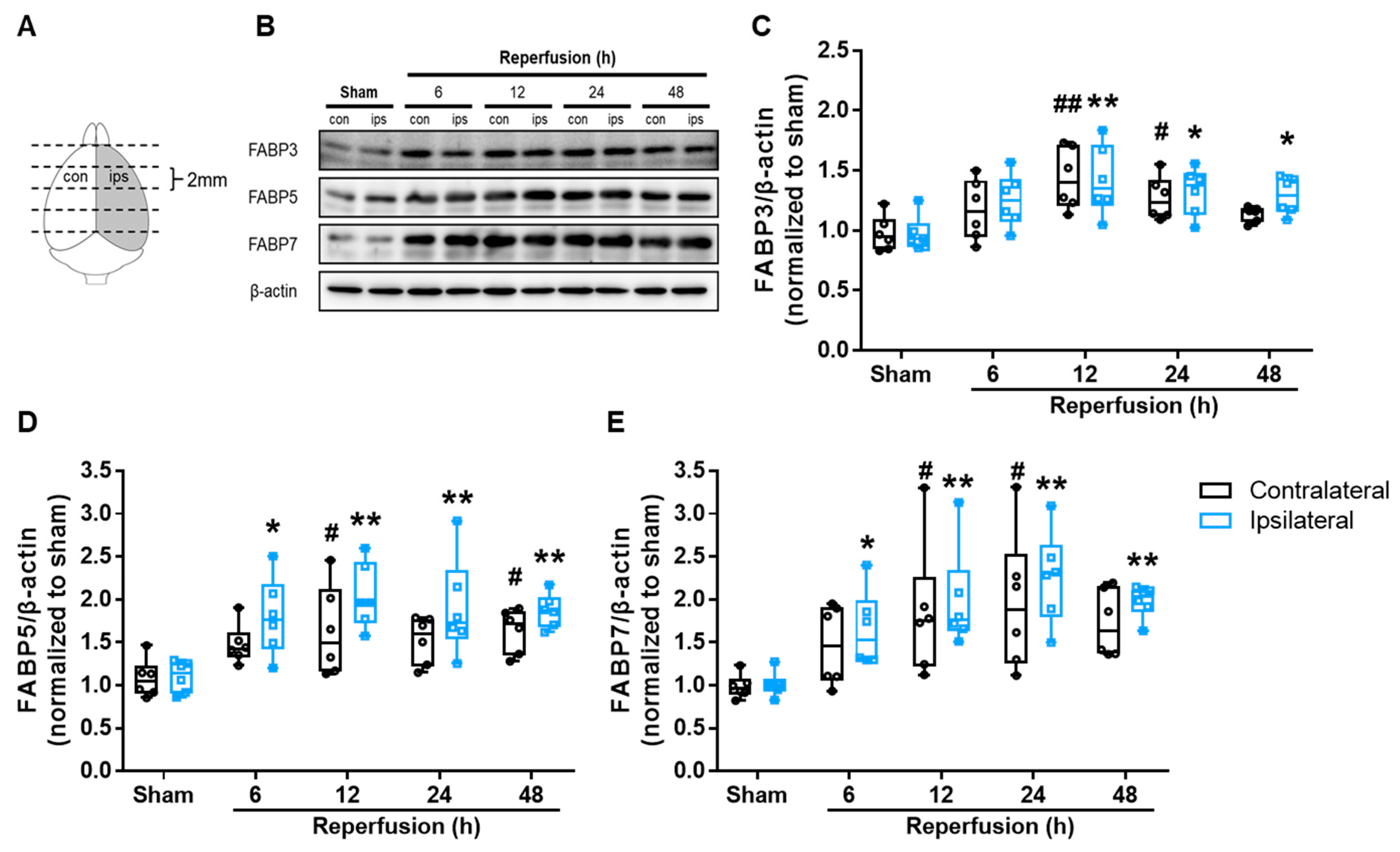
3.3. I/R Induced FABP3, FABP5 and FABP7 Protein Expression in Mouse Brains
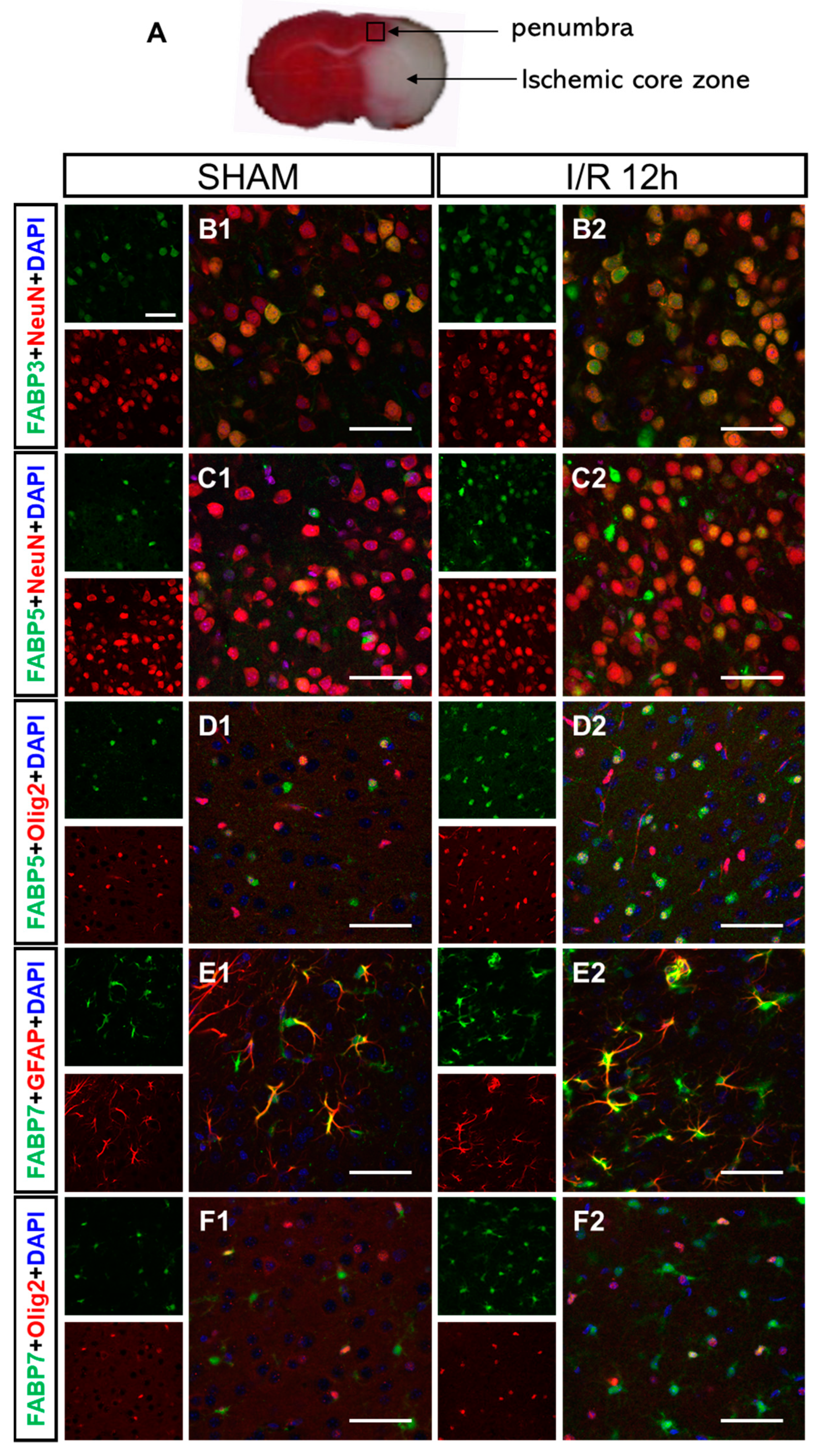
3.4. I/R-Induced FABPs Were Expressed in Specific Cells in the Cortex
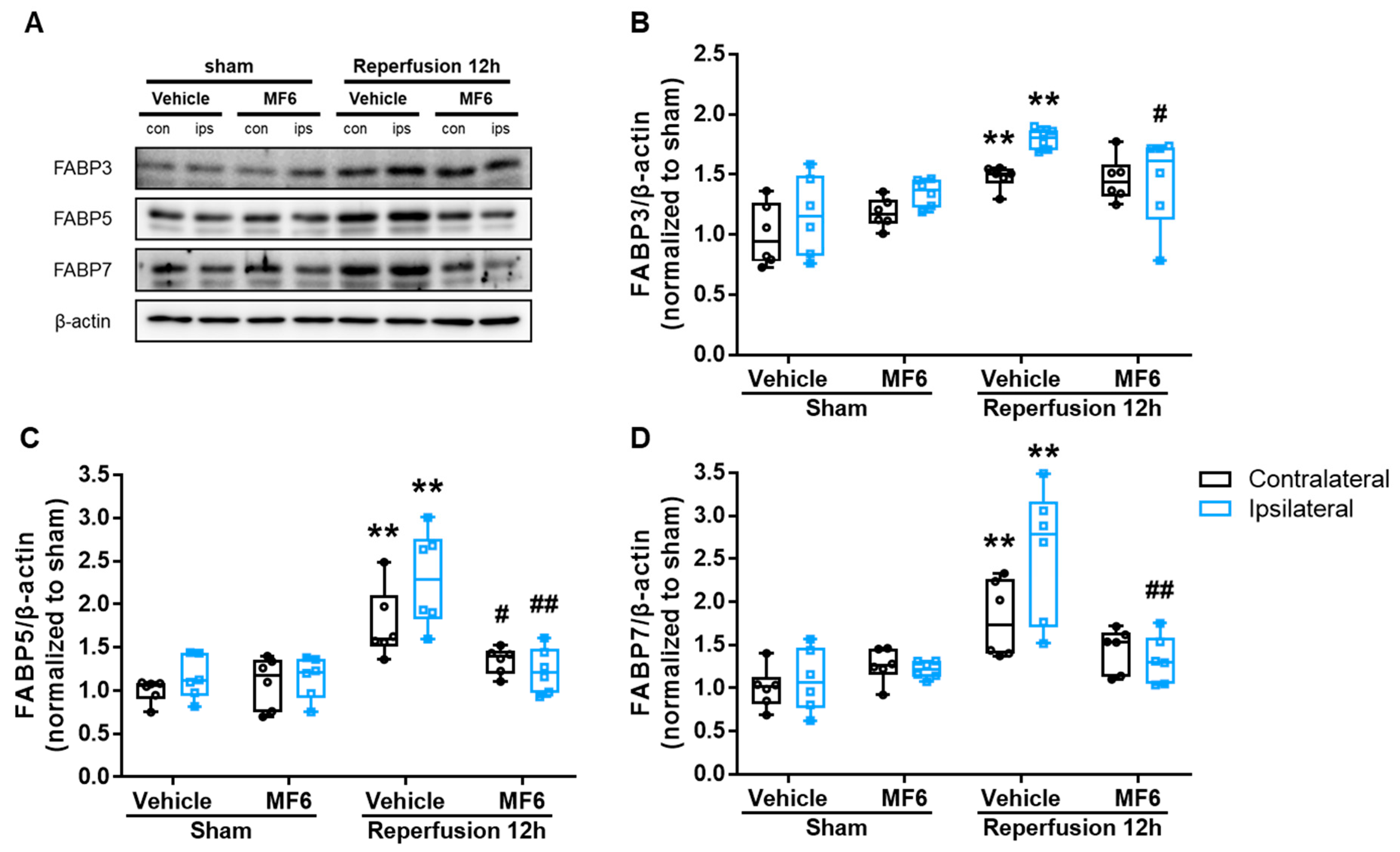
3.5. MF6 Suppressed FABP3, FABP5 and FABP7 Protein Upregulation in I/R Mouse Brains
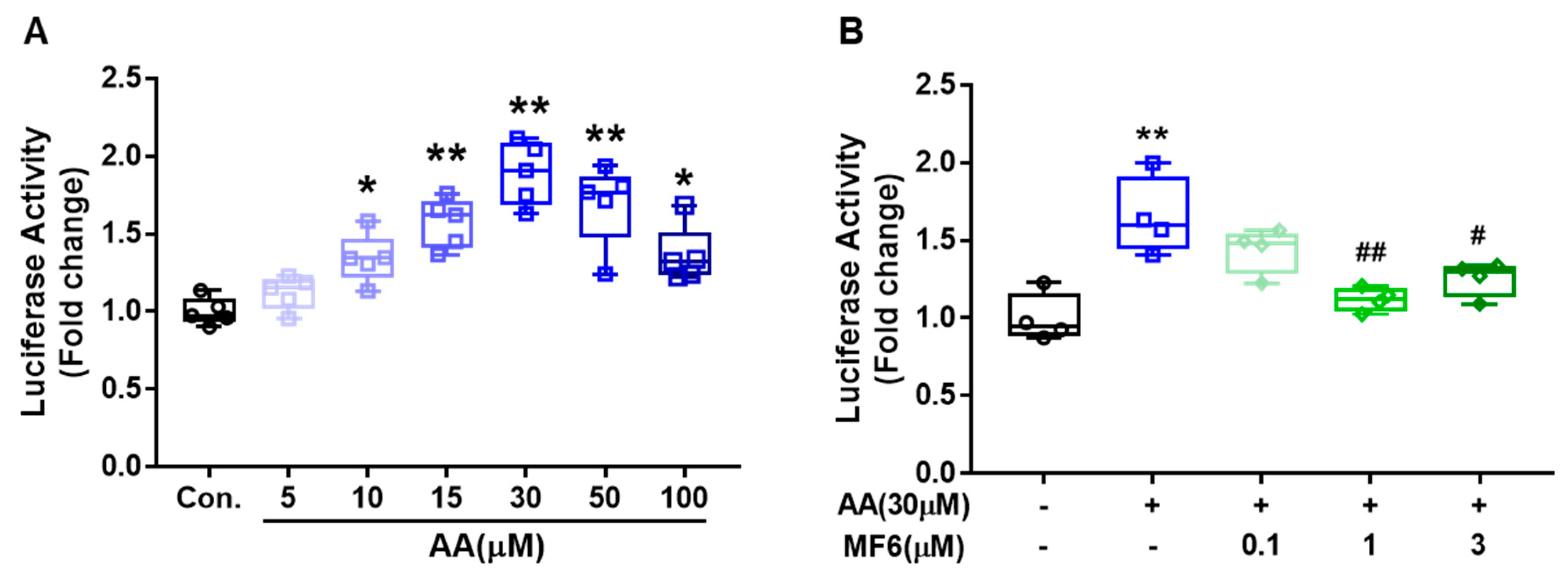
3.6. MF6 Prevented AA-Induced FABP5 Upregulation in SH-SY5Y Cells
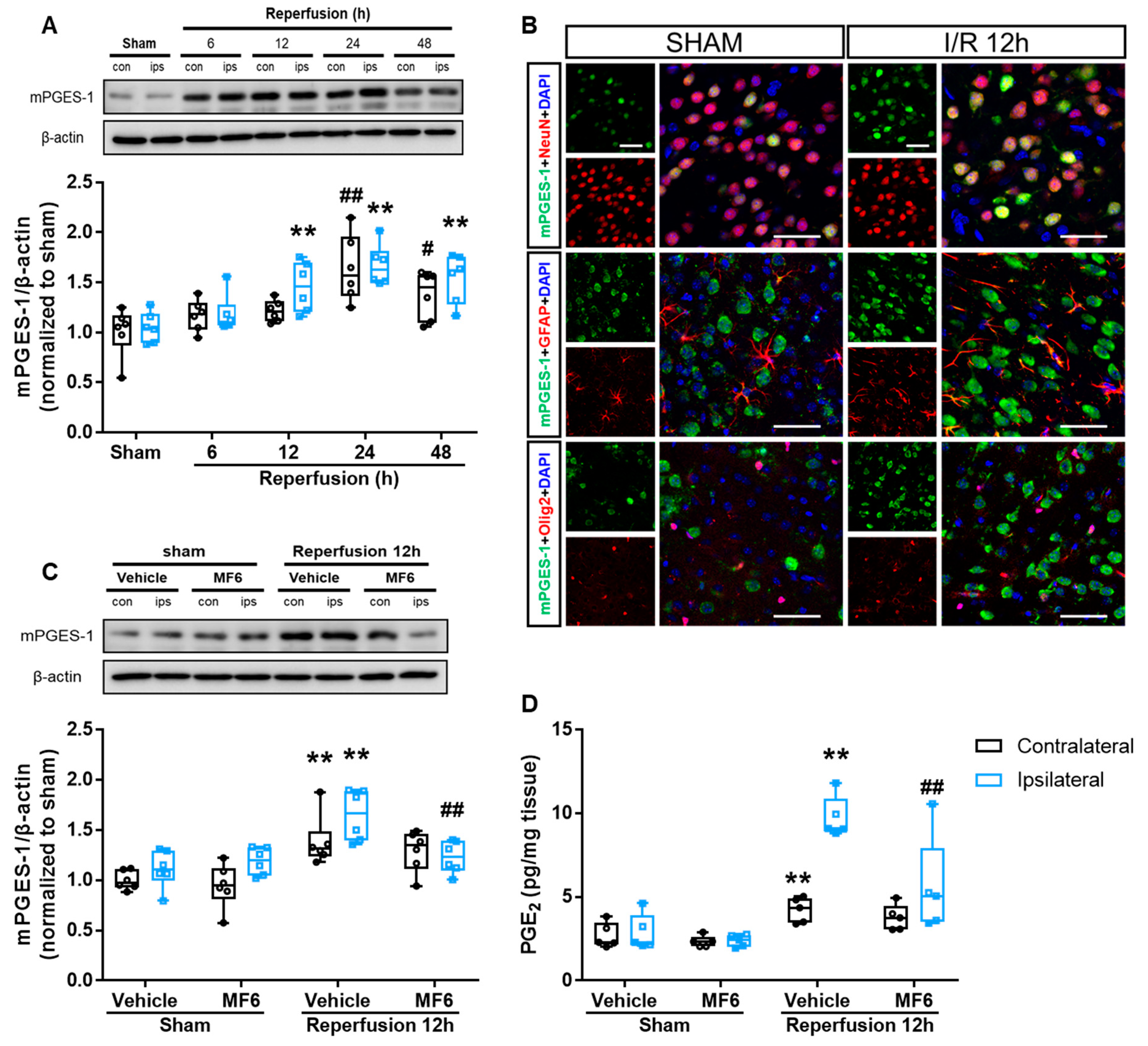
3.7. MF6 Suppressed the Microsomal Prostaglandin E Synthase-1 (mPGES-1)–PGE2 Signaling Pathway in I/R Mouse Brains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Terasaki, Y.; Liu, Y.; Hayakawa, K.; Pham, L.D.; Lo, E.H.; Ji, X.; Arai, K. Mechanisms of Neurovascular Dysfunction in Acute Ischemic Brain. Curr. Med. Chem. 2014, 21, 2035–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouns, R.; De Deyn, P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Matsumata, M.; Inada, H.; Osumi, N. Fatty acid binding proteins and the nervous system: Their impact on mental conditions. Neurosci. Res. 2016, 102, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Amiri, M.; Yousefnia, S.; Seyed Forootan, F.; Peymani, M.; Ghaedi, K.; Nasr Esfahani, M.H. Diverse roles of fatty acid binding proteins (FABPs) in development and pathogenesis of cancers. Gene 2018, 676, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, C.; Ohnishi, T.; Maekawa, M.; Watanabe, A.; Ohba, H.; Arai, R.; Iwayama, Y.; Hisano, Y.; Toyota, T.; Toyoshima, M.; et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum. Mol. Genet. 2015, 24, 2409. [Google Scholar] [CrossRef] [Green Version]
- Owada, Y. Fatty acid binding protein: Localization and functional significance in the brain. Tohoku J. Exp. Med. 2008, 214, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.Z.; Mita, R.; Beaulieu, M.; Gao, Z.; Godbout, R. Fatty acid binding proteins in brain development and disease. Int. J. Dev. Biol. 2010, 54, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Chiasserini, D.; Biscetti, L.; Eusebi, P.; Salvadori, N.; Frattini, G.; Simoni, S.; De Roeck, N.; Tambasco, N.; Stoops, E.; Vanderstichele, H.; et al. Differential role of CSF fatty acid binding protein 3, alpha-synuclein, and Alzheimer’s disease core biomarkers in Lewy body disorders and Alzheimer’s dementia. Alzheimers Res. Ther. 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Steinacker, P.; Bahn, E.; Bibl, M.; Brechlin, P.; Schlossmacher, M.G.; Locascio, J.J.; Wiltfang, J.; Kretzschmar, H.A.; Poser, S.; et al. Serum heart-type fatty acid-binding protein and cerebrospinal fluid tau: Marker candidates for dementia with Lewy bodies. Neurodegener. Dis. 2007, 4, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Wilar, G.; Kawahata, I.; Cheng, A.; Fukunaga, K. Impaired Acquisition of Nicotine-Induced Conditioned Place Preference in Fatty Acid-Binding Protein 3 Null Mice. Mol. Neurobiol. 2021, 58, 2030–2045. [Google Scholar] [CrossRef]
- Jia, W.; Kawahata, I.; Cheng, A.; Fukunaga, K. The Role of CaMKII and ERK Signaling in Addiction. Int. J. Mol. Sci. 2021, 22, 3189. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Kobayashi, Y.; Onozato, M.; Owada, Y.; Fukunaga, K. FABP3 protein promotes alpha-synuclein oligomerization associated with 1-methyl-1,2,3,6-tetrahydropiridine-induced neurotoxicity. J. Biol. Chem. 2014, 289, 18957–18965. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.; Geng, L.; Zhang, F.; Shu, L.; Wei, L.; Yeung, P.K.K.; Lam, K.S.L.; Chung, S.K.; Chang, J.; Vanhoutte, P.M.; et al. Adipocyte fatty acid-binding protein exacerbates cerebral ischaemia injury by disrupting the blood-brain barrier. Eur. Heart J. 2020, 41, 3169–3180. [Google Scholar] [CrossRef]
- Shi, M.; Huang, R.S.; Guo, F.; Li, L.Z.; Feng, Y.H.; Wei, Z.J.; Zhou, L.; Ma, L.; Fu, P. Pharmacological inhibition of fatty acid-binding protein 4 (FABP4) protects against renal ischemia-reperfusion injury. RSC Adv. 2018, 8, 15207–15214. [Google Scholar] [CrossRef] [Green Version]
