
Oxidoborates Templated by Cationic Nickel(II) Complexes and Self-Assembled from B(OH)3


1
School of Natural Sciences, Bangor University, Bangor LL57 2UW, UK
2
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
*
Author to whom correspondence should be addressed.
†
Current address: Chemistry Department, College of Science, University of Thi-Qar, Nasiryah 0096442, Iraq.
Inorganics 2021, 9(9), 68; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090068
Submission received: 19 August 2021
/
Revised: 27 August 2021
/
Accepted: 30 August 2021
/
Published: 31 August 2021
(This article belongs to the Special Issue Boron Chemistry: Fundamentals and Applications)
Abstract
:Several oxidoborates, self-assembled from B(OH)3 and templated by cationic Ni(II) coordination compounds, were synthesized by crystallization from aqueous solution. These include the ionic compounds trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O (1), s-[Ni(dien)2][B5O6(OH)4]2 (dien = N-(2-aminoethyl)-1,2-ethanediamine (2), trans-[Ni(dmen)2(H2O)2] [B5O6(OH)4]2.2H2O (dmen = N,N-dimethyl-1,2-diaminoethane) (3), [Ni(HEen)2][B5O6(OH)4]2 (HEen = N-(2-hydroxyethyl)-1,2-diaminoethane) (4), [Ni(AEN)][B5O6(OH)4].H2O (AEN = 1-(3-azapropyl) -2,4-dimethyl-1,5,8-triazaocta-2,4-dienato(1-)) (5), trans-[Ni(dach)2(H2O)2][Ni(dach)2] [B7O9(OH)5]2.4H2O (dach = 1,2-diaminocyclohexane) (6), and the neutral species trans-[Ni(en)(H2O)2{B6O7(OH)6}].H2O (7) (en = 1,2-diaminoethane), and [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (8). Compounds 1–8 were characterized by single-crystal XRD studies and by IR spectroscopy and 2, 4–7 were also characterized by thermal (TGA/DSC) methods and powder XDR studies. The solid-state structures of all compounds show extensive stabilizing H-bond interactions, important for their formation, and also display a range of gross structural features: 1 has an insular tetraborate(2-) anion, 2–5 have insular pentaborate(1-) anions, 6 has an insular heptaborate(2-) anion (‘O+’ isomer), whilst 7 and 8 have hexaborate(2-) anions directly coordinated to their Ni(II) centers, as bidentate or tridentate ligands, respectively. The Ni(II) centers are either octahedral (1–4, 7, 8) or square-planar (5), and compound 6 has both octahedral and square-planar metal geometries present within the structure as a double salt. Magnetic susceptibility measurements were undertaken on all compounds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Synthetic borates (oxidoborates is the IUPAC recommended name for this class of compounds [1]) show great structural diversity and their structures complement the variety of structural motifs observed in borate minerals [2,3,4,5]. Borate minerals have large scale economic importance [6,7,8] (e.g., as vitreous, agriculture, fire retardant products) and some of these minerals, and their synthetic counterparts, have also found more specialist uses [9,10,11,12,13,14,15,16,17,18] (e.g., as fluorescent, optical SHG and wide band-gap semi-conductor materials).
Structurally, borates salts consist of metallic or non-metallic cationic centers and hydroxyoxidoborate units, with variable degrees of condensation, as either insular anions or as anionic 1-D chains, 2-D layers or 3-D networks [2,3,4,5]. Oxidoborate materials can be synthesized from aqueous solution or from solid-state or solvothermal methods and the latter non-aqueous methods often lead to the formation of the more highly condensed oxidoborate structures [5]. In aqueous solution, oxidoborate speciation is pH and boron-concentration dependent [19,20], and a dynamic combinatorial library (DCL) [21] of oxidoborate anions co-exist in rapidly attained aqueous equilibria.
Many oxidoborate anions have been successfully crystallized from aqueous solution using B(OH)3 and an appropriate templating cation, e.g., triborate(1-), tetraborate(2-), pentaborate(1-), hexaborate(2-), heptaborate(2-) (two isomers), heptaborate(3-), octaborate(2-) (two isomers), nonaborate(3-), tetradecaborate(4-), and pentadecaborate(3-) anions [5]. These salts are selectively templated from the solution by the cations present in self-assembly processes [22,23,24]. However, pentaborate(1-) salts often crystallize out of such solutions and this is particularly so if the cation is relatively small and has a charge of +1 [5]. This process primarily occurs since the [B5O6(OH)4]− anion is ideally shaped to form giant H-bonded lattices through anion–anion interactions with ‘cavities’ large enough to be occupied by the small cations [25,26,27,28].
We are interested in expanding the structural diversity of isolated and coordinated oxidoborate chemistry and have developed a strategy of incorporating relatively highly charged (>+1) and/or cations, with the potential of forming multiple H-bond interactions, into the aqueous DCL of borate anions so that they can template and crystal engineer novel structures [29,30]. Using labile cationic transition-metal complexes [31] introduces a further DCL of potential cations into reaction medium and leads to the possibility of oxidoborate anions entering the primary coordination shell of the metal, as O-donor ligands.
With this in the forefront of our mind, we have recently investigated the reactions of labile complexes of Zn(II) [32,33,34,35] and Cu(II) [36,37,38,39,40] and have prepared novel species containing coordinated [B2O3(OH)2]2− [39], [B5O6(OH)4]− [37], [B6O7(OH)6]2− [34,35,36,37,38], [B12O18(OH)6]6− [32,35] and [B20O32(OH)8]12− [39] ligands. Ni(II) complexes and their reactions with B(OH)3 have received little attention to date and reported oxidoborate species are restricted to an unusual hexadentate tris(aminoethoxy)hexaborate(2-) anion complex, [Ni{(H2NCH2CH2O)3B6O7(OH)3}] [41], and a series of salts containing the insular pentaborate(1-) [42,43,44,45,46,47] or heptaborate(2-) [33,48] anions.
