
Somatic Functional Deletions of Upstream Open Reading Frame-Associated Initiation and Termination Codons in Human Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Definition of TLSs and uORF-Associated Genomic Positions
2.2. Variation Data Obtained from GDC Data Portal
2.3. Calculation of Read Coverage
2.4. Identification of Somatic Variants at uAUG, aTIS, and uStop Codons
2.5. Filtering of Raw Variability Data
2.6. Selection of uORF-Associated Variants for Functional Testing
2.7. Cell Lines and Plasmids
2.8. Dual Luciferase Reporter Assays
2.9. RNA Preparation and qRT-PCR
3. Results
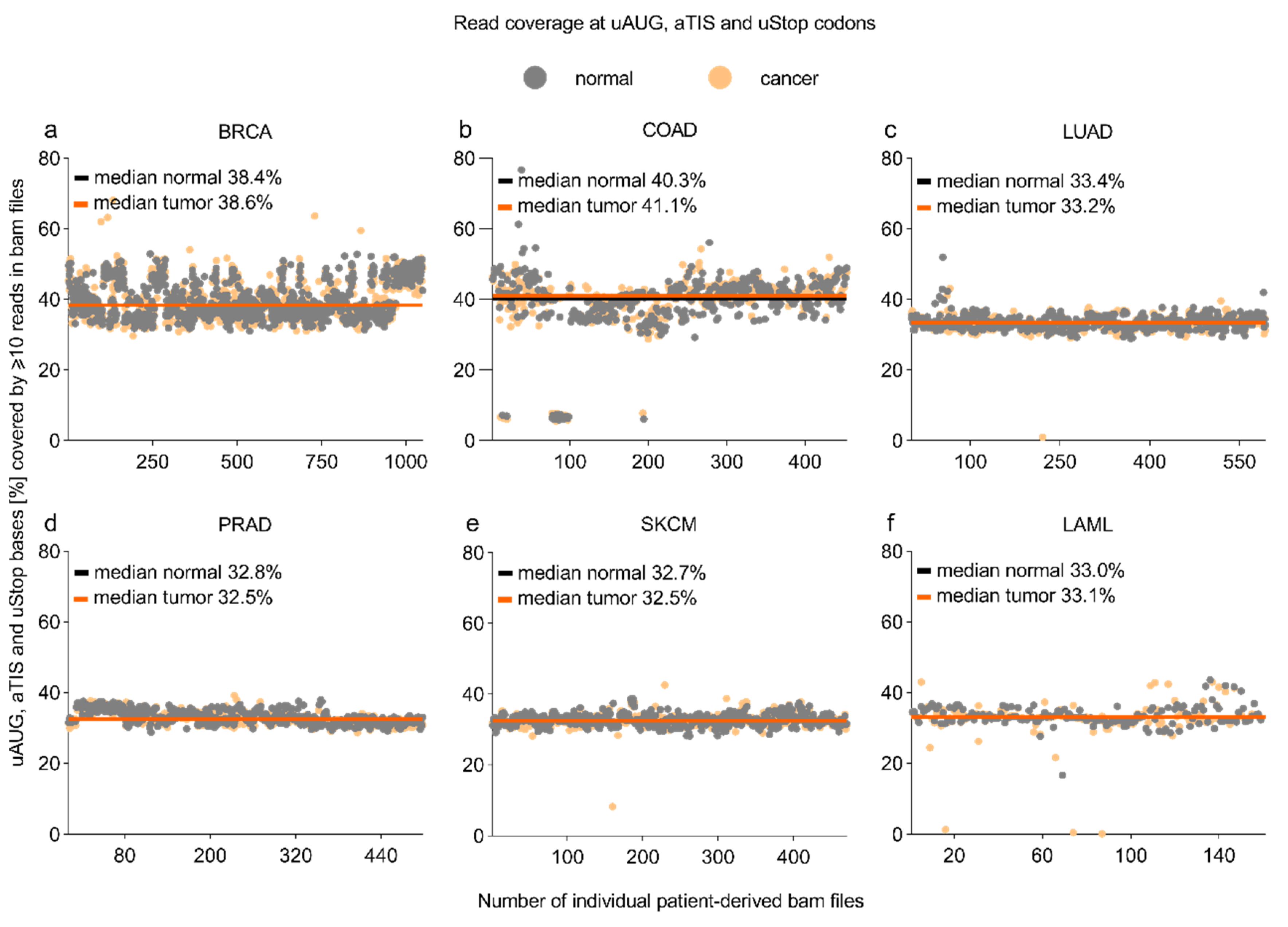
3.1. Read Coverage at uAUG, aTIS, and uStop Codons in TCGA-Derived Whole-Exome Sequencing Datasets
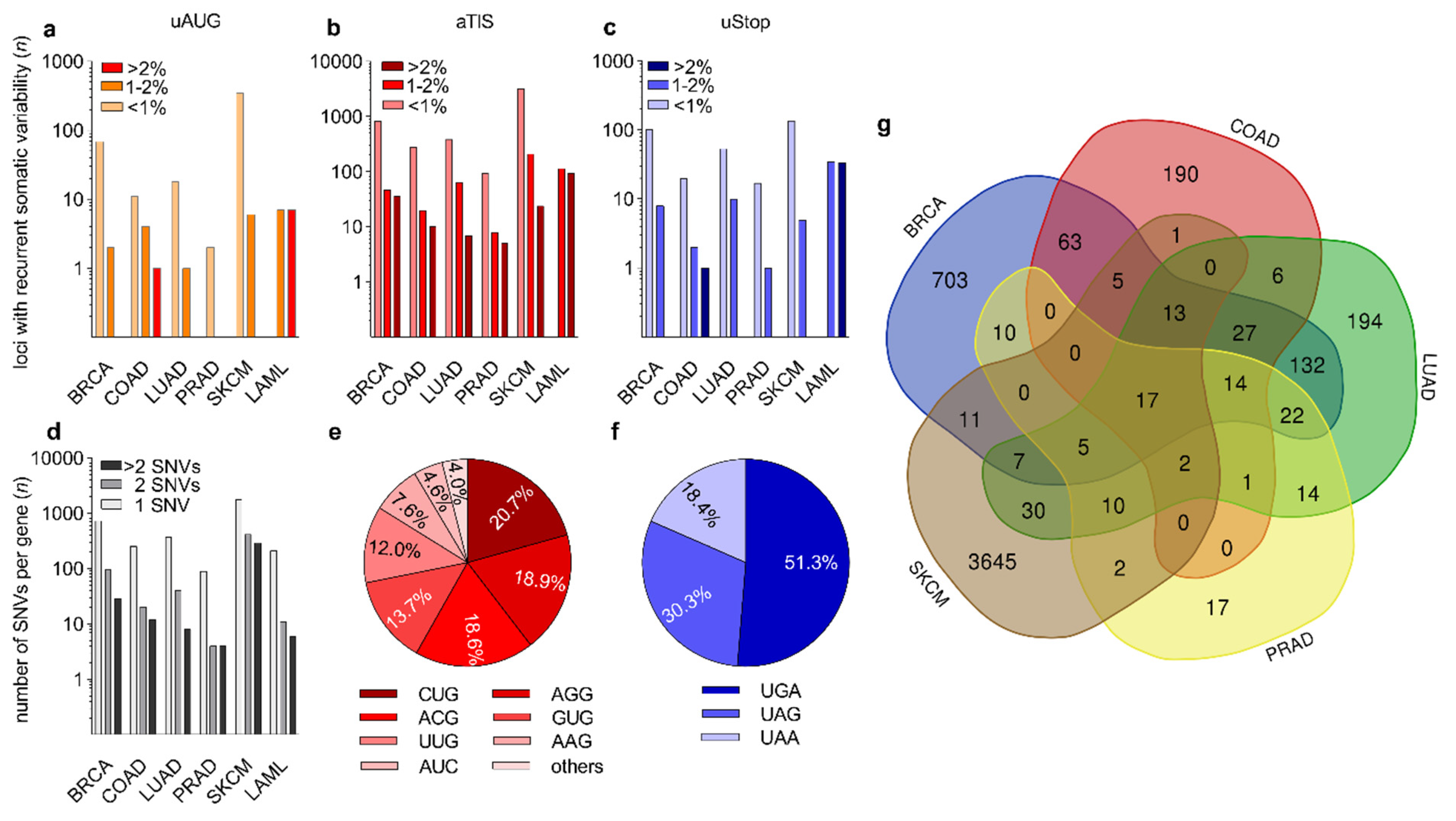
3.2. Identification of Recurrent Somatic Genetic Variation at uAUG, aTIS, and uStop Codons
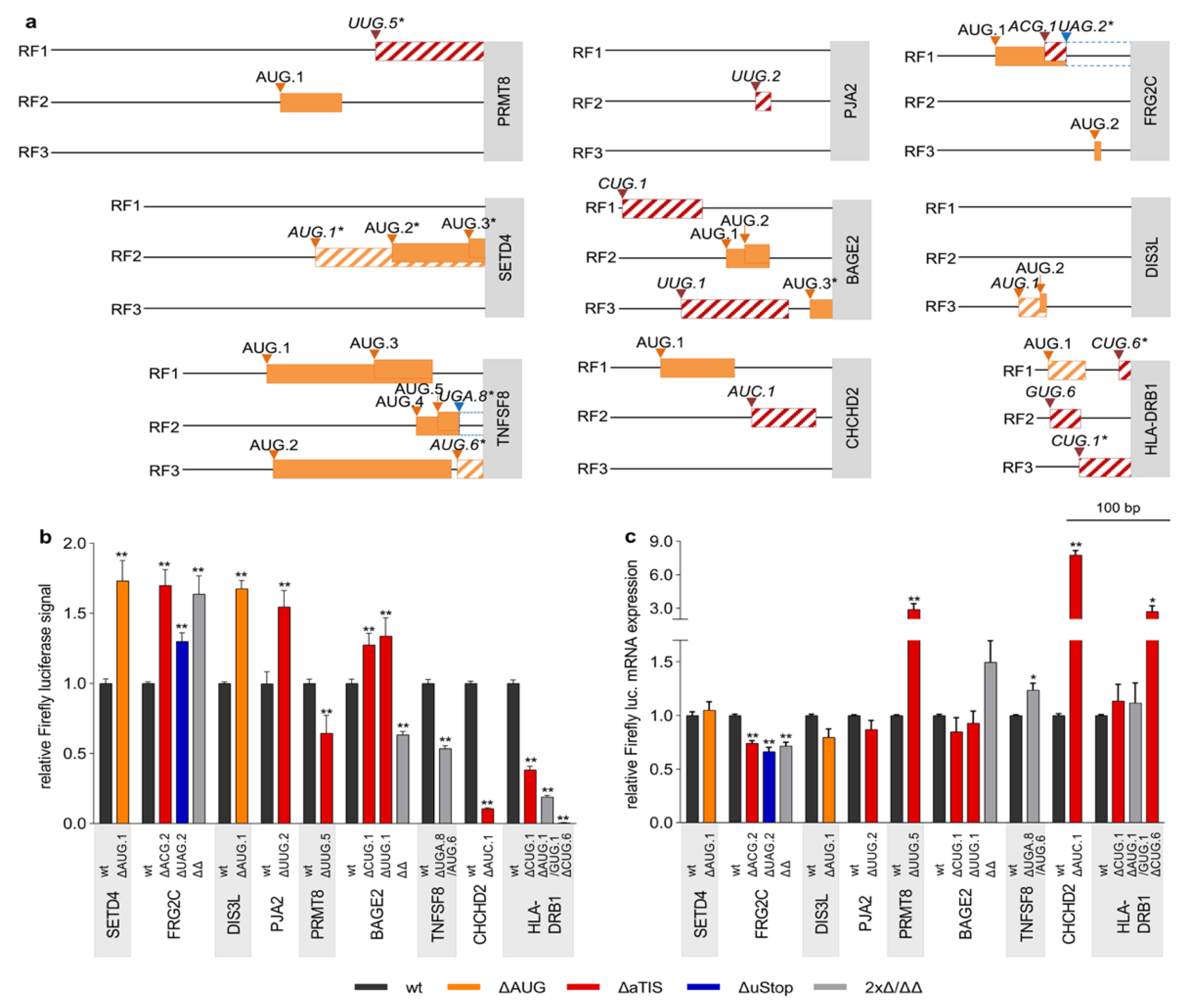
3.3. Defective Translational Regulation by uORF-Associated Somatic SNVs
4. Discussion
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Liu, B.; Lee, S.; Huang, S.X.; Shen, B.; Qian, S.B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E2424–E2432. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Brunner, A.D.; Cogan, J.Z.; Nuñez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; Leonetti, M.D.; et al. Pervasive functional translation of noncanonical human open reading frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wan, J.; Liu, B.; Ma, M.; Shen, B.; Qian, S.B. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2015, 12, 147–153. [Google Scholar] [CrossRef]
- Fritsch, C.; Herrmann, A.; Nothnagel, M.; Szafranski, K.; Huse, K.; Schumann, F.; Schreiber, S.; Platzer, M.; Krawczak, M.; Hampe, J.; et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012, 22, 2208–2218. [Google Scholar] [CrossRef] [Green Version]
- Young, S.K.; Wek, R.C. Upstream open reading frames differentially regulate genespecific translation in the integrated stress response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wethmar, K.; Schulz, J.; Muro, E.M.; Talyan, S.; Andrade-Navarro, M.A.; Leutz, A. Comprehensive translational control of tyrosine kinase expression by upstream open reading frames. Oncogene 2016, 35, 1736–1742. [Google Scholar] [CrossRef] [Green Version]