- Beniyama, Y.; Matsuno, K.; Miyachi, H. Structure-guided design, synthesis and in vitro evaluation of a series of pyrazole-based fatty acid binding protein (FABP) 3 ligands. Bioorg. Med. Chem. Lett. 2013, 23, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Cheng, A.; Yabuki, Y.; Takahata, I.; Miyachi, H.; Fukunaga, K. Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology 2019, 150, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Haga, H.; Yamada, R.; Izumi, H.; Shinoda, Y.; Kawahata, I.; Miyachi, H.; Fukunaga, K. Novel fatty acid-binding protein 3 ligand inhibits dopaminergic neuronal death and improves motor and cognitive impairments in Parkinson’s disease model mice. Pharmacol. Biochem. Behav. 2020, 191, 172891. [Google Scholar] [CrossRef]
- Cheng, A.; Wang, Y.; Shinoda, Y.; Kawahata, I.; Yamamoto, T.; Jia, W.; Yamamoto, H.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Fatty acid-binding protein 7 triggers α-synuclein oligomerization in glial cells and oligodendrocytes associated with oxidative stress. Acta Pharmacol. Sin. 2021, 0, 1–11. [Google Scholar]
- Cheng, A.; Shinoda, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Development of FABP3 ligands that inhibit arachidonic acid-induced alpha-synuclein oligomerization. Brain Res. 2019, 1707, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, Y.; Wang, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Analysis of binding affinity and docking of novel fatty acid-binding protein (FABP) ligands. J. Pharmacol. Sci. 2020, 143, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Izumi, H.; Shinoda, Y.; Fukunaga, K. Neuroprotective effects of protein tyrosine phosphatase 1B inhibitor on cerebral ischemia/reperfusion in mice. Brain Res. 2018, 1694, 1–12. [Google Scholar] [CrossRef]
- Sun, M.; Shinoda, Y.; Fukunaga, K. KY-226 Protects Blood-brain Barrier Function Through the Akt/FoxO1 Signaling Pathway in Brain Ischemia. Neuroscience 2019, 399, 89–102. [Google Scholar] [CrossRef]