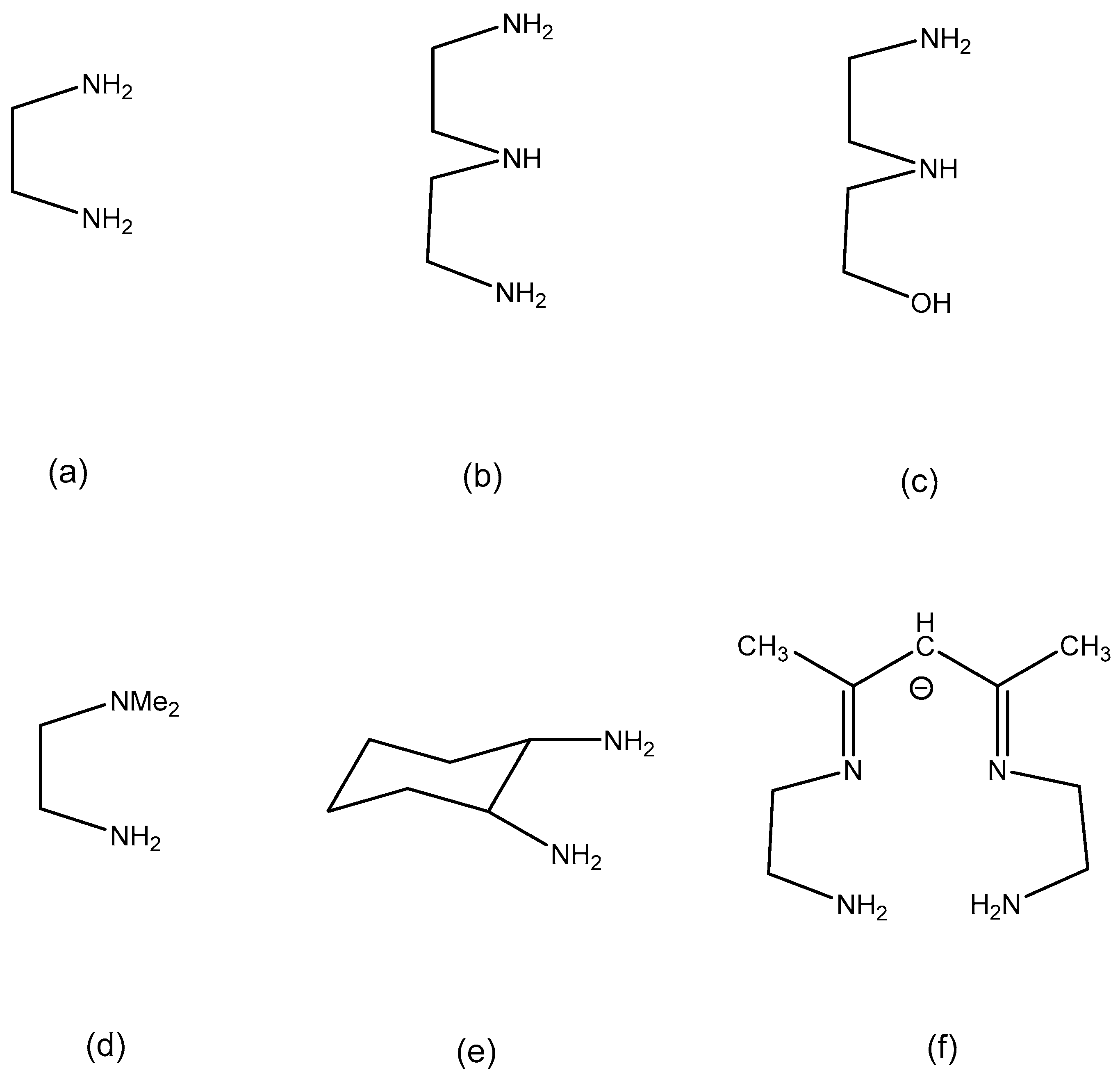
Ni(II) complexes have a d8 electronic configuration and should also be relatively labile [31]. In this manuscript we report on the use coordination complexes of Ni(II) containing N-donor ligands NH3, en, dien, dmen, HEen, AEN, dach (see Figure 1 for ligand structures and abbreviations) to synthesize eight new oxidoborate compounds. Their single-crystal structures and their spectroscopic and physical properties are reported.
2. Results and Discussion
2.1. Synthesis and General Discussion
The Ni(II) complex oxidoborates 1–8 were prepared in acceptable yields as a crystalline solids from the reaction of a Ni(II) complex cation hydroxide salt with boric acid in a ratio of either 1:5 or 1:10 (Scheme 1). The Ni(II) complex cation hydroxides were obtained in situ from the corresponding salts containing either chloride or sulfate anions. Anion exchange (from Cl−) was used for 2, 4, 5, 6, and 8 and stoichiometric reactions with Ag2O (from Cl−) was used for 1 and 3 and a stoichiometric reaction with Ba(OH)2.8H2O (from SO42−) was used for 7.
