
Cyclodextrin Dispersion of Mebendazole and Flubendazole Improves In Vitro Antiproliferative Activity

 ,
, .jpg) , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
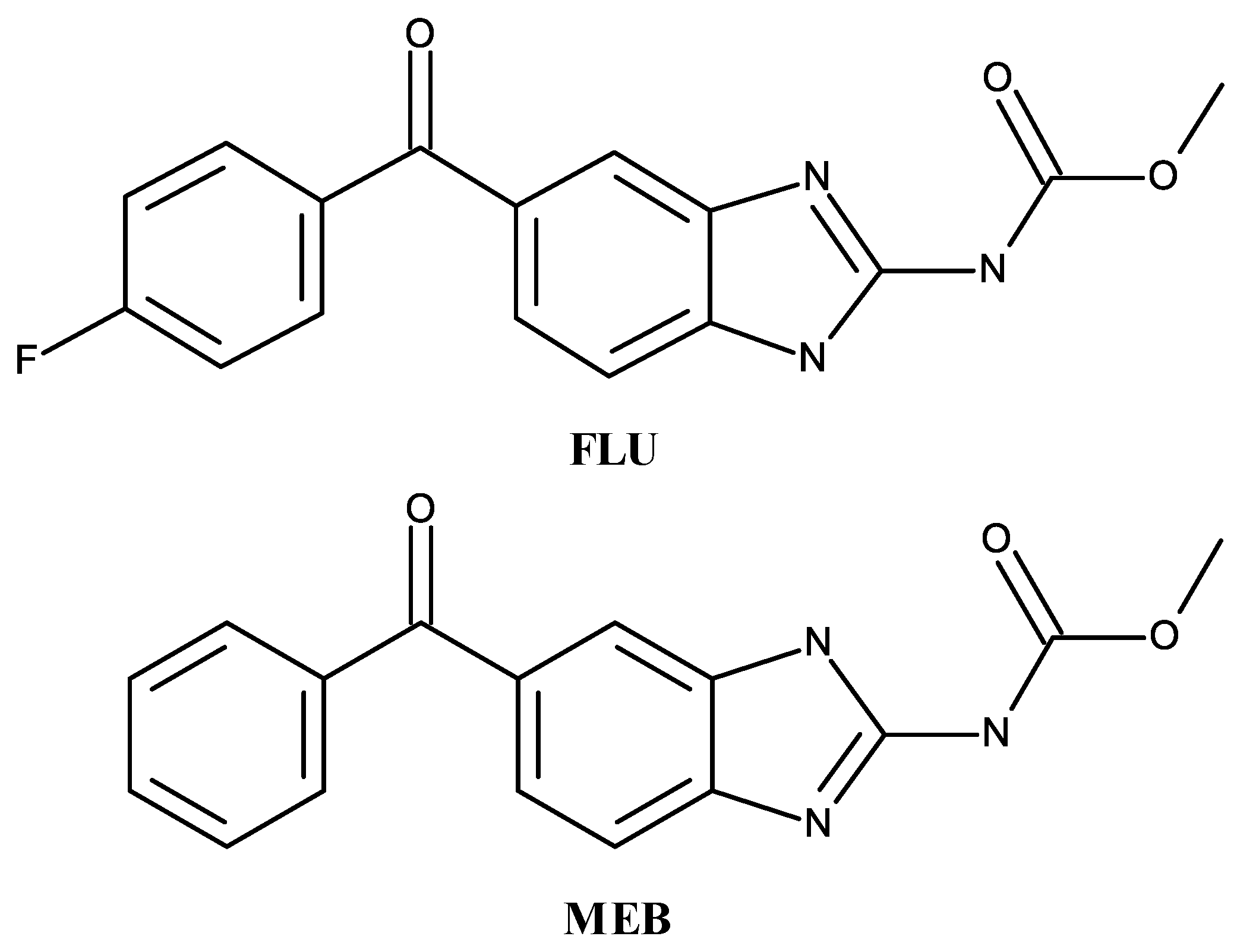
2.1. Preparation of Binary Conjugates
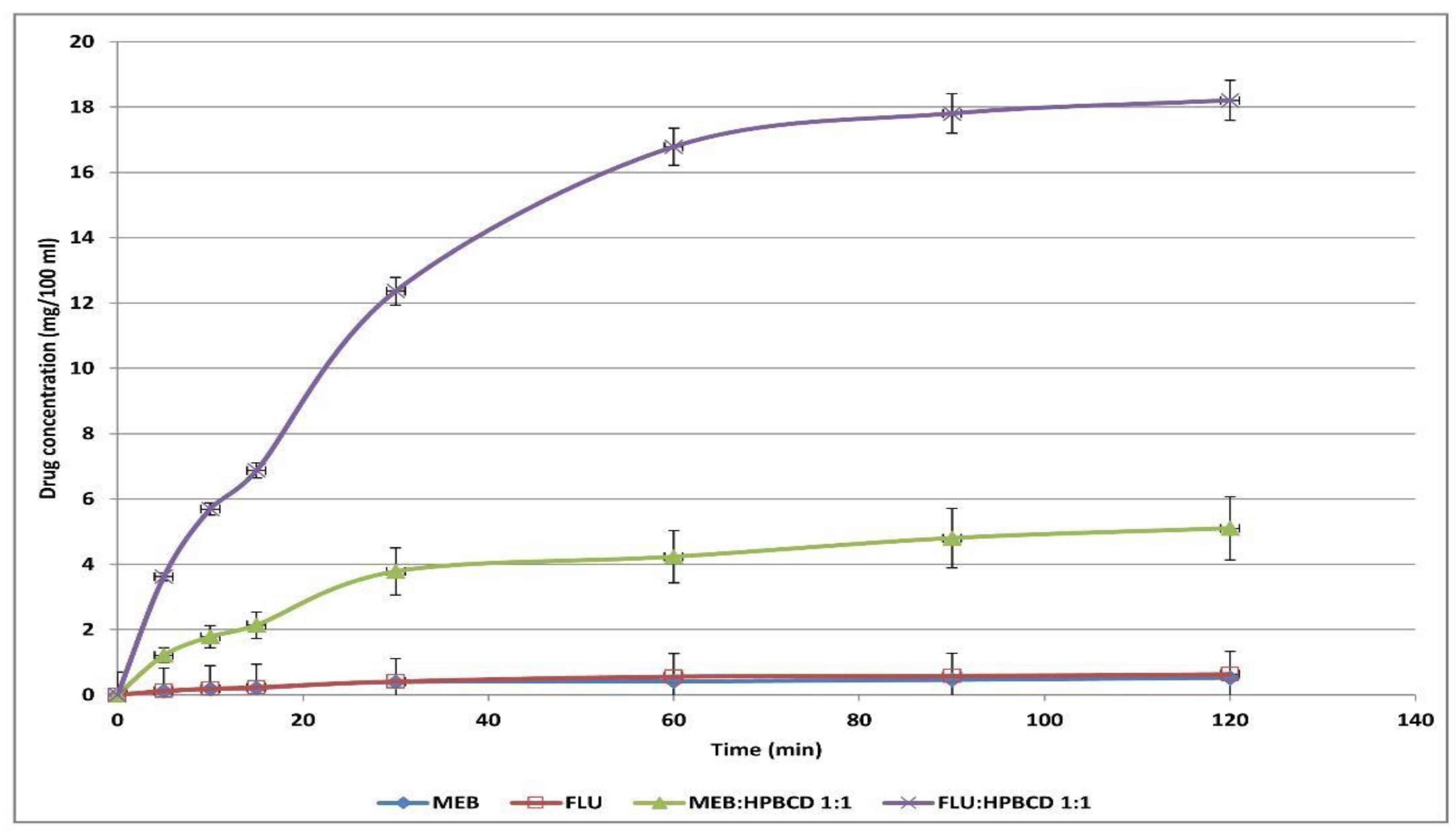
2.2. In Vitro Dissolution Studies