- Yabuki, Y.; Fukunaga, K. Oral administration of glutathione improves memory deficits following transient brain ischemia by reducing brain oxidative stress. Neuroscience 2013, 250, 394–407. [Google Scholar] [CrossRef]
- Boneva, N.B.; Mori, Y.; Kaplamadzhiev, D.B.; Kikuchi, H.; Zhu, H.; Kikuchi, M.; Tonchev, A.B.; Yamashima, T. Differential expression of FABP 3, 5, 7 in infantile and adult monkey cerebellum. Neurosci. Res. 2010, 68, 94–102. [Google Scholar] [CrossRef]
- Boneva, N.B.; Kaplamadzhiev, D.B.; Sahara, S.; Kikuchi, H.; Pyko, I.V.; Kikuchi, M.; Tonchev, A.B.; Yamashima, T. Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus 2011, 21, 162–171. [Google Scholar] [CrossRef]
- Armstrong, E.H.; Goswami, D.; Griffin, P.R.; Noy, N.; Ortlund, E.A. Structural Basis for Ligand Regulation of the Fatty Acid-binding Protein 5, Peroxisome Proliferator-activated Receptor beta/delta (FABP5-PPAR beta/delta) Signaling Pathway. J. Biol. Chem. 2014, 289, 14941–14954. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.; Kannan-Thulasiraman, P.; Noy, N. Involvement of Fatty Acid Binding Protein 5 and PPAR beta/delta in Prostate Cancer Cell Growth. PPAR Res. 2010, 2010, 234629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda-Matsuo, Y.; Ota, A.; Fukada, T.; Uematsu, S.; Akira, S.; Sasaki, Y. Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 11790–11795. [Google Scholar] [CrossRef] [Green Version]
- Matsumata, M.; Sakayori, N.; Maekawa, M.; Owada, Y.; Yoshikawa, T.; Osumi, N. The Effects of Fabp7 and Fabp5 on Postnatal Hippocampal Neurogenesis in the Mouse. Stem Cells 2012, 30, 1532–1543. [Google Scholar] [CrossRef]
- Sharifi, K.; Ebrahimi, M.; Kagawa, Y.; Islam, A.; Tuerxun, T.; Yasumoto, Y.; Hara, T.; Yamamoto, Y.; Miyazaki, H.; Tokuda, N.; et al. Differential expression and regulatory roles of FABP5 and FABP7 in oligodendrocyte lineage cells. Cell Tissue Res. 2013, 354, 683–695. [Google Scholar] [CrossRef]