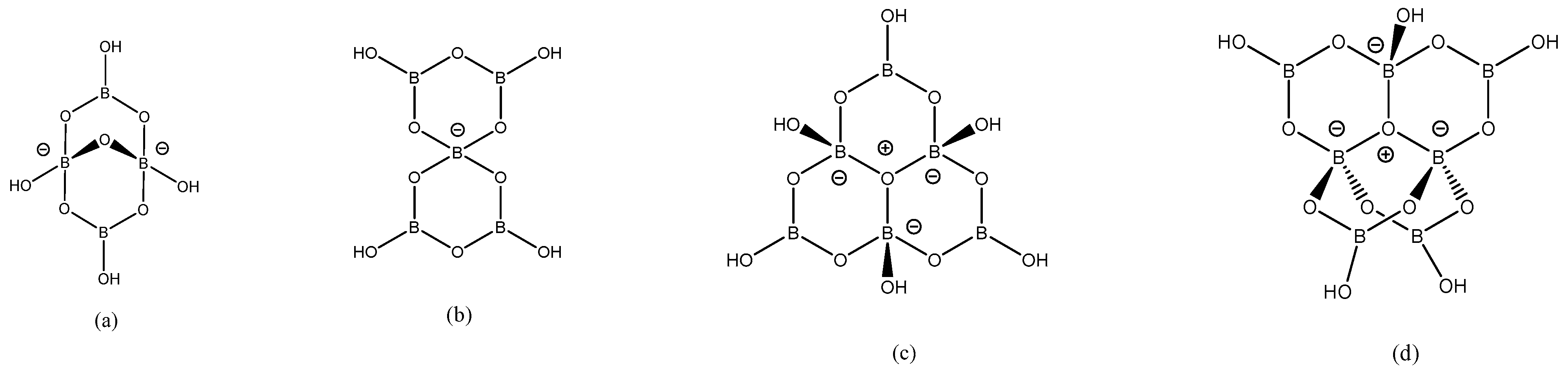
The new compounds were identified by XRD single-crystal studies (Section 2.2), IR spectroscopic analysis and accompanied by thermal decomposition analysis (TGA and DSC) and powder XRD studies for 2, 4–7 (Section 2.3). Six of these eight compounds contain insular oxidoborate anions and two compounds contain coordinated hexaborate(2-) anions; the oxidoborate anions found in these new compounds are drawn out in Figure 2. The coordination geometry around the Ni(II) centers are either octahedral (1–4, 7, 8) or square-planar (5) and compound 6 has both octahedral and square-planar metal geometries present within the structure as a double salt. The new Ni(II) oxidoborate complexes are coloured with those with square-planar centres (5 and 6) are orange/red with the others having blueish hues. All compounds were stable in the solid state were insoluble in organic solvents and decomposed by aqueous solution (see 11B NMR, Section 2.3). Since the products arise through self-assembly processes that we believe are associated with H-bonded structure directing effects induced by the cations present [21,22,23,24], these interactions are discussed in detail in Section 2.2.2.
2.2. Crystallographic Studies on 1–8 and Their Solid-State H-Bonding Interactions
2.2.1. General Considerations for 1–6 and the Structures of 7 and 8
Selected crystallographic data for 1–8 can be found in Experimental Methods (Section 3) and full crystallographic details are available as Supplementary Materials. Compounds 1–6 are all anionic compounds containing discrete insular cationic Ni(II) coordination complexes with N-donor or O-donor ligands, partnered with anionic oxidopolyborate anions. The cations in 2–6 are as found in their precursor complexes whilst 1 contains the partially aquated trans-[Ni(NH3)4(H2O)2]2+ cation. The cationic charges are neutralized by the insular oxiodopolyborate anions [B4O5(OH)4]2−, [B5O6(OH)4]−, or [B7O9(OH)5]2− for 1, 2–5 and 6, respectively. Compounds 1, 3, 5 and 6 also contain one, two, one and four interstitial H2O molecules per formula unit, respectively. The dien ligand in 2, and the HEen ligand in 4 are both disordered over two positions. Compounds 7 and 8 are uncharged coordination compounds with [B6O7(OH)6]2- coordinated as O-donor ligands to octahedral Ni(II) centres either as a bidentate or a tridentate ligand in 7 or 8, respectively. Compounds 7 and 8 have one and five interstitial H2O molecules, respectively, and are also partially aquated. There is some disorder of the interstitial H2O molecules in both 7 and 8 and some additional disorder in the en ligand and the dmen ligands of 7 and 8. Compound 6 is unusual and is best formulated as a double salt. It contains both octahedral trans-[Ni(dach)2(H2O)2]2+ and square-planar [Ni(dach)2]2+ cations partnered by two crystallographically equivalent [B7O9(OH)5]2− anions and four interstitial H2O molecules. The dach ligands are also disordered in 6 but have chair conformations with their amino groups equatorial and with one dach ligand in each cation having R,R stereochemistry and the other S,S. Interestingly, two of these interstitial H2O molecules (O22) in 6 are in ‘axial’ positions in the square-planar complex at 3.1517(13) Å (T = 0.61 [49]). In the octahedral complex, these two H2O ligands (O21) are at 2.1422(15) Å (T = 0.98). A structurally related (but d9) Cu(II) compound, prepared by an identical method [37], contains a square-planar (also with very long axial H2O ‘bonds’, T = 0.70) and a Jahn–Teller distorted axially elongated octahedral complexes (T = 0.80). Structurally, the insular [B7O9(OH)5]2- dianion is previously known to adopt either a ‘chain’ or ‘O+’ isomeric forms [28,50,51,52] and the ‘O+’ isomer is present in 6. The new Ni(II) oxidoborates complex salts 1–6 all contain cations and anions that are known in other salts and the gross structures of the component ions, bond distances and internuclear angles for the component ions in 1–6 are as expected and need no further comment [5,53,54,55]. The uncharged complexes 7 and 8 are structurally novel although similar compounds have been observed in Co(II), Cu(II) and Zn(II) oxidoborate chemistry [34,35,36,37,38,56,57,58]. Compounds 7 and 8 contain isolated species [35,37,38] rather than polymer 1-D coordination chains [34,36,38] and these two structures will be discussed in more detail below.