- McGillivray, P.; Ault, R.; Pawashe, M.; Kitchen, R.; Balasubramanian, S.; Gerstein, M. A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Res. 2018, 46, 3326–3338. [Google Scholar] [CrossRef]
- Ivanov, I.P.; Firth, A.E.; Michel, A.M.; Atkins, J.F.; Baranov, P.V. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011, 39, 4220–4234. [Google Scholar] [CrossRef]
- Johnstone, T.G.; Bazzini, A.A.; Giraldez, A.J. Upstream ORF s are prevalent translational repressors in vertebrates. EMBO J. 2016, 35, 706–723. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [Green Version]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Zhang, M.Q. CART Classification of Human 5’ UTR sequences. Genome Res. 2000, 10, 1807–1816. [Google Scholar] [CrossRef]
- Cao, X.; Slavoff, S.A. Non-AUG start codons: Expanding and regulating the small and alternative ORFeome. Exp. Cell Res. 2020, 391, 1–5. [Google Scholar] [CrossRef]
- Baird, T.D.; Palam, L.R.; Fusakio, M.E.; Willy, J.A.; Davis, C.M.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKα. Mol. Biol. Cell 2014, 25, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Giess, A.; Torres Cleuren, Y.N.; Tjeldnes, H.; Krause, M.; Bizuayehu, T.T.; Hiensch, S.; Okon, A.; Wagner, C.R.; Valen, E. Profiling of Small Ribosomal Subunits Reveals Modes and Regulation of Translation Initiation. Cell Rep. 2020, 31, 1–10. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of eukaryotic translation initiation and principle of its Regulation. Annu. Rev. Biochem. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wethmar, K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Fernandes, R.; Romão, L. Translational Regulation by Upstream Open Reading Frames and Human Diseases. In The mRNA Metabolism in Human Disease; Romão, L., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 99–116. ISBN 978-3-030-19966-1. [Google Scholar]
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Roberts, A.M.; Quaife, N.M.; Schafer, S.; Rackham, O.; et al. Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Somers, J.; Pöyry, T.; Willis, A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, J.; Mah, N.; Neuenschwander, M.; Kischka, T.; Ratei, R.; Schlag, P.M.; Castaños-Vélez, E.; Fichtner, I.; Tunn, P.U.; Denkert, C.; et al. Loss-of-function uORF mutations in human malignancies. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Pizzinga, M.; Harvey, R.F.; Garland, G.D.; Mordue, R.; Dezi, V.; Ramakrishna, M.; Sfakianos, A.; Monti, M.; Mulroney, T.E.; Poyry, T.; et al. The cell stress response: Extreme times call for post-transcriptional measures. Wiley Interdiscip. Rev. RNA 2020, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Akulich, K.A.; Sinitcyn, P.G.; Makeeva, D.S.; Andreev, D.E.; Terenin, I.M.; Anisimova, A.S.; Shatsky, I.N.; Dmitriev, S.E. A novel uORF-based regulatory mechanism controls translation of the human MDM2 and eIF2D mRNAs during stress. Biochimie 2019, 157, 92–101. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, Y.; Xu, Y.; Zhao, Y.; Hua, R.; Wang, K.; Sun, M.; Li, Y.; Yang, S.; Zhang, X.J.; et al. Loss-of-function mutations of an inhibitory upstream ORF in the human hairless transcript cause Marie Unna hereditary hypotrichosis. Nat. Genet. 2009, 41, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wiestner, A.; Schlemper, R.J.; van der Maas, A.P.; Skoda, R.C. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat. Genet. 1998, 18, 49–52. [Google Scholar] [CrossRef]
- Liu, L.; Dilworth, D.; Gao, L.; Monzon, J.; Summers, A.; Lassam, N.; Hogg, D. Mutation of the CDKN2A 5′ UTR creates an aberrant initiation codon and predisposes to melanoma. Nat. Genet. 1999, 21, 128–132. [Google Scholar] [CrossRef]
- Occhi, G.; Regazzo, D.; Trivellin, G.; Boaretto, F.; Ciato, D.; Bobisse, S.; Ferasin, S.; Cetani, F.; Pardi, E.; Korbonits, M.; et al. A Novel Mutation in the Upstream Open Reading Frame of the CDKN1B Gene Causes a MEN4 Phenotype. PLoS Genet. 2013, 9, 1–11. [Google Scholar] [CrossRef]
- Baek, I.C.; Kim, J.K.; Cho, K.H.; Cha, D.S.; Cho, J.W.; Park, J.K.; Song, C.W.; Yoon, S.K. A novel mutation in Hr causes abnormal hair follicle morphogenesis in hairpoor mouse, an animal model for Marie Unna Hereditary Hypotrichosis. Mamm. Genome 2009, 20, 350–358. [Google Scholar] [CrossRef]
- Wethmar, K.; Bégay, V.; Smink, J.J.; Zaragoza, K.; Wiesenthal, V.; Dörken, B.; Calkhoven, C.F.; Leutz, A. C/EBPβΔuORF mice—A genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010, 24, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Chen, Y.J.; Liu, Y.W.; Lin, K.Y.; Chen, S.W.; Lin, C.Y.; Lu, Y.C.; Hsu, P.C.; Lee, S.C.; Tsai, H.J. Transgenic zebrafish model to study translational control mediated by upstream open reading frame of human