- Song, G.X.; Shen, Y.H.; Liu, Y.Q.; Sun, W.; Miao, L.P.; Zhou, L.J.; Liu, H.L.; Yang, R.; Kong, X.Q.; Cao, K.J.; et al. Overexpression of FABP3 promotes apoptosis through inducing mitochondrial impairment in embryonic cancer cells. J. Cell. Biochem. 2012, 113, 3701–3708. [Google Scholar] [CrossRef]
- Zhuang, L.; Li, C.; Chen, Q.; Jin, Q.; Wu, L.; Lu, L.; Yan, X.; Chen, K. Fatty acid-binding protein 3 contributes to ischemic heart injury by regulating cardiac myocyte apoptosis and MAPK pathways. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H971–h984. [Google Scholar] [CrossRef]
- Kawahata, I.; Bousset, L.; Melki, R.; Fukunaga, K. Fatty Acid-Binding Protein 3 is Critical for α-Synuclein Uptake and MPP+-Induced Mitochondrial Dysfunction in Cultured Dopaminergic Neurons. Int. J. Mol. Sci. 2019, 20, 5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.X.; Zhang, M.M.; Mori, Y.; Yao, C.J.; Larsen, C.P.; Yamashima, T.; Zhou, L.F. Cellular Localization of Epidermal-Type and Brain-Type Fatty Acid-Binding Proteins in Adult Hippocampus and Their Response to Cerebral Ischemia. Hippocampus 2010, 20, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Brady, J.D.; Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 2007, 10, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Roy-O’Reilly, M.; McCullough, L.D. Astrocytes fuel the fire of lymphocyte toxicity after stroke. Proc. Natl. Acad. Sci. USA 2017, 114, 425–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, M.; Yamamoto, Y.; Sharifi, K.; Kida, H.; Kagawa, Y.; Yasumoto, Y.; Islam, A.; Miyazaki, H.; Shimamoto, C.; Maekawa, M.; et al. Astrocyte-expressed FABP7 regulates dendritic morphology and excitatory synaptic function of cortical neurons. Glia 2016, 64, 48–62. [Google Scholar] [CrossRef]
- Senbokuya, N.; Yoshioka, H.; Yagi, T.; Owada, Y.; Kinouchi, H. Effects of FABP7 on functional recovery after spinal cord injury in adult mice. J. Neurosurg. Spine 2019, 31, 291–297. [Google Scholar] [CrossRef]