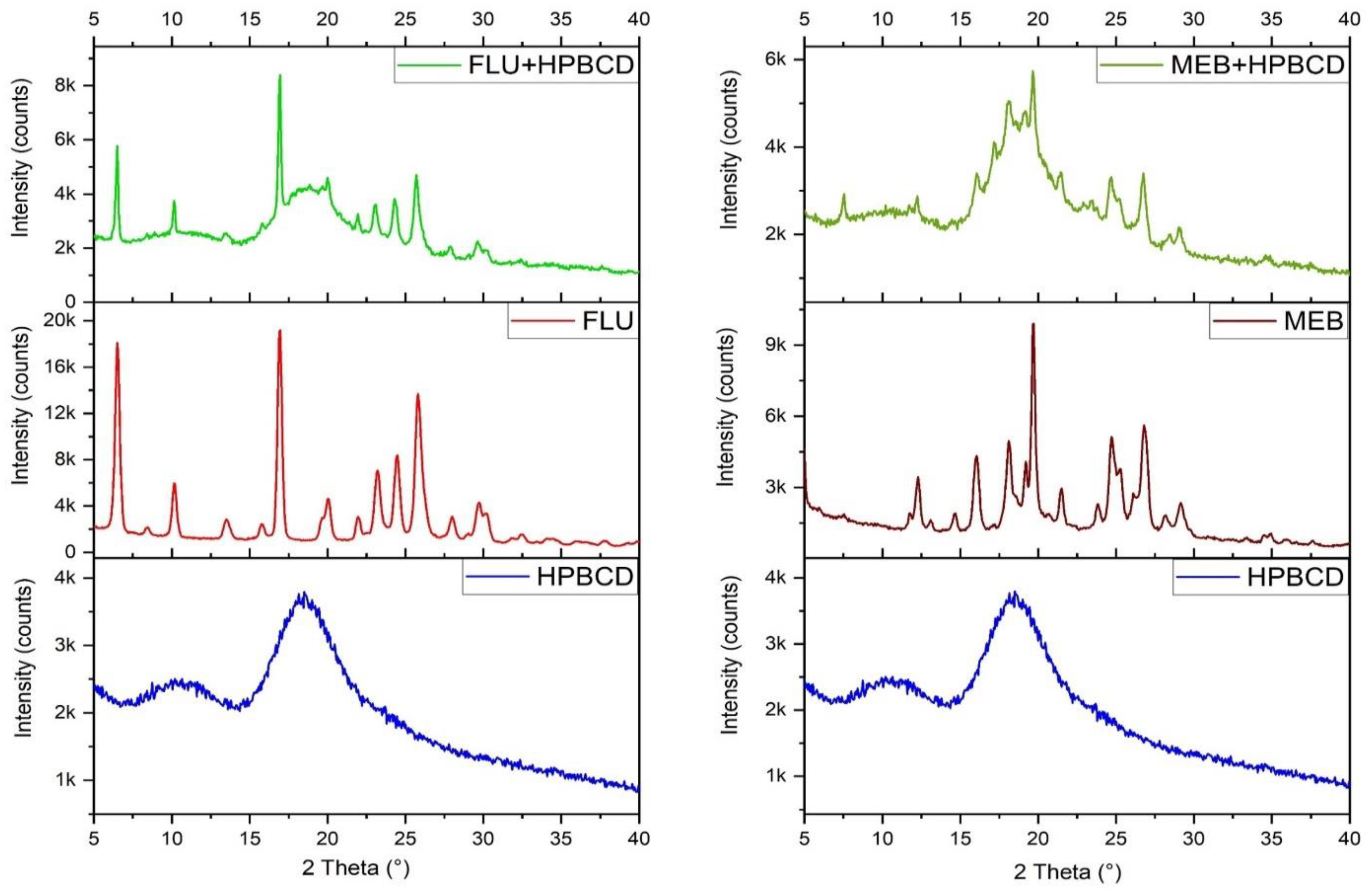
2.3. X-ray Diffraction
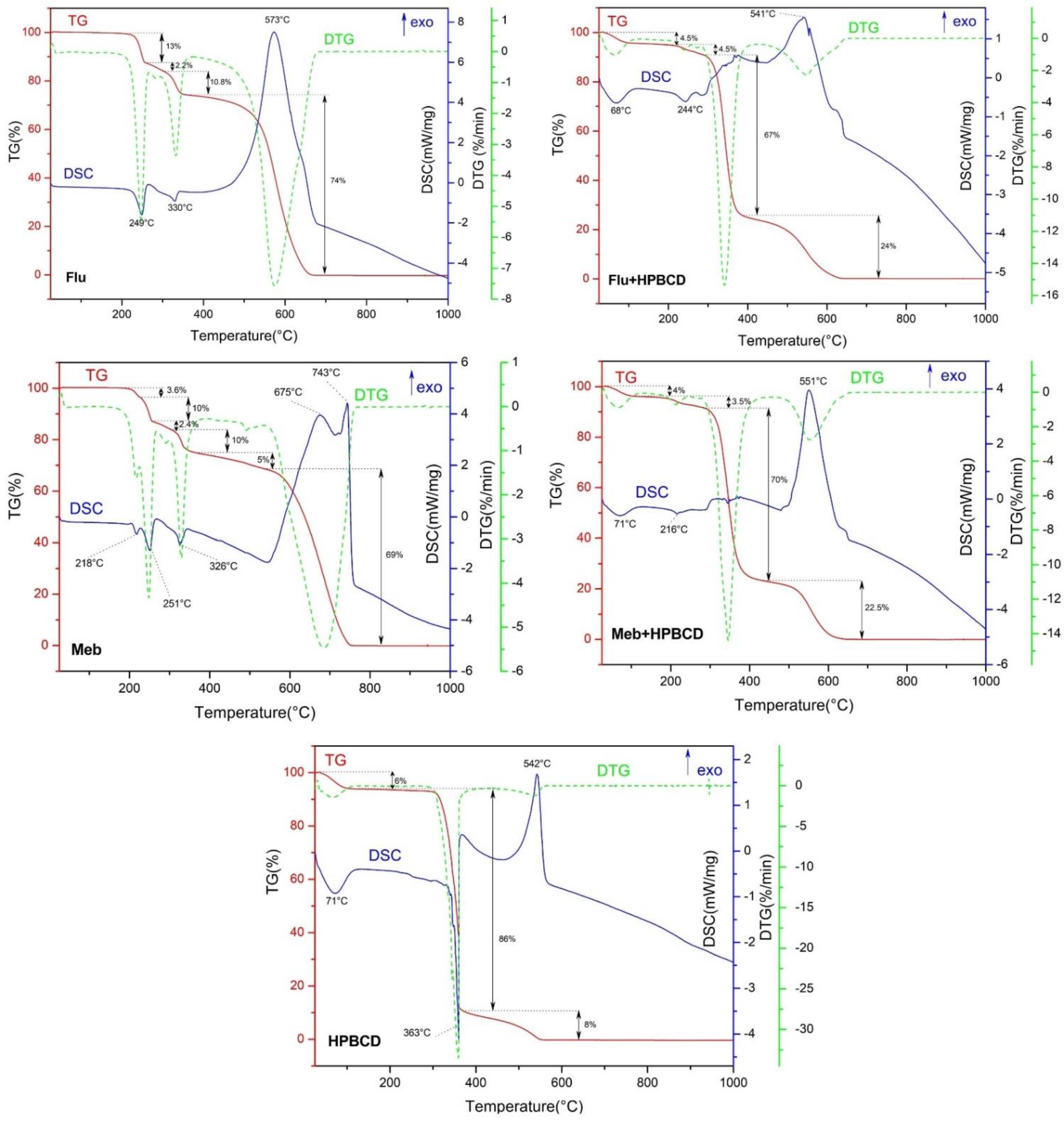
2.4. Thermal Analysis
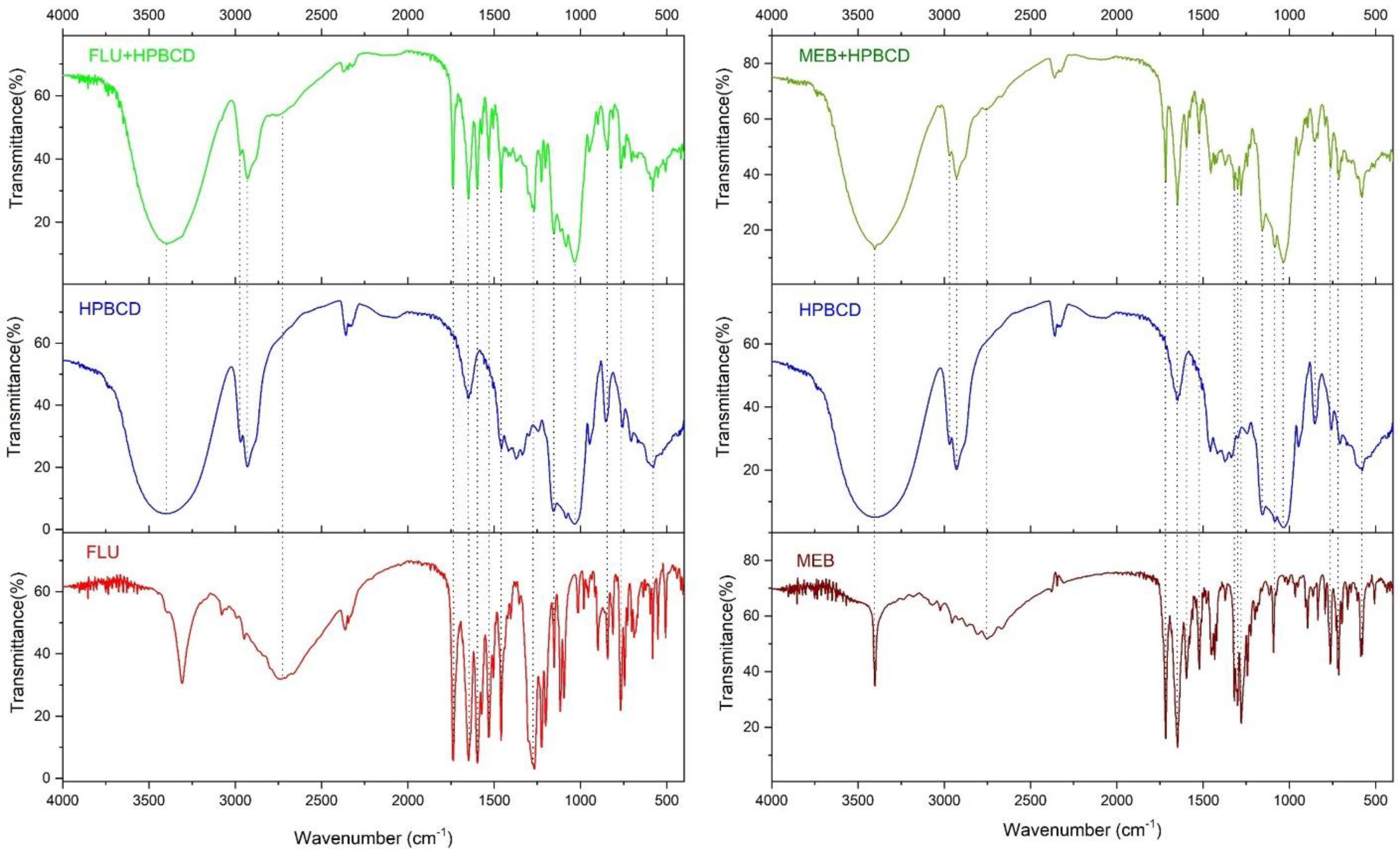
2.5. FTIR Spectrometry
2.6. Cell Culture
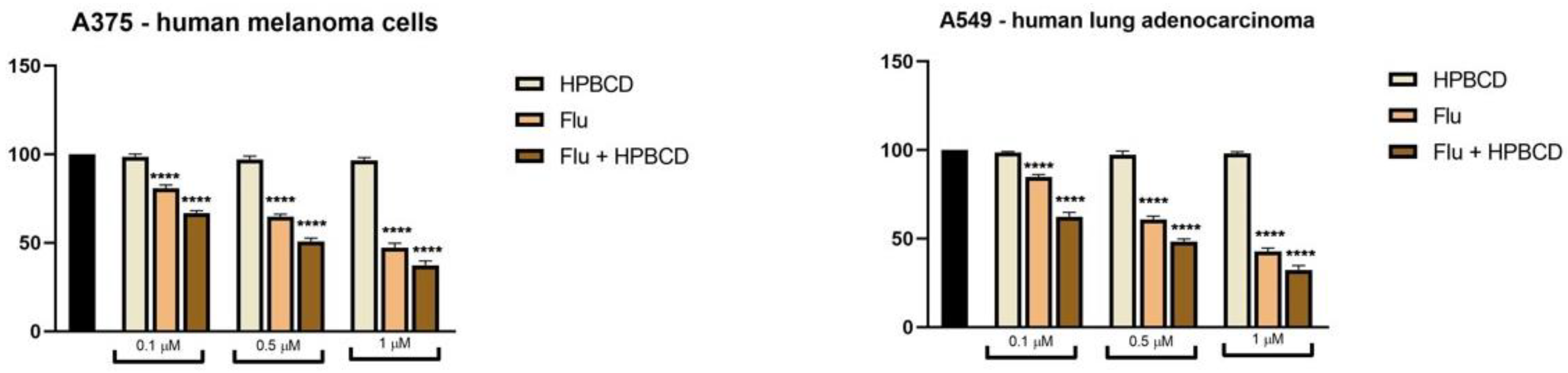
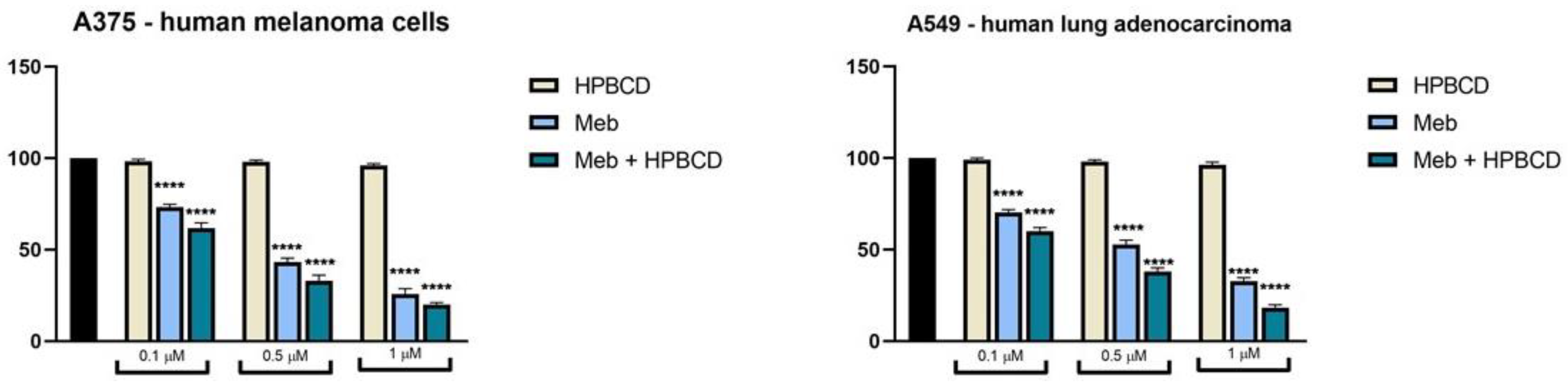