chop gene. Nucleic Acids Res. 2011, 39, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Hampf, M.; Gossen, M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006, 356, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.L.; Hsieh, A.C. The Untranslated Regions of mRNAs in Cancer. Trends Cancer 2019, 5, 245–262. [Google Scholar] [CrossRef] [Green Version]
- García-Nieto, P.E.; Morrison, A.J.; Fraser, H.B. The somatic mutation landscape of the human body. Genome Biol. 2019, 20, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Milanese, J.-S.; Wang, E. Germline mutations and their clinical applications in cancer. Breast Cancer Manag. 2019, 8, 1–4. [Google Scholar] [CrossRef]
- Lawrence, M.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Spealman, P.; Naik, A.W.; May, G.E.; Kuersten, S.; Freeberg, L.; Murphy, R.F.; McManus, J. Conserved non-AUG uORFs revealed by a novel regression analysis of ribosome profiling data. Genome Res. 2018, 28, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Andreev, D.E.; O’connor, P.B.; Fahey, C.; Kenny, E.M.; Terenin, I.M.; Dmitriev, S.E.; Cormican, P.; Morris, D.W.; Shatsky, I.N.; Baranov, P.V. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. Elife 2015, 4, 1–21. [Google Scholar] [CrossRef]
- Palmer, C.J.; Galan-Caridad, J.M.; Weisberg, S.P.; Lei, L.; Esquilin, J.M.; Croft, G.F.; Wainwright, B.; Canoll, P.; Owens, D.M.; Reizis, B. Zfx facilitates tumorigenesis caused by activation of the Hedgehog pathway. Cancer Res. 2014, 74, 5914–5924. [Google Scholar] [CrossRef] [Green Version]
- Faria, J.A.Q.A.; Corrêa, N.C.R.; de Andrade, C.; de Angelis Campos, A.C.; dos Santos Samuel de Almeida, R.; Rodrigues, T.S.; Miranda de Goes, A.; Gomes, D.A.; Silva, F.P. SET domain-containing Protein 4 (SETD4) is a Newly Identified Cytosolic and Nuclear Lysine Methyltransferase involved in Breast Cancer Cell Proliferation. J. Cancer Sci. 2013, 29, 997–1003. [Google Scholar]
- Feng, X.; Lu, H.; Yue, J.; Shettigar, M.; Liu, J.; Denzin, L.K.; Shen, Z. Deletion of Mouse Setd4 Promotes the Recovery of Hematopoietic Failure. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 779–792. [Google Scholar] [CrossRef]
- Shukla, S.; Bjerke, G.A.; Muhlrad, D.; Yi, R.; Parker, R. The ribonuclease PARN controls the levels of specific miRNAs that contribute to p53 regulation. Mol. Cell. 2019, 73, 1204–1216. [Google Scholar] [CrossRef] [Green Version]