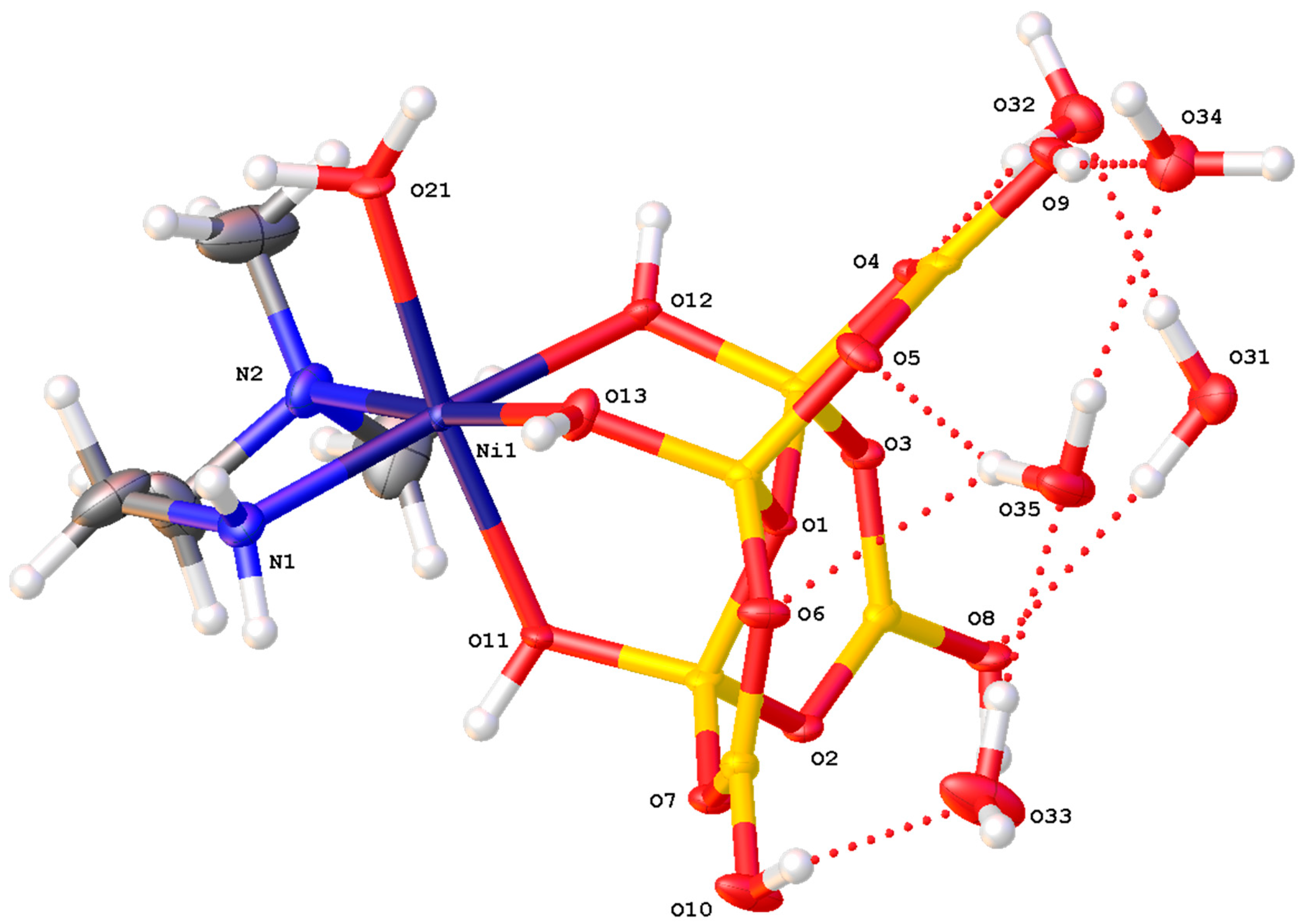
A drawing of the structure of trans-[Ni(en)(H2O)2{B6O7(OH)6}].H2O (7), with selected crystallographic numbering, can be found in Figure 3. Compound 7 is a distorted neutral octahedral Ni(II) complex with trans H2O ligands (containing O21 and O22), a disordered 1,2-diaminoethane ligand and a bidentate [B6O7(OH)6]2− ligand that is coordinated through two hydroxy groups (containing O8 and O9). A single disordered water of crystallization containing O31 (s.o.f. 0.47(2)) or O31B (s.o.f. 0.53(2)) is also shown. The O21-Ni1-O22 angle is 171.67(12)o and the average Ni-(OH2) distance is 2.089(3) Å. This average distance is slightly longer than the average oxidoborate O8/O9-Ni1 distance of 2.065(3) Å. The chelating en ligand significantly distorts the Ni(II) centre where the Ni1-N1 and Ni1-N2 distances are 2.086(4) and 2.085(4) Å, respectively. The N1-Ni1-N2 angle is 82.80(16)o and the oxidoborate/en ligand O8-Ni1-N2 and O9-Ni1-N1 angles are both significantly <180o, whilst the O8-Ni1-O9 angle is 90.48(14)o. These Ni-N and Ni-(OH2) bond lengths are within the expected ranges for Ni(II) complexes [53,54,55] but this is the first example of a Ni-O bondlength arising from an oxidoborate ligand and it is not significantly different from other N-O bondlengths. The B-O bondlengths and OBO and BOB angles of the hexaborate(2-) ligand are in accord with previously reported data for this ligand including three very long B-O bondlengths (1.498(6)–1.538(6)o; av. 1.517(6)o) to the three-coordinate O atom (O1) [28,29,48,51,52]. There is an intramolecular H-bond from an oxidoborate hydroxy group (containing O10) and the coordinated aqua ligand (containing O21), O10H10…O21, with a O10…O21 distance of 2.704(5) Å and a O10-H10-O21 angle of 144.9o (Figure 3). This H-bond is part of two R11(8) rings [59] and both rings involve the Ni(II) centre and two Ni-O coordinate bonds. There is a similar H-bond interaction in the only other known example of a bidentate hexaborate(2-) complex, (NH4)2[Zn(H2O)2(B6O7(OH)6}2], but here, interestingly, the H-bond originates from the aqua ligand and the uncoordinated hydroxy site of the oxidoborate is the acceptor [35].