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Vargas, M.R. FABP7 upregulation induces a neurotoxic phenotype in astrocytes. Glia 2020, 68, 2693–2704. [Google Scholar] [CrossRef]
- Arai, Y.; Funatsu, N.; Numayama-Tsuruta, K.; Nomura, T.; Nakamura, S.; Osumi, N. Role of Fabp7, a downstream gene of Pax6, in the maintenance of neuroepithelial cells during early embryonic development of the rat cortex. J. Neurosci. 2005, 25, 9752–9761. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, R.; Sakayori, N.; Matsumata, M.; Owada, Y.; Wakamatsu, Y.; Osumi, N. Fatty acid binding protein (Fabp7) is involved in the maintenance of neural stem/progenitor cells, survival of neurons and maturation of astrocytes. Neurosci. Res. 2011, 71, E127. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, Y.; Chen, S.C.; Zhao, T.; Huang, Q.; Hu, Z.L.; Xia, N.Z.; Liu, J.J.; Chen, W.J.; Zhang, Y.; et al. Metabolite changes in the ipsilateral and contralateral cerebral hemispheres in rats with middle cerebral artery occlusion. Neural Regen. Res. 2017, 12, 931–937. [Google Scholar] [PubMed]
- Xu, Z.N.S.; Lee, R.J.; Chu, S.S.; Yao, A.N.; Paun, M.K.; Murphy, S.P.; Mourad, P.D. Evidence of Changes in Brain Tissue Stiffness After Ischemic Stroke Derived From Ultrasound-Based Elastography. J. Ultras Med. 2013, 32, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Gangishetti, U.; Christina Howell, J.; Perrin, R.J.; Louneva, N.; Watts, K.D.; Kollhoff, A.; Grossman, M.; Wolk, D.A.; Shaw, L.M.; Morris, J.C.; et al. Non-beta-amyloid/tau cerebrospinal fluid markers inform staging and progression in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 98. [Google Scholar] [CrossRef]
- Sepe, F.N.; Chiasserini, D.; Parnetti, L. Role of FABP3 as biomarker in Alzheimer’s disease and synucleinopathies. Future Neurol. 2018, 13, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, C.E.; Veerhuis, R.; De Vente, J.; Verhey, F.R.J.; Vreeling, F.; van Boxtel, M.P.J.; Glatz, J.F.C.; Pelsers, M.A.L. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 2011, 18, 865–871. [Google Scholar] [CrossRef]