- Hinnebusch, A.G. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005, 59, 407–450. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fu, Y.; Wang, Z.; Zhou, S.; Sun, Y.; Wu, Y.; Xu, A. HLA-DRB1 may be antagonistically regulated by the coordinately evolved promoter and 3′-UTR under stabilizing selection. PLoS ONE 2011, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Crivello, P.; Ahci, M.; Maaßen, F.; Wossidlo, N.; Arrieta-Bolaños, E.; Heinold, A.; Lange, V.; Falkenburg, J.H.F.; Horn, P.A.; Fleischhauer, K.; et al. Multiple Knockout of Classical HLA Class II β-Chains by CRISPR/Cas9 Genome Editing Driven by a Single Guide RNA. J. Immunol. 2019, 202, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Wang, H.; Chen, W.; Sun, Z.; Chen, J.; Xu, Y.; Weng, M.; Shi, Q.; Ma, D.; Miao, C. Ubiquitylation of MFHAS1 by the ubiquitin ligase praja2 promotes M1 macrophage polarization by activating JNK and p38 pathways. Cell Death Dis. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Shi, Q.Q.; Zhu, M.M.; Shen, J.; Wang, H.H.; Ma, D.; Miao, C.H. MFHAS1 Is Associated with Sepsis and Stimulates TLR2/NF-κB Signaling Pathway Following Negative Regulation. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Xu, Y.; Zhong, J.; Wang, H.; Weng, M.; Cheng, Q.; Wu, Q.; Sun, Z.; Jiang, H.; Zhu, M.; et al. MFHAS1 promotes colorectal cancer progress by regulating polarization of tumor-associated macrophages via STAT6 signaling pathway. Oncotarget 2016, 7, 78726–78735. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage polarization in the development and progression of ovarian cancers: An overview. Front. Oncol. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage Polarization: Anti-Cancer Strategies to Target Tumor-Associated Macrophage in Breast Cancer. J. Cell. Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef]
- Lee, D.S.M.; Park, J.; Kromer, A.; Baras, A.; Rader, D.J.; Ritchie, M.D.; Ghanem, L.R.; Barash, Y. Disrupting upstream translation in mRNAs is associated with human disease. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Slavoff, S.A.; Mitchell, A.J.; Schwaid, A.G.; Cabili, M.N.; Ma, J.; Levin, J.Z.; Karger, A.D.; Budnik, B.A.; Rinn, J.L.; Saghatelian, A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2013, 9, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.J.; Rothnagel, J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef]
- Dmitriev, S.E.; Vladimirov, D.O.; Lashkevich, K.A. A Quick Guide to Small-Molecule Inhibitors of Eukaryotic Protein Synthesis. Biochemistry 2020, 85, 1389–1421. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| uORF-Associated Nucleotides in hg38 uAUG: 190,878 aTIS: 2,515,399 uStop: 624,157 | |||||||
|---|---|---|---|---|---|---|---|
| Type of Cancer | BRCA | COAD | LUAD | PRAD | SKCM | LAML | All |
| Patients, n | 1044 | 433 | 569 | 498 | 470 | 149 | 3163 |
| filters to identify SNVs ≥10 sequencing reads in tumor and normal BAM file ≥3 alternative reads in tumor VCF file SNV alters/deletes uAUG or aTIS or deletes uStop codon ≥1 TV includes the SNV | |||||||
| All SNVs | 7278 | 8756 | 8161 | 2017 | 26051 | 1913 | 48491 |
| Affected patients, % | 99.8 | 99.1 | 100 | 99.6 | 100 | 100 | 99.7 |
| additional filters to identify somatic SNVs ≥4 x higher alt/ref read ratio in tumor ≥2 patients with same somatic mutation | |||||||
| Recurrent somatic SNVs | 1029 | 339 | 494 | 114 | 3748 | 258 | 5277 |
| Affected patients, % | 68.0 | 67.4 | 68.0 | 38.8 | 86.8 | 75.2 | 66.5 |
| Variants in ≥1% of pts. | 94 | 37 | 75 | 14 | 241 | 258 | 567 |
| Affected patients, % | 35.1 | 40.4 | 31.1 | 20.9 | 61.7 | 75.2 | 38.7 |
| SNVs w/o dbSNP entry | 80 | 45 | 56 | 3 | 2112 | 72 | 2363 |
| % of recurrent som. SNVs | 7.5 | 13.3 | 11.3 | 2.6 | 56.4 | 27.9 | 44.8 |
| Gene Symbol RefSeq ID | Genomic Position | Codon Variant | Functional Effect | Type of Cancer (% of Patients) | uORF Size in bp | # of uAUG in TLS | uStop to CDS Distance (bp) | Kozak Context | TV with uORF |
|---|---|---|---|---|---|---|---|---|---|
| ASNS NM_001352496.2 | chr7: 97928300 | AUG.2 > GUG | uAUG > aTIS | BRCA (0.4) | 303 | 4 | 74 | ++ | 1/7 |
| BAGE2 NM_182482.2 | chr21: 10413537 | CUG.1 > CCG | loss of aTIS | COAD (2.1); LUAD (1.4); LAML (1.3); SKCM (0.9); PRAD (0.6); BRCA (1.0) | 78 | 3 | 126 | + | 5/5 |
| BAGE2 NM_182482.2 | chr21: 10413594 | UUG.1 > GUG | syn aTIS | SKCM (1.9); BRCA (1.3); PRAD (0.8); COAD (0.6) | 105 | 3 | 41 | − | 5/5 |
| CFH NM_000186.3 | chr1: 196651949 | UUG.3 > UUU/ GUG.5 > UUG | loss of aTIS/ syn aTIS | BRCA (2.2) | 39/138 | 1 | 132/31 | + | 2/2 |
| CHCHD2 NM_001320327.1 | chr7: 56106490 | AUC.1 > AUU | loss of aTIS | SKCM (8.1) | 63 | 1 | 16 | − | 2/2 |
| DIS3L NM_133375.4 | chr15: 66294990 | AUG.1 > AUU | uAUG > aTIS | SKCM (0.9) | 27 | 2 | 83 | ++ | 10/13 |
| ESRRG NM_001438.4 | chr1: 216723407 | GUG.1 > GGG | loss of aTIS | BRCA (3.0) | 39 | 1 | 70 | + | 4/20 |
| FANK1 NM_145235.5 | chr10: 125896600 | AGG.2 > ACG | syn aTIS | COAD (1.6) | 105 | 0 | 44 | + | 2/3 |
| FRG2C NM_001124759.3 | chr3: 75664297 | ACG.2 > AUG | aTIS > uAUG | LAML (6.0); SKCM (2.1); LUAD (0.7); PRAD (0.4) | 21 | 3 | 63 | + | 1/1 |
| HLA-DRB1 NM_002124.3 | chr6: 32589821 | AUG.1 > AUA/ GUG.1 > AUG | uAUG > aTIS/ aTIS > uAUG | LAML (1.3); LUAD (1.1); PRAD (0.6); BRCA (0.6); SKCM (0.4) | 36/30 | 1 | 45/49 | + | 2/4 |
| HLA-DRB1 NM_002124.3 | chr6: 32589752 | CUG.1 > CUC | loss of GTG | COAD (1.6); SKCM (1.3); LUAD (0.5); BRCA (0.2) | 66 | 1 | −13 | + | 2/4 |
| HLA-DRB1 NM_002124.3 | chr6: 32589795 | CUG.6 > GUG | syn aTIS | LAML (2.7); LUAD (0.3); BRCA (0.3) | 813 | 1 | −801 | − | 2/4 |
| NDST3 NM_004784.3 | chr4: 118053872 | AUG.4 > AUU | uAUG > aTIS | SKCM (1.1) | 21 | 4 | 20 | + | 1/1 |
| PJA2 NM_014819.4 | chr5: 109383504 | UUG.2 > UUU | loss of aTIS | LAML (2.0); LUAD (1.4); BRCA (1.3); PRAD (0.4) | 15 | 0 | 58 | + | 1/1 |
| PRKCQ NM_001282644.2 | chr10: 6515109 | UUG.2 > UUU | loss of aTIS | LAML (3.4); BRCA (2.8); LUAD (1.8); PRAD (0.6); SKCM (0.4) | 156 | 1 | −74 | + | 4/7 |
| PRMT8 NM_019854.5 | chr12: 3491521 | UUG.5 > GUG | syn aTIS | BRCA (1.7) | 1290 | 1 | −1185 | ++ | 1/2 |
| RBBP4 NM_001135256.1 | chr1: 32651992 | AUG.3 > UUG | uAUG > aTIS | LAML (2.0) | 12 | 3 | −1 | − | 1/3 |
| ROPN1 NM_017578.4 | chr3: 123980570 | GUG.6 > GGG | loss of aTIS | BRCA (2.2); LAML (1.3) | 54 | 4 | 36 | − | 1/3 |
| SETD4 NM_001007261.2 | chr21: 36058888 | AUG.1 > UUG | uAUG > aTIS | LAML (1.3) | 174 | 3 | −8 | + | 4/4 |
| SYNPR NM_144642.5 | chr3: 63443357 | GUG.2 > GGG | loss of aTIS | BRCA (8.2); COAD (4.4); LAML (3.4) | 30 | 1 | 57 | − | 1/2 |
| TEDDM1 NM_172000.4 | chr1: 182400520 | AUG.2 > UUG | uAUG > aTIS | LAML (2.7) | 39 | 2 | −4 | + | 1/1 |
| TMIGD3 NM_001081976.2 | chr1: 111563996 | UUG.1 > GUG | syn aTIS | LAML (4.0); LUAD (0.7); PRAD (0.4); BRCA (0.3) | 138 | 0 | −94 | + | 2/3 |
| TNFSF8 NM_001244.3 | chr9: 114930326 | AUG.6 > GUG | uAUG > aTIS | LAML (1.4) | 147 | 6 | −147 | + | 2/2 |
| Gene Symbol RefSeq ID | Genomic Position | Codon Variant | New CDS Overlap | Type of Cancer (% of Patients) | # of uAUG in TLS | # of in-Frame uAUG + aTIS | uStop to CDS Distance in bp | New uStop to CDS Distance in bp | TV with uORF |
|---|---|---|---|---|---|---|---|---|---|
| ARPP21 NM_198399.2 | chr3: 35681735 | UAA.5 > UUA | yes | LAML (1.3) | 3 | 1 + 5 | 15 | −285 | 8/8 |
| FANK1 NM_145235.5 | chr10: 125896600 | UAG.1 > UAC | yes | COAD (1.6) | 0 | 0 + 3 | 42 | −1080 | 2/3 |
| FRG2C NM_001124759.3 | chr3: 75664315 | UAG.2 > UGG | yes | LAML (2.7); SKCM (0.6); PRAD (0.6) | 3 | 0 + 3 | 63 | −912 | 1/1 |
| NDST3 NM_004784.3 | chr4: 118053862 | UAA.5 > UAU | no | LAML (2.7) | 4 | 0 + 1 | 47 | +24 | 1/1 |
| OLIG3 NM_175747.2 | chr6: 137494221 | UAA.3 > AAA | no | LAML (2.7) | 0 | 0 + 8 | 49 | +12 | 1/1 |
| ROPN1 NM_017578.4 | chr3: 123980570 | UGA.5 > GGA | yes | BRCA (2.2); LAML (1.3) | 4 | 0 + 9 | 86 | −141 | 1/3 |
| TNFSF8 NM_001244.3 | chr9: 114930326 | UGA.8 > UGG | yes | LAML (1.3) | 6 | 2 + 8 | 22 | −240 | 2/2 |
| ZNF596 NM_001287256.1 | chr8: 240836 | UAG.1 > UUG | no | LAML (3.4) | 1 | 0 + 5 | 58 | +51 | 7/7 |
| ZSCAN21 NM_145914.3 | chr7: 100056915 | UAA.1 > UUA | yes | LAML (3.4) | 1 | 1 + 0 | 90 | −1935 | 4/4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jürgens, L.; Manske, F.; Hubert, E.; Kischka, T.; Flötotto, L.; Klaas, O.; Shabardina, V.; Schliemann, C.; Makalowski, W.; Wethmar, K. Somatic Functional Deletions of Upstream Open Reading Frame-Associated Initiation and Termination Codons in Human Cancer. Biomedicines 2021, 9, 618. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060618
Jürgens L, Manske F, Hubert E, Kischka T, Flötotto L, Klaas O, Shabardina V, Schliemann C, Makalowski W, Wethmar K. Somatic Functional Deletions of Upstream Open Reading Frame-Associated Initiation and Termination Codons in Human Cancer. Biomedicines. 2021; 9(6):618. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060618
Chicago/Turabian StyleJürgens, Lara, Felix Manske, Elvira Hubert, Tabea Kischka, Lea Flötotto, Oliver Klaas, Victoria Shabardina, Christoph Schliemann, Wojciech Makalowski, and Klaus Wethmar. 2021. "Somatic Functional Deletions of Upstream Open Reading Frame-Associated Initiation and Termination Codons in Human Cancer" Biomedicines 9, no. 6: 618. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060618