2.7. Cell Viability Assessment
2.8. Molecular Docking Analysis
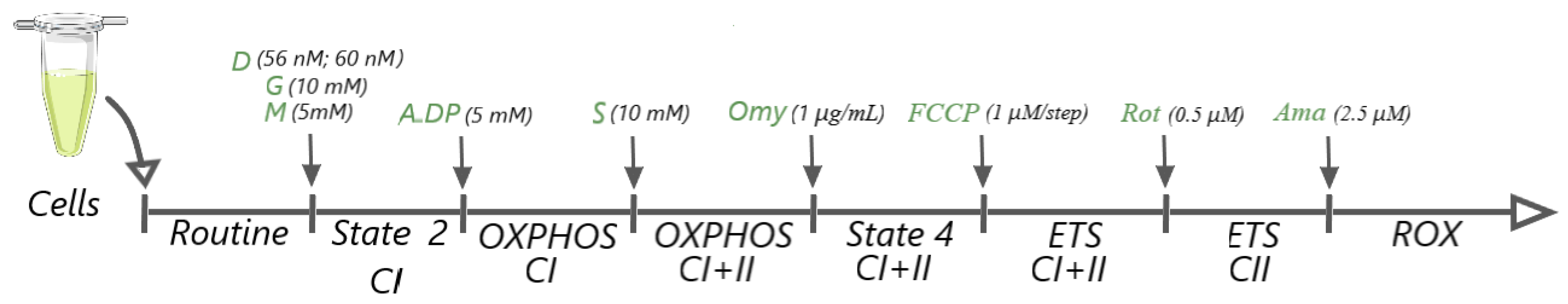
2.9. High-Resolution Respirometry Studies in Permeabilized Cells
- State 2CI: the basal respiration in a non-phosphorylating state; mitochondrial respiration dependent on CI (NADH-generating) substrates.