- Sebastian Monasor, L.; Muller, S.A.; Colombo, A.V.; Tanrioever, G.; Konig, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 2020, 9, e54083. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-alpha, TGF-beta and IFN-gamma. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Rao, D.; Perraud, A.L.; Schmitz, C.; Gally, F. Cigarette smoke inhibits LPS-induced FABP5 expression by preventing c-Jun binding to the FABP5 promoter. PLoS ONE 2017, 12, e0178021. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.A.; Yeung, S.C.; Ip, M.S.M.; Mak, J.C.W. Effects of intermittent hypoxia on A-/E-FABP expression in human aortic endothelial cells. Int. J. Cardiol. 2010, 145, 396–398. [Google Scholar] [CrossRef] [Green Version]
- Kamizato, K.; Sato, S.; Shil, S.K.; Umaru, B.A.; Kagawa, Y.; Yamamoto, Y.; Ogata, M.; Yasumoto, Y.; Okuyama, Y.; Ishii, N.; et al. The role of fatty acid binding protein 7 in spinal cord astrocytes in a mouse model of experimental autoimmune encephalomyelitis. Neuroscience 2019, 409, 120–129. [Google Scholar] [CrossRef]
- MacDougall, G.; Anderton, R.S.; Mastaglia, F.L.; Knuckey, N.W.; Meloni, B.P. Proteomic analysis of cortical neuronal cultures treated with poly-arginine peptide-18 (R18) and exposed to glutamic acid excitotoxicity. Mol. Brain 2019, 12. [Google Scholar] [CrossRef]
- Cheng, A.; Kawahata, I.; Fukunaga, K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines 2020, 8, 635. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid. Redox Sign. 2011, 14, 1889–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Bobinski, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Levi, L.; Casadesus, G.; Kunos, G.; Noy, N. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in the brain. J. Biol. Chem. 2014, 289, 12748–12758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blitek, A.; Szymanska, M. Peroxisome proliferator-activated receptor beta/delta and gamma agonists differentially affect prostaglandin E2 and cytokine synthesis and nutrient transporter expression in porcine trophoblast cells during implantation. Theriogenology 2020, 152, 36–46. [Google Scholar] [CrossRef]