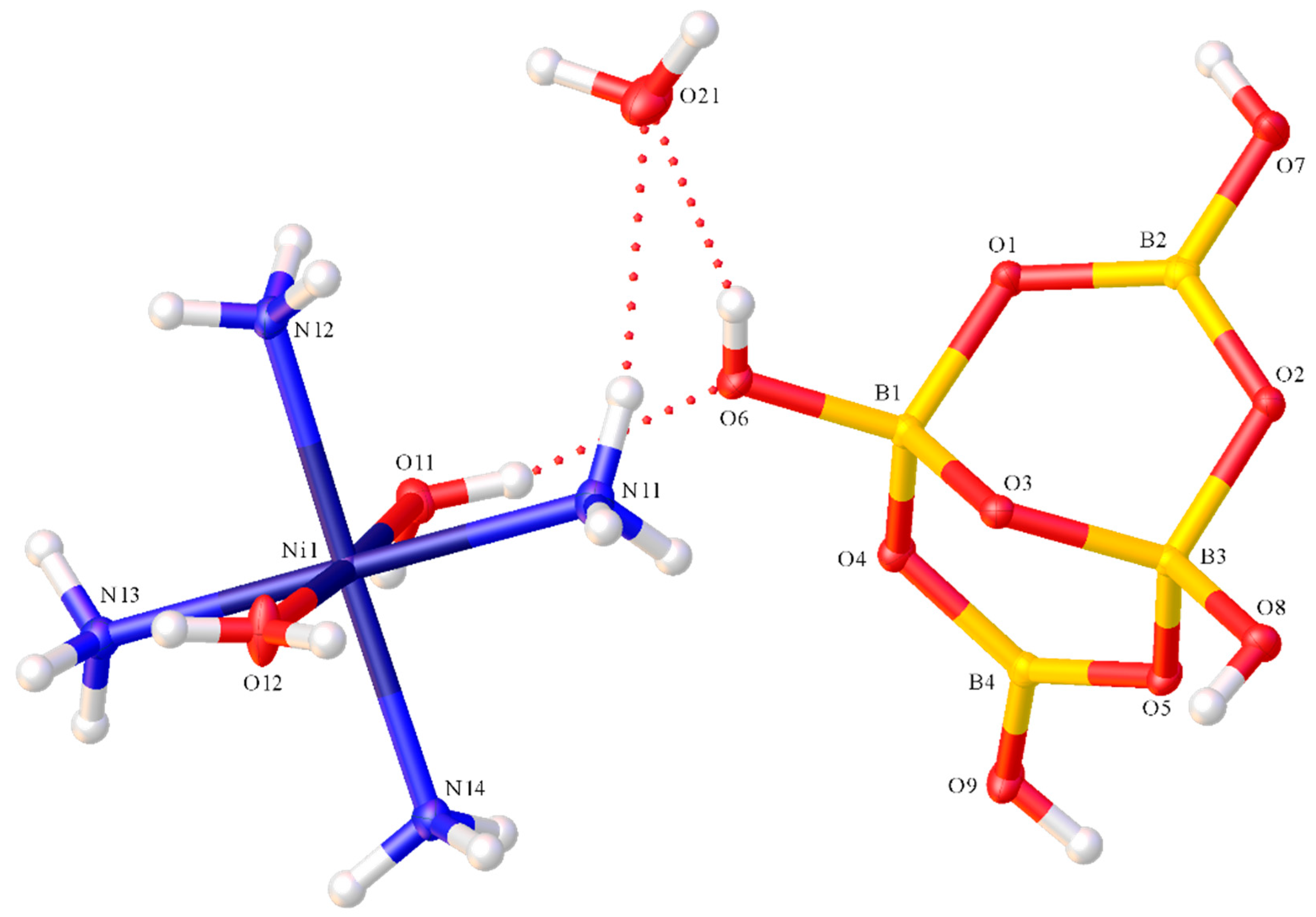
A drawing of the structure of [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (8), with selected crystallographic numbering, is shown in Figure 4. Compound 8 is a distorted neutral octahedral Ni(II) complex with disordered aqua ligand (containing O21) and dmen ligands (0.496(5) s.o.f shown) and a tridentate [B6O7(OH)6]2− ligand coordinated through three hydroxy groups (containing O11, O12 and O13). The five waters of crystallization contain O31-O35. The Ni1-O21 distance for the aqua ligand (2.131(11) Å) is significantly longer than the Ni-O distances associated with the three hydroxy groups of the tridentate oxidoborate ligand (2.0415(17)–2.0978(17) Å; av. 2.078(2) Å) but this difference was not significant for 7. The N1-Ni1-N2 angle associated with the chelating dmen ligand is 84.5(12)o and angles between the three O-donor atoms of the fac-hexaborate ligand range from 84.26(7)–90.66(6)o; av. 86.94(8)o. The aqua ligand with O21 is trans to oxidoborate O11 with an angle of 173.2(3)o, and the other trans angles are significantly <180.0o. The Ni1-N distances of 2.031(11) Å (N1) and 2.13(2) Å (N2) are significantly different but there is more steric demand at N2 as it is substituted by the two methyl groups. Overall, the bondlengths and angles associated with the Ni(II) centre and those within the hexaborate(2-) ligand in 8 are in accord with comparable data for 7.