- OXPHOSCI: the active phosphorylating respiration in the presence of CI substrates.
- OXPHOSCI+CII: the active maximal phosphorylating respiration in the presence of CI and CII substrates.
- State 4CI+CII: the basal/leak respiration dependent on both CI and CII substrates in a non-phosphorylating state—after inhibition of ATP synthase.
- ETSCI+CII: mitochondrial respiration in a fully uncoupled state that reveals the maximal electron transport system capacity:
- ROX: the non-mitochondrial residual oxygen consumption—after CI and CIII inhibition by rotenone and antimycin A, respectively.
Data Acquisition and Statistical Analysis
2.10. HET-CAM Assay
3. Results and Discussion
3.1. In Vitro Dissolution Test
3.2. X-ray Diffraction
3.3. Thermal Analysis
3.4. FTIR Spectroscopy
3.5. FLU and MEB Exert Dose-Dependent Cytotoxic Effects
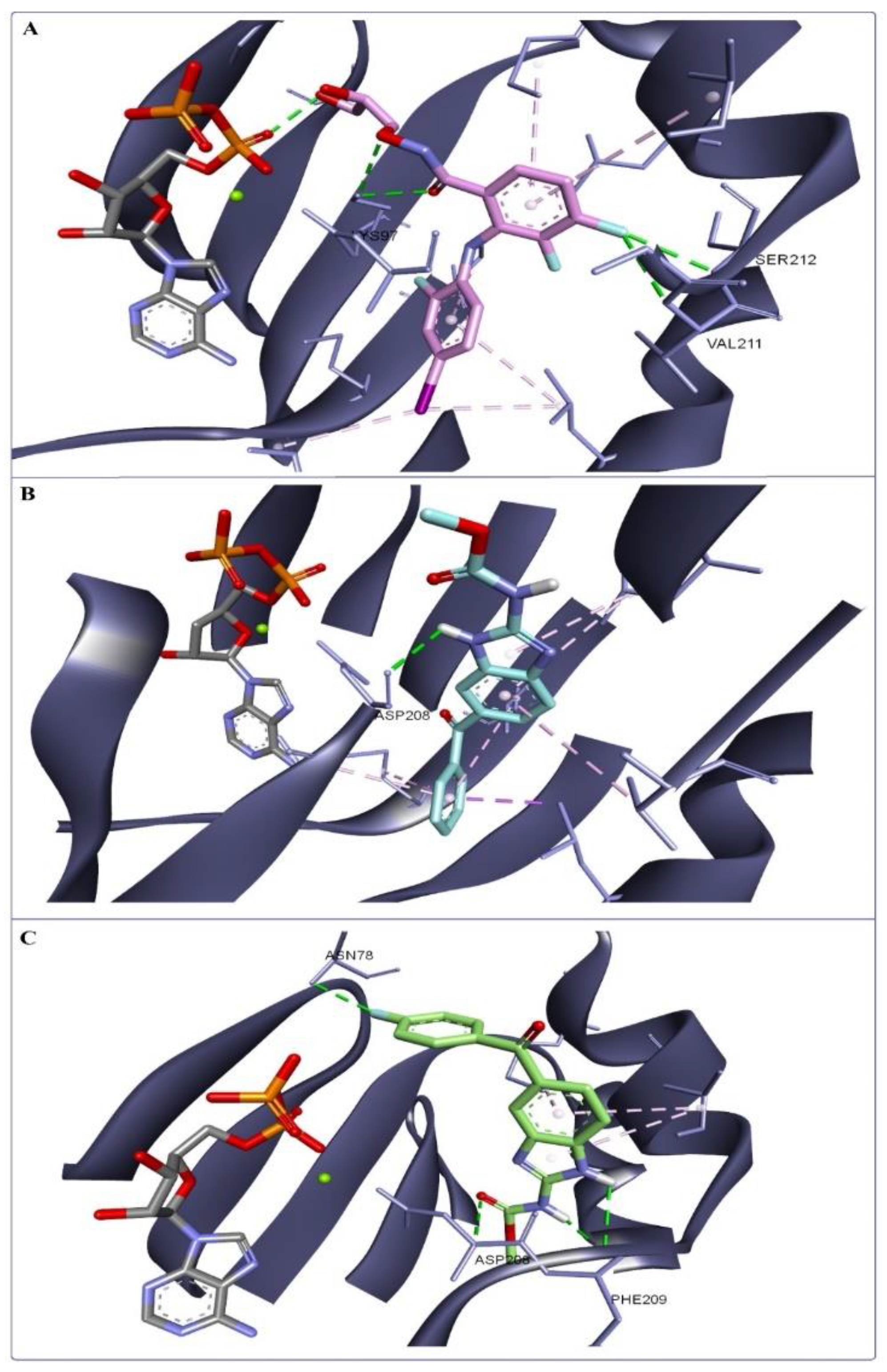
3.6. Molecular Docking
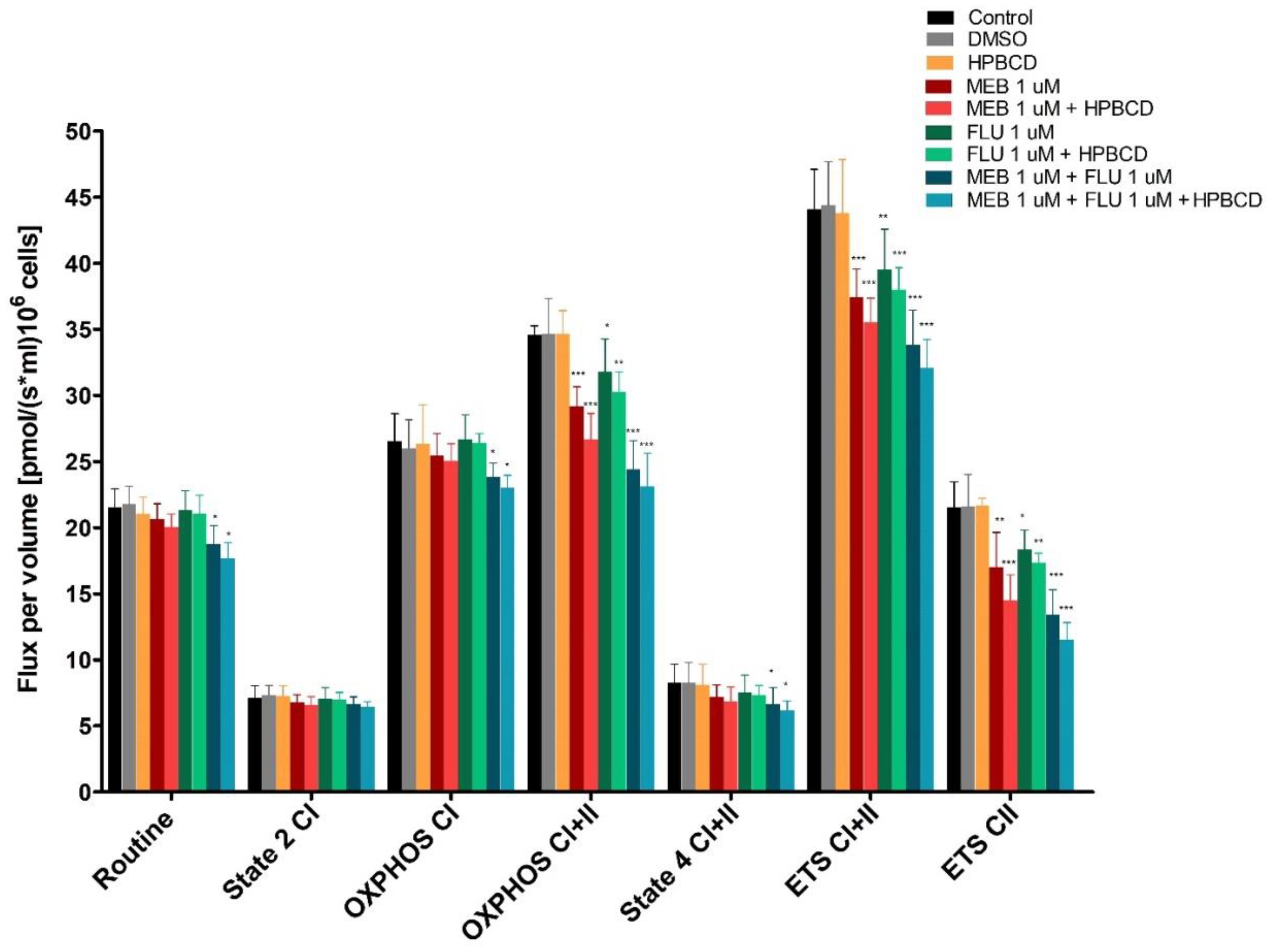
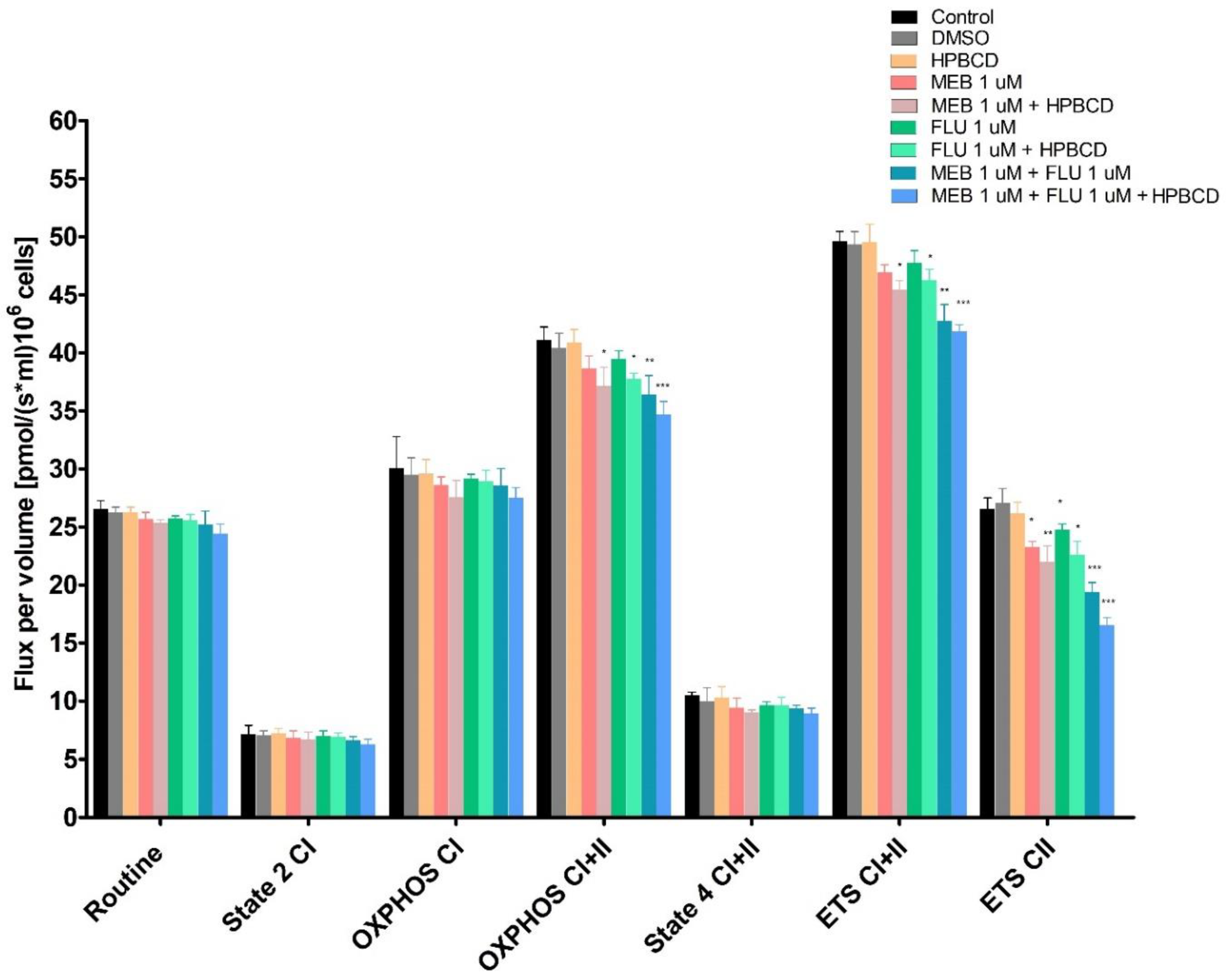
3.7. High-Resolution Respirometry Studies in Permeabilized Cells
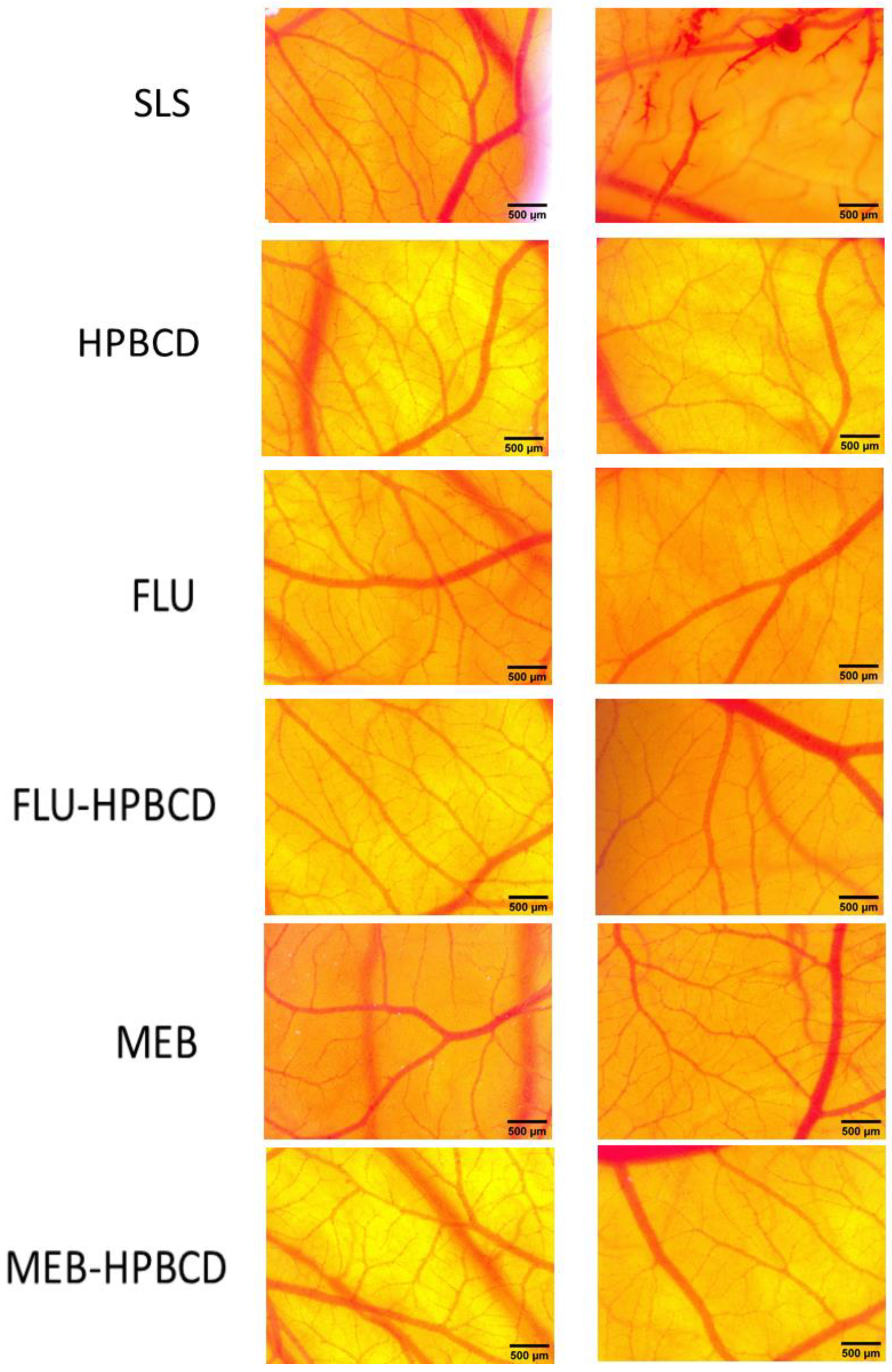
3.8. CAM Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Geary, T.G.; MacKenzie, C.D.; Silber, S.A. Flubendazole as a macrofilaricide: History and background. PLoS Negl. Trop. Dis. 2019, 13, e0006436. [Google Scholar] [CrossRef] [Green Version]
- Laclette, J.; Guerra, G.; Zetina, C. Inhibition of tubulin polymerization by Mebendazole. Biochem. Biophys. Res. Commun. 1980, 92, 417–423. [Google Scholar] [CrossRef]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature. Cancers 2019, 11, 1284. [Google Scholar] [CrossRef] [Green Version]
- Popović, D.J.; Poša, M.; Popović, K.J.; Kolarović, J.; Popović, J.K.; Banović, P. Application of a widely-used tropical anti-worm agent, mebendazole, in modern oncology. Trop. J. Pharm. Res. 2017, 16, 2555–2562. [Google Scholar] [CrossRef]
- Huang, H.; He, Q.; Guo, B.; Xu, X.; Wu, Y.; Li, X. Progress in Redirecting Antiparasitic Drugs for Cancer Treatment. Drug Des. Dev. Ther. 2021, 15, 2747–2767. [Google Scholar] [CrossRef]
- Son, D.-S.; Lee, E.-S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, e29. [Google Scholar] [CrossRef] [PubMed]
- Nath, J.; Paul, R.; Ghosh, S.K.; Paul, J.; Singha, B.; Debnath, N. Drug repurposing and relabeling for cancer therapy: Emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 2020, 258, 118189. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443. [Google Scholar] [CrossRef]
- Williamson, T.; de Abreu, M.C.; Trembath, D.G.; Brayton, C.; Kang, B.; Mendes, T.B.; de Assumpção, P.P.; Cerutti, J.M.; Riggins, G.J. Mebendazole disrupts stromal desmoplasia and tumorigenesis in two models of pancreatic cancer. Oncotarget 2021, 12, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, C.G.; Williamson, T.; Riggins, G.J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neuro-Oncol. 2018, 140, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.R.; Selvin, T.; Blom, K.; Rubin, J.; Berglund, M.; Jarvius, M.; Lenhammar, L.; Parrow, V.; Loskog, A.; Fryknäs, M.; et al. Mebendazole is unique among tubulin-active drugs in activating the MEK–ERK pathway. Sci. Rep. 2020, 10, 13124. [Google Scholar] [CrossRef]
- Xie, X.; Cai, X.; Tang, Y.; Jiang, C.; Zhou, F.; Yang, L.; Liu, Z.; Wang, L.; Zhao, H.; Zhao, C.; et al. Flubendazole Elicits Antitumor Effects by Inhibiting STAT3 and Activating Autophagy in Non-small Cell Lung Cancer. Front. Cell Dev. Biol. 2021, 9, 2260. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.-J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.-J.; Zheng, F.-M.; Xu, J.; Xu, L.-Z.; Long, Z.-J.; et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-Y.; Jung, B.-K.; Hong, S.-J. Albendazole and Mebendazole as Anti-Parasitic and Anti-Cancer Agents: An Update. Korean J. Parasitol. 2021, 59, 189–225. [Google Scholar] [CrossRef] [PubMed]
- Santiago, T.; de la Torre-Iglesias, P.M.; Torrado, G.; Torrado, S.; Bolas, F.; Garcia, J.J. Enhanced bioavailability and anthelmintic efficacy of mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des. Dev. Ther. 2014, 8, 1467–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, S.; Garg, T.; Rath, G.; Murthy, R.R.; Goyal, A.K. Enhancing the bioavailability of mebendazole by integrating the principles solid dispersion and nanocrystal techniques, for safe and effective management of human echinococcosis. Artif. Cells Nanomed. Biotechnol. 2016, 44, 937–942. [Google Scholar] [CrossRef]
- Studenovský, M.; Rumlerová, A.; Kostka, L.; Etrych, T. HPMA-Based Polymer Conjugates for Repurposed Drug Mebendazole and Other Imidazole-Based Therapeutics. Polymers 2021, 13, 2530. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, X.; Yao, X.; Cai, Y. Synthesis of chloromethyl benzimidazole grafted polybenzimidazole with enhanced UV absorption and its effect as a stabilizer on the ultraviolet-aging behaviors of poly(vinyl chloride) films. J. Appl. Polym. Sci. 2019, 136, 47626. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Tichý, T.; Vávra, J.; Dash, R.P.; Slusher, C.E.; Gadiano, A.J.; Wu, Y.; Jančařík, A.; Tenora, L.; Monincová, L.; et al. N-Substituted Prodrugs of Mebendazole Provide Improved Aqueous Solubility and Oral Bioavailability in Mice and Dogs. J. Med. Chem. 2018, 61, 3918–3929. [Google Scholar] [CrossRef]
- Real, D.; Leonardi, D.; Williams, R.O.; Repka, M.A.; Salomon, C.J. Solving the Delivery Problems of Triclabendazole Using Cyclodextrins. AAPS PharmSciTech 2018, 19, 2311–2321. [Google Scholar] [CrossRef]
- Saidman, E.; Chattah, A.K.; Aragón, L.; Sancho, M.; Camí, G.; Garnero, C.; Longhi, M. Inclusion complexes of β-cyclodextrin and polymorphs of mebendazole: Physicochemical characterization. Eur. J. Pharm. Sci. 2019, 127, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Lahiani-Skiba, M.; Coquard, A.; Bounoure, F.; Vérité, P.; Arnaud, P.; Skiba, M. Mebendazole complexes with various cyclodextrins: Preparation and physicochemical characterization. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 197–201. [Google Scholar] [CrossRef]
- Ceballos, L.; Moreno, L.; Torrado, J.J.; Lanusse, C.; Álvarez, L. Exploring flubendazole formulations for use in sheep. Pharmacokinetic evaluation of a cyclodextrin-based solution. BMC Vet. Res. 2012, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, M.; Agha, B.; Rothweiler, F.; Löschmann, N.; Voges, Y.; Mittelbronn, M.; Starzetz, T.; Harter, P.N.; Abhari, B.A.; Fulda, S.; et al. Identification of flubendazole as potential anti-neuroblastoma compound in a large cell line screen. Sci. Rep. 2015, 5, 8202. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, L.; Alvarez, L.; Mackenzie, C.; Geary, T.; Lanusse, C. Pharmacokinetic comparison of different flubendazole formulations in pigs: A further contribution to its development as a macrofilaricide molecule. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Priotti, J.; Baglioni, M.V.; García, A.; Rico, M.J.; Leonardi, D.; Lamas, M.C.; Márquez, M.M. Repositioning of Anti-parasitic Drugs in Cyclodextrin Inclusion Complexes for Treatment of Triple-Negative Breast Cancer. AAPS PharmSciTech 2018, 19, 3734–3741. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Press, M.F.; Lenz, H.-J. EGFR, HER2 and VEGF Pathways: Validated targets for cancer treatment. Drugs 2007, 67, 2045–2075. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Macașoi, I.; Pavel, I.Z.; Moacă, A.E.; Avram, Ș.; David, V.L.; Coricovac, D.; Mioc, A.; Spandidos, D.A.; Tsatsakis, A.; Șoica, C.; et al. Mechanistic investigations of antitumor activity of a Rhodamine B-oleanolic acid derivative bioconjugate. Oncol. Rep. 2020, 44, 1169–1183. [Google Scholar] [CrossRef]
- Jianu, C.; Stoin, D.; Cocan, I.; David, I.; Pop, G.; Lukinich-Gruia, A.; Mioc, M.; Mioc, A.; Șoica, C.; Muntean, D.; et al. In Silico and in Vitro Evaluation of the Antimicrobial and Antioxidant Potential of Mentha × smithiana R. GRAHAM Essential Oil from Western Romania. Foods 2021, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.; Steck, W. A modified HET–CAM assay approach to the assessment of anti-irritant properties of plant extracts. Food Chem. Toxicol. 2000, 38, 867–872. [Google Scholar] [CrossRef]
- Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) ICCVAM-Recommended Test Method Protocol: Hen’s Egg Test-Chorioallantoic Membrane. 2010. Available online: http://iccvam.niehs.nih.gov/ (accessed on 12 October 2021).
- Kishore, A.S.; Surekha, P.A.; Sekhar, P.V.R.; Srinivas, A.; Murthy, P.B. Hen Egg Chorioallantoic Membrane Bioassay: An in Vitro Alternative to Draize Eye Irritation Test for Pesticide Screening. Int. J. Toxicol. 2008, 27, 449–453. [Google Scholar] [CrossRef]
- Coricovac, D.; Farcas, C.; Nica, C.; Pinzaru, I.; Simu, S.; Stoian, D.; Soica, C.; Proks, M.; Avram, S.; Navolan, D.; et al. Ethinylestradiol and Levonorgestrel as Active Agents in Normal Skin, and Pathological Conditions Induced by UVB Exposure: In Vitro and In Ovo Assessments. Int. J. Mol. Sci. 2018, 19, 3600. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Fact Sheets-Cancer. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 12 October 2021).
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An Emerging Approach in Drug Discovery. In Drug Repurposing: Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020; Available online: https://www.intechopen.com/chapters/72744 (accessed on 12 October 2021).
- Su, J.; Chen, J.; Li, L.; Li, B.; Shi, L.; Zhang, H.; Ding, X. Preparation of Natural Borneol/2-Hydroxypropyl-β-cyclodextrin Inclusion Complex and Its Effect on the Absorption of Tetramethylpyrazine Phosphate in Mouse. Chem. Pharm. Bull. 2012, 60, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topal, B.; Altindal, D.; Gümüşderelioğlu, M. Melatonin/HPβCD complex: Microwave synthesis, integration with chitosan scaffolds and inhibitory effects on MG-63CELLS. Int. J. Pharm. 2015, 496, 801–811. [Google Scholar] [CrossRef]
- Vialpando, M.; Smulders, S.; Bone, S.; Jager, C.; Vodak, D.; Van Speybroeck, M.; Verheyen, L.; Backx, K.; Boeykens, P.; Brewster, M.E.; et al. Evaluation of Three Amorphous Drug Delivery Technologies to Improve the Oral Absorption of Flubendazole. J. Pharm. Sci. 2016, 105, 2782–2793. [Google Scholar] [CrossRef] [Green Version]
- Bezzon, V.D.; Ferreira, F.F.; Smith, P.; Benmore, C.J.; Byrn, S.R.; de Araujo, G.L.B. Amorphous dispersions of flubendazole in hydroxypropyl methylcellulose: Formulation stability assisted by pair distribution function analysis. Int. J. Pharm. 2021, 600, 120500. [Google Scholar] [CrossRef] [PubMed]
- Vigh, T.; Démuth, B.; Balogh, A.; Galata, D.L.; Van Assche, I.; Mackie, C.; Vialpando, M.; Van Hove, B.; Psathas, P.; Borbás, E.; et al. Oral bioavailability enhancement of flubendazole by developing nanofibrous solid dosage forms. Drug Dev. Ind. Pharm. 2017, 43, 1126–1133. [Google Scholar] [CrossRef]
- Swanepoel, E.; Liebenberg, W.; Devarakonda, B.; De Villiers, M.M. Developing a discriminating dissolution test for three mebendazole polymorphs based on solubility differences. Die Pharm. 2003, 58, 117–121. [Google Scholar]
- Camí, G.E.; Brusau, E.V.; Narda, G.E.; Maggio, R.M. Dual approach for concomitant monitoring of dissolution and transformation at solid-state. Mebendazole salts case study. J. Drug Deliv. Sci. Technol. 2020, 55, 101344. [Google Scholar] [CrossRef]
- Geng, Q.; Li, T.; Wang, X.; Chu, W.; Cai, M.; Xie, J.; Ni, H. The mechanism of bensulfuron-methyl complexation with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin and effect on soil adsorption and bio-activity. Sci. Rep. 2019, 9, 1882. [Google Scholar] [CrossRef]
- Kumar, R.; Sinha, V.R.; Dahiya, L.; Singh, G.; Sarwal, A. Impact of cyclodextrin derivatives on systemic release of duloxetine HCl via buccal route. Drug Dev. Ind. Pharm. 2020, 46, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jiang, J.-Y.; Liu, Y.-Y.; Fu, Y.; Zhao, L.-X.; Li, C.-Y.; Ye, F. Enhanced Solubility, Stability, and Herbicidal Activity of the Herbicide Diuron by Complex Formation with β-Cyclodextrin. Polymers 2019, 11, 1396. [Google Scholar] [CrossRef] [Green Version]
- Al-Nahary, T.T.; El-Ries, M.A.N.; Mohamed, G.G.; Attia, A.K.; Mabkhot, Y.; Haroun, M.; Barakat, A. Multiclass analysis on repaglinide, flubendazole, robenidine hydrochloride and danofloxacin drugs. Arab. J. Chem. 2013, 6, 131–144. [Google Scholar] [CrossRef]
- Holanda, B.B.; Alarcon, R.T.; Guerra, R.B.; Rinaldo, D.; Spazzini, F.C.; Castro, R.A.; Bannach, G. Investigation of thermal degradation products of mebendazole by thermal and spectroscopic analysis. J. Anal. Appl. Pyrolysis 2018, 135, 76–84. [Google Scholar] [CrossRef]
- Garbuio, A.Q.P.; Hanashiro, T.; Markman, B.E.; Fonseca, F.A.; Perazzo, F.F.; Rosa, P.C.P. Evaluation and study of mebendazole polymorphs present in raw materials and tablets available in the Brazilian pharmaceutical market. J. Appl. Pharm. Sci. 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- de Villiers, M.M.; Terblanche, R.J.; Liebenberg, W.; Swanepoel, E.; Dekker, T.G.; Song, M. Variable-temperature X-ray powder diffraction analysis of the crystal transformation of the pharmaceutically preferred polymorph C of mebendazole. J. Pharm. Biomed. Anal. 2005, 38, 435–441. [Google Scholar] [CrossRef]
- Calvo, N.L.; Kaufman, T.S.; Maggio, R.M. Mebendazole crystal forms in tablet formulations. An ATR-FTIR/chemometrics approach to polymorph assignment. J. Pharm. Biomed. Anal. 2016, 122, 157–165. [Google Scholar] [CrossRef]
- Devine, R.; Martin, D.; Kinsella, G.; Findlay, J.; Stephens, J. Characterization of an aryl piperazine/2-hydroxypropyl-β-cyclodextrin association, a complex with antidiabetic potential. Results Chem. 2020, 2, 100026. [Google Scholar] [CrossRef]
- Rojas-Aguirre, Y.; Yépez-Mulia, L.; Castillo, I.; López-Vallejo, F.; Soria-Arteche, O.; Hernández-Campos, A.; Castillo, R.; Hernández-Luis, F. Studies on 6-chloro-5-(1-naphthyloxy)-2-(trifluoromethyl)-1H-benzimidazole/2-hydroxypropyl-β-cyclodextrin association: Characterization, molecular modeling studies, and in vivo anthelminthic activity. Bioorg. Med. Chem. 2011, 19, 789–797. [Google Scholar] [CrossRef]
- Kumar, R.; Sinha, V.; Dahiya, L.; Sarwal, A. Transdermal delivery of duloxetine-sulfobutylether-β-cyclodextrin complex for effective management of depression. Int. J. Pharm. 2021, 594, 120129. [Google Scholar] [CrossRef]
- El-Maradny, H.; Mortada, S.; Kamel, O.; Hikal, A. Characterization of ternary complexes of meloxicam-HPβCD and PVP or L-arginine prepared by the spray-drying technique. Acta Pharm. 2008, 58, 455–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araujo, G.L.B.; Ferreira, F.F.; Bernardes, C.E.S.; Sato, J.A.P.; Gil, O.M.; De Faria, D.L.A.; Loebenberg, R.; Byrn, S.R.; Ghisleni, D.D.M.; Bou-Chacra, N.A.; et al. A New Thermodynamically Favored Flubendazole/Maleic Acid Binary Crystal Form: Structure, Energetics, and in Silico PBPK Model-Based Investigation. Cryst. Growth Des. 2018, 18, 2377–2386. [Google Scholar] [CrossRef]
- Castillo, J.A.; Palomo-Canales, J.; Garcia, J.J.; Lastres, J.L.; Bolas, F.; Torrado, J.J. Preparation and Characterization of Albendazole β-Cyclodextrin Complexes. Drug Dev. Ind. Pharm. 1999, 25, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Ehteda, A.; Galettis, P.; Chu, S.W.L.; Pillai, K.; Morris, D.L. Complexation of Albendazole with Hydroxypropyl-β-Cyclodextrin Significantly Improves its Pharmacokinetic Profile, Cell Cytotoxicity and Antitumor Efficacy in Nude Mice. Anticancer Res. 2012, 32, 3659–3666. [Google Scholar]
- Fernandes, C.M.; Veiga, F. Effect of the Hydrophobic Nature of Triacetyl-β-cyclodextrin on the Complexation with Nicardipine Hydrochloride: Physicochemical and Dissolution Properties of the Kneaded and Spray-dried Complexes. Chem. Pharm. Bull. 2002, 50, 1597–1602. [Google Scholar] [CrossRef] [Green Version]
- Soica, C.; Gyeresi, A.; Frentiu, B.; Dehelean, C.; Aluas, M. Preparation and physico-chemical characterization of chlorthalidone- hydroxypropyl-β-cyclodextrin binary systems. Rev. Chim. 2007, 58, 606–611. [Google Scholar]
- Pourgholami, M.H.; Wangoo, K.T.; Morris, D.L. Albendazole-cyclodextrin complex: Enhanced cytotoxicity in ovarian cancer cells. Anticancer Res. 2008, 28, 2775–2779. [Google Scholar] [PubMed]
- Matencio, A.; Hoti, G.; Monfared, Y.; Rezayat, A.; Pedrazzo, A.; Caldera, F.; Trotta, F. Cyclodextrin Monomers and Polymers for Drug Activity Enhancement. Polymers 2021, 13, 1684. [Google Scholar] [CrossRef] [PubMed]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.-Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro-Oncology 2014, 17, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Pang, X.; Lian, W.; Xu, L.; Wang, J.; Jia, H.; Zhang, B.; Liu, A.-L.; Du, G.-H. Discovery of VEGFR2 inhibitors by integrating naïve Bayesian classification, molecular docking and drug screening approaches. RSC Adv. 2018, 8, 5286–5297. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Yang, L.; Yao, Y.; Xu, L.; Xiang, Y.; Zhao, H.; Wang, L.; Zuo, Z.; Huang, X.; Zhao, C. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J. Exp. Clin. Cancer Res. 2019, 38, 519. [Google Scholar] [CrossRef] [Green Version]
- Elissondo, M.C.; Ceballos, L.; Alvarez, L.; Bruni, S.S.; Lanusse, C.; Denegri, G. Flubendazole and ivermectin in vitro combination therapy produces a marked effect on Echinococcus granulosus protoscoleces and metacestodes. Parasitol. Res. 2009, 105, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.S.; Ghanim, A.M.H.; Saber, S. Mebendazole augments sensitivity to sorafenib by targeting MAPK and BCL-2 signalling in n-nitrosodiethylamine-induced murine hepatocellular carcinoma. Sci. Rep. 2019, 9, 19095. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, P.C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.-S.; Fang, H.-B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget 2017, 8, 12576–12595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, V.; Pedro, J.-S.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Macasoi, I.; Mioc, A.; Mioc, M.; Racoviceanu, R.; Soica, I.; Chevereșan, A.; Dehelean, C.; Dumitrașcu, V. Targeting Mitochondria through the Use of Mitocans as Emerging Anticancer Agents. Curr. Med. Chem. 2019, 27, 5730–5757. [Google Scholar] [CrossRef] [PubMed]
- Bossche, H.V.D.; Janssen, P.A. The biochemical mechanism of action of the antinematodal drug tetramisole. Biochem. Pharmacol. 1969, 18, 35–42. [Google Scholar] [CrossRef]
- Aminzadeh-Gohari, S.; Weber, D.D.; Catalano, L.; Feichtinger, R.G.; Kofler, B.; Lang, R. Targeting Mitochondria in Melanoma. Biomolecules 2020, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luepke, N. Hen’s egg chorioallantoic membrane test for irritation potential. Food Chem. Toxicol. 1985, 23, 287–291. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB ID | Grid Box Center Coordinates | Grid Box Size | Conformers Generated Per Ligand |
|---|---|---|---|---|
| Apoptosis regulator Bcl-2 (Bcl-2) | 2W3L | center_x = 38.8892 center_y = 27.0675 center_z = −12.5145 | size_x = 13.7411 size_y = 11.5648 size_z = 9.6457 | 8 |
| Apoptosis regulator Bcl-X (Bcl-XL) | 2YXJ | center_x = −8.8801 center_y = −14.5418 center_z = 10.0100 | size_x = 13.4584 size_y = 20.4060 size_z = 9.6457 | 8 |
| Epidermal growth factor receptor 1 (EGFR1) | 4RJ8 | center_x = −53.5443 center_y = 1.1587 center_z = −25.1500 | size_x = 10.5346 size_y = 10.7549 size_z = 13.7560 | 8 |
| Vascular endothelial growth factor receptor 2 | 3VHK | center_x = −27.5445 center_y = 26.9531 center_z = 16.6738 | size_x = 10.0752 size_y = 11.0246 size_z = 11.6961 | 8 |
| RAC-alpha serine/threonine-protein kinase (AKT1) | 3CQU | center_x = 5.2027 center_y = 2.2676 center_z = 17.5609 | size_x = 14.7068 size_y = 14.4124 size_z = 9.3509 | 8 |
| Mammalian target of rapamycin- target of rapamycin complex subunit LST8 (mTOR-LST8) | 4JT5 | center_x = 52.1512 center_y = −0.7990 center_z = −49.2709 | size_x = 14.3371 size_y = 11.0246 size_z = 810.8186 | 8 |
| Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform (PI3Kγ) | 4FA6 | center_x = 45.2920 center_y = 14.3911 center_z = 31.0579 | size_x = 14.6994 size_y = 10.7549 size_z = 10.8186 | 8 |
| Dual specificity mitogen-activated protein kinase 1 (MEK1) | 3EQG | center_x = −3.5569 center_y = 57.7644 center_z = 34.5863 | size_x = 14.7068 size_y = 10.9225 size_z = 9.7322 | 8 |
| Mitogen-activated protein kinase 1 (ERK2) | 4G6N | center_x = 21.5960 center_y = 5.8329 center_z = 15.3508 | size_x = 14.4689 size_y = 11.4524 size_z = 10.8186 | 8 |
| Bcl-2 | Bcl-XL | EGFR1 | VEGFR2 | AKT1 | mTOR | PI3K | MEK1 | ERK2 | |
|---|---|---|---|---|---|---|---|---|---|
| Free Binding Energy ∆G (kcal/mol) | |||||||||
| Native ligand | −10.3 | −10 | −8.8 | −9.7 | −10.5 | −8.5 | −9.3 | −8.7 | −7.5 |
| FLU | −7 | −8.3 | −7.5 | −9.6 | −8.9 | −8.2 | −8.2 | −9.4 | −7.7 |
| MEB | −7 | −7.9 | −7.5 | −9.3 | −9.1 | −8.7 | −8 | −9.3 | −7.7 |
| Samples | Irritation Score | Type of Effect |
|---|---|---|
| SLS 0.5% | 18.56 | Strong irritant |
| HPBCD 10 mM | 0.0 | Non-irritant |
| FLU 10 mM | 0.0 | Non-irritant |
| FLU-HPBCD 10 mM | 0.0 | Non-irritant |
| MEB 10 mM | 0.0 | Non-irritant |
| MEB-HPBCD | 0.0 | Non-irritant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minda, D.; Mioc, A.; Banciu, C.; Soica, C.; Racoviceanu, R.; Mioc, M.; Macasoi, I.; Avram, S.; Voicu, A.; Motoc, A.; et al. Cyclodextrin Dispersion of Mebendazole and Flubendazole Improves In Vitro Antiproliferative Activity. Processes 2021, 9, 2185. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9122185
Minda D, Mioc A, Banciu C, Soica C, Racoviceanu R, Mioc M, Macasoi I, Avram S, Voicu A, Motoc A, et al. Cyclodextrin Dispersion of Mebendazole and Flubendazole Improves In Vitro Antiproliferative Activity. Processes. 2021; 9(12):2185. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9122185
Chicago/Turabian StyleMinda, Daliana, Alexandra Mioc, Christian Banciu, Codruța Soica, Roxana Racoviceanu, Marius Mioc, Ioana Macasoi, Stefana Avram, Adrian Voicu, Andrei Motoc, and et al. 2021. "Cyclodextrin Dispersion of Mebendazole and Flubendazole Improves In Vitro Antiproliferative Activity" Processes 9, no. 12: 2185. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9122185