- de Oliveira, A.C.; Candelario-Jalil, E.; Bhatia, H.S.; Lieb, K.; Hull, M.; Fiebich, B.L. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: Evidence for uncoupled regulation of mPGES-1 and COX-2. Glia 2008, 56, 844–855. [Google Scholar] [CrossRef]
- Ikeda-Matsuo, Y. The Role of mPGES-1 in Inflammatory Brain Diseases. Biol. Pharm. Bull. 2017, 40, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, D.; Falcone, J.; Kanjiya, M.P.; Park, S.H.; Carbonetti, G.; Studholme, K.; Gomez, M.; Lu, Y.; Elmes, M.W.; Smietalo, N.; et al. Fatty acid-binding protein 5 controls microsomal prostaglandin E synthase 1 (mPGES-1) induction during inflammation. J. Biol. Chem. 2018, 293, 5295–5306. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Ridder, D.A.; Schwaninger, M. NF-kappaB signaling in cerebral ischemia. Neuroscience 2009, 158, 995–1006. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Kawahata, I.; Degawa, T.; Ikeda-Matsuo, Y.; Sun, M.; Han, F.; Fukunaga, K. Fatty Acid-Binding Proteins Aggravate Cerebral Ischemia-Reperfusion Injury in Mice. Biomedicines 2021, 9, 529. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050529
Guo Q, Kawahata I, Degawa T, Ikeda-Matsuo Y, Sun M, Han F, Fukunaga K. Fatty Acid-Binding Proteins Aggravate Cerebral Ischemia-Reperfusion Injury in Mice. Biomedicines. 2021; 9(5):529. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050529
Chicago/Turabian StyleGuo, Qingyun, Ichiro Kawahata, Tomohide Degawa, Yuri Ikeda-Matsuo, Meiling Sun, Feng Han, and Kohji Fukunaga. 2021. "Fatty Acid-Binding Proteins Aggravate Cerebral Ischemia-Reperfusion Injury in Mice" Biomedicines 9, no. 5: 529. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050529