2.2.2. Solid-State H-Bonding Interactions in 1–8
Compounds 1–8 are self-assembled and crystallized from the various oxidoborate and the Ni(II) amine complexes that are each in equilibrium in the reacting aqueous solution [19,20,31]. There is a strong preference for the oxidoborate anion to enter the primary coordination shell in related Cu(II) and Zn(II) chemistry with the formation of energetically favourable O-donor coordinate bonds [32,34,35,36,37,38,39,40,58]. Interestingly, Ni(II) salts are described as labile, although in practice they are often considerably less labile than corresponding Cu(II) and Zn(II) complexes [31]. In accord with this, the Ni(II) cations used in this study generally remain more or less intact in salts 1–6 after prolonged crystallization from aqueous solution, with formation of oxidoborate O-donor bonds only observed in compounds 7 and 8. H-bond interactions also appear to be of paramount importance in driving this crystallization process and these interactions are described in detail for each compound in the following paragraphs.
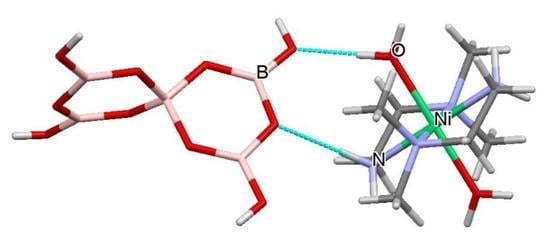
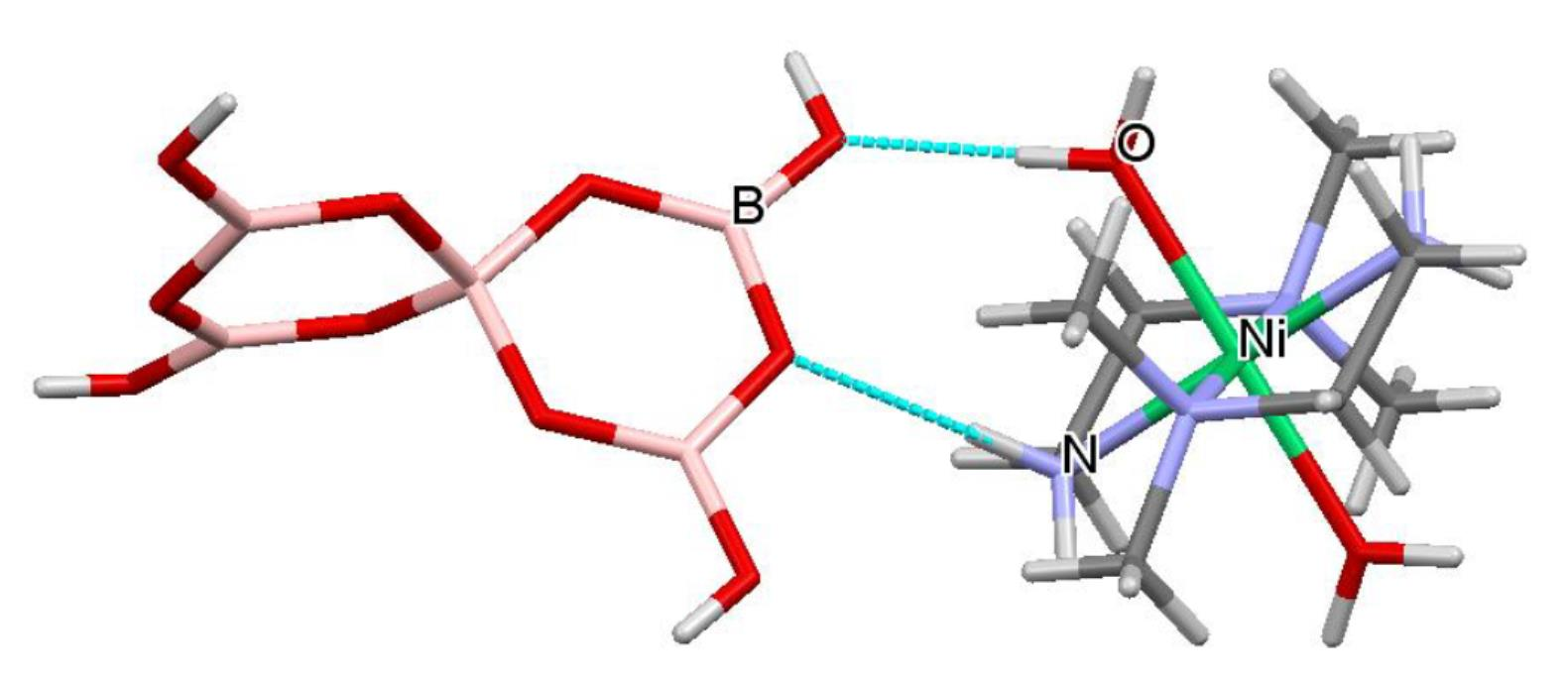
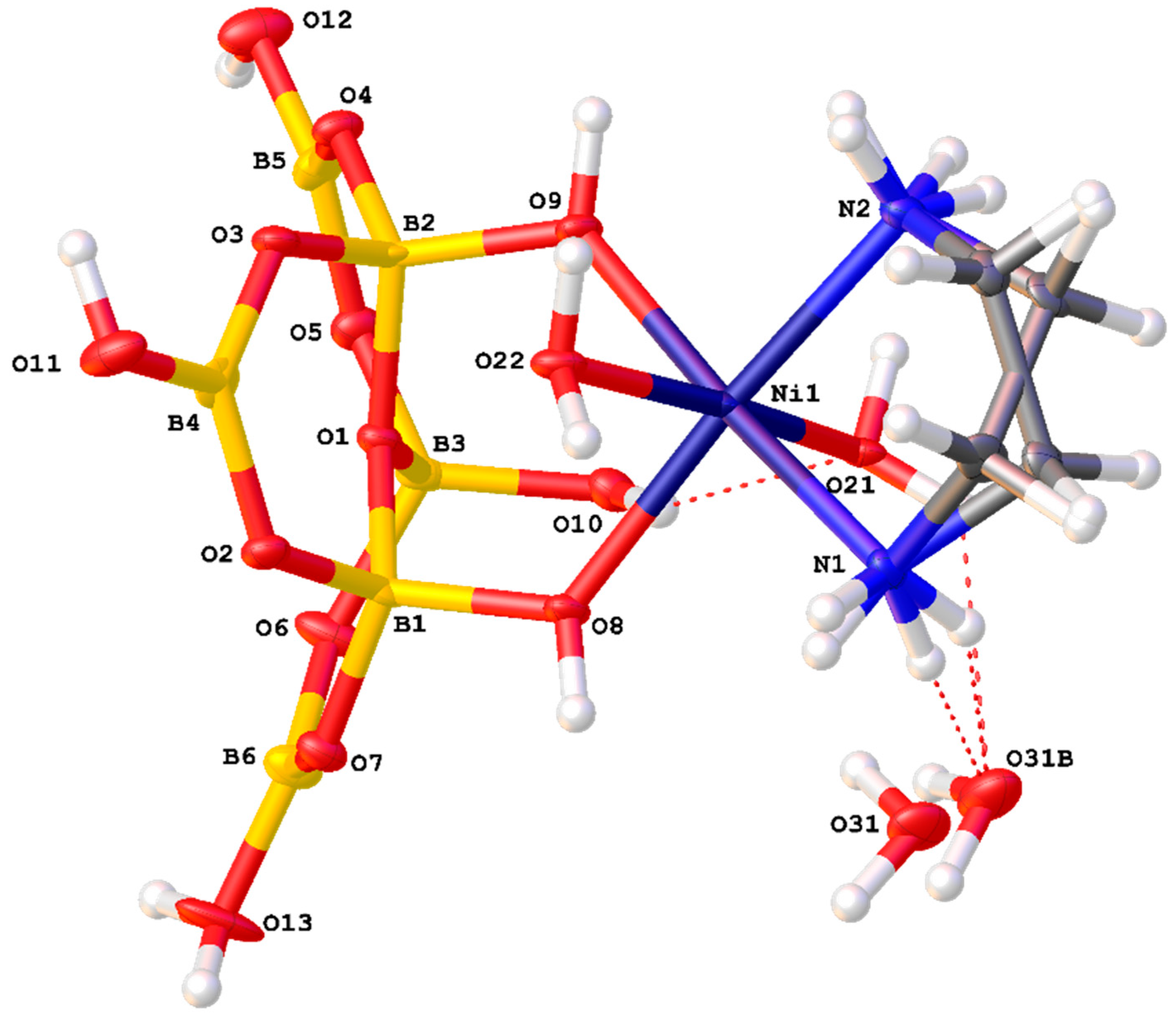
A drawing of the components of the formula unit of 1, showing important atomic numbering, is shown in Figure 5. The cations, anions and interstitial H2O molecules have numerous H-bond donor and/or acceptor sites and all potential H-bond donor sites are used in the solid-state structure. The dotted red lines in Figure 5 illustrate H-bond interactions which link cation to anion (O11H11…O6), cation to interstitial H2O (N11H11…O21), and anion to interstitial H2O (O6H6…O21) forming a R22(8) ring incorporating the Ni(II) centre. These interactions as well as anion–anion interactions template the formation of the tetraborate salt. Full details of all H-bond interactions are available in the Supplementary Materials. Siveav and co-workers [46] have analysed H-bond interactions in tetraborate(2-) systems using the terminology of Zolatarev et al. [60] but we prefer the complimentary and more widely known Etter symbolism [59] in this manuscript. R22(8) interactions are very important at stabilizing hydroxyoxidopolyborate anions and three of the four boron based hydroxy centres are involved in such interactions: observed in R22(8) interactions are very important at stabilizing hydroxyoxidopolyborate anions and three of the four boron based hydroxy centres are involved in such interactions with O8-H8…O5′ and O9′-H9′…O3 paired in a R22(8) ring and O7-H7…O1′ and O7′-H7′…O1 forming a reciprocal pair in another R22(8) ring. The fourth boron based hydroxy group O6H6 is a donor to a H2O molecule, O21 (Figure 5) forming an unusual R33(8) ring {Ni1-O11A-H11A…O6-H6…O21…H11E-N11} and is also part of a larger R44(12) ring {O6-H6…O21-H21A…O3′-B1′-O6′-H6′…O21′-H21A’…O3-B1-} (Figure 6). Since the cation is involved in templating the structure its H-bond interactions are very important and these include O11-H11A…O6, O11-H11B…O8, O12-H12A…O4, O12-H12B…O2, N14-H14A…O8, N12-H12D…O7 and N12-H12C…O7 which originate from the aqua or ammine ligands. Further patterns are difficult to visualize although R22(8) {Ni1-N12-H12C….O7-B2-O2…H12B-O12-} and a C22(10) {Ni1-O11-H11A…O6-B1-O3-B3-O8…H14A’-N14′-} are discernable.
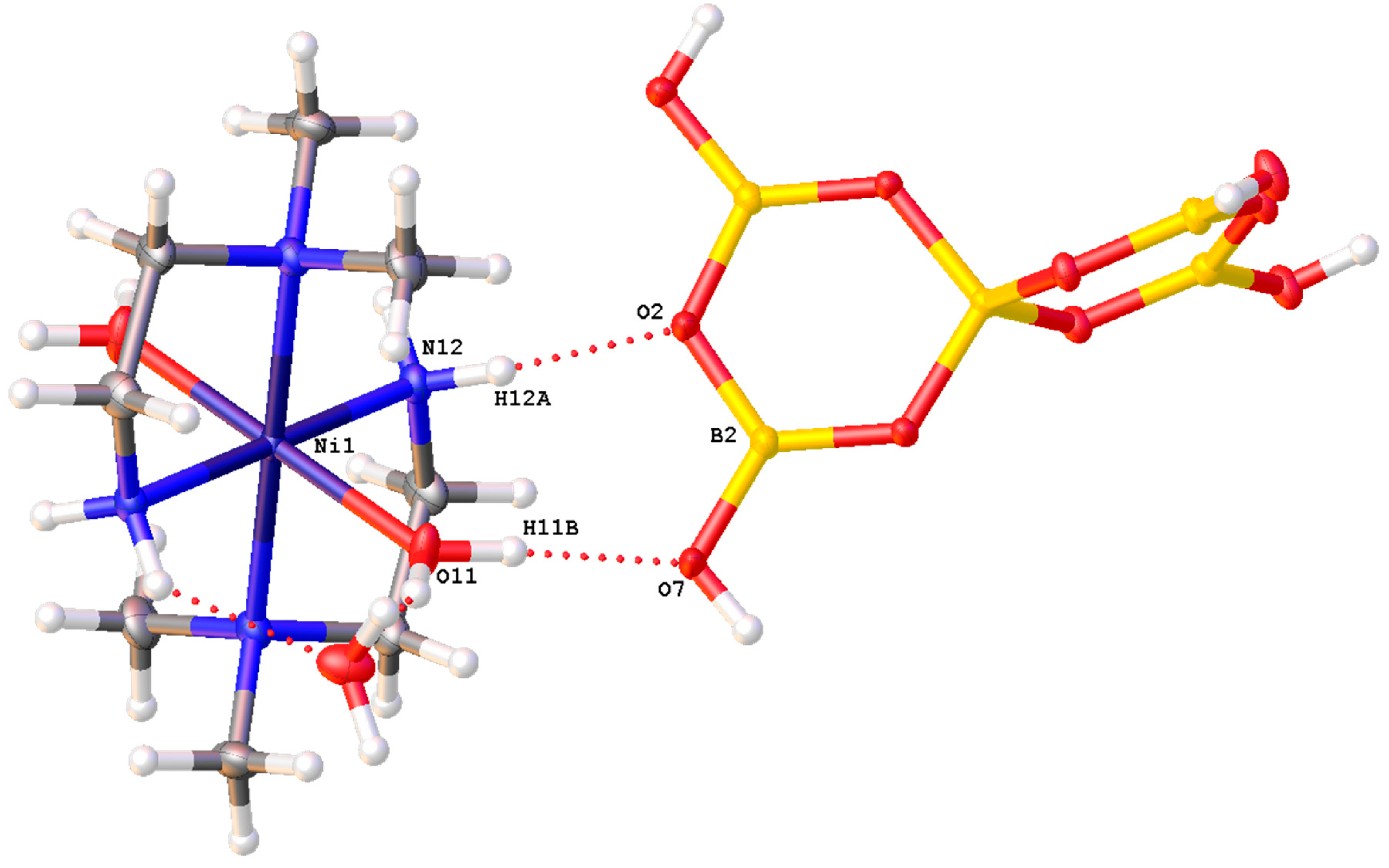
Compounds 2–5 are all pentaborate(1-) salts and anion–anion H-bond interactions are well known to strongly influence their formation [5,25,26,27,28] since each pentaborate(1-) anion has four donor H-bond sites that are sterically arranged to form 3D H-bonded anionic giant structures, in which the cations occupy vacant ‘open-sites’ in the anionic lattice. The anion–anion interactions are conveniently described using a combination of ‘acceptor-sites nomenclature’, as defined by Schubert [26], and Etter terminology [59]. Compounds 2–4 have very similar anion–anion interactions and the four acceptor sites can be described as α,α,α,β with the α-site interactions reciprocal R22(8) and the β-interactions reciprocal R22(12); both types of interactions are common in pentaborate(1-) structures [5,27]. The anion interactions in 5 are different with three pentaborate(1-) acceptor sites (α,α,γ), all reciprocal R22(8), and an interstitial H2O acceptor site. The cations in 2–5 are situated within the anionic lattices and have the potential to be H-bond donors (and acceptors) which can further stabilize the anionic lattices and help to template their self-assembly. Examples of such interactions, which involve novel ring systems that incorporate the Ni(II) centres, ligands interstitial molecules and pentaborate(1-) anions, are R44(14) {Ni1-N3B-H3B…O10-O1OH…O9-O9H…O4-B1-O1-B2-O7…H2B-N2B-} (2) (includes three pentaborate(1-) anions), R22(8) {Ni1-O11-H11B…O7-B2-O2…H12A-N12-} (3) (one pentaborate(1-) anion), R22(8) {Ni1-N11-H11D…O11-B2-O7…H11-O11-} (4) (one pentaborate(1-) anion) and R22(8) {Ni1-N11-H11D…O1-B2-O7…H11-O11-} (4) (one pentaborate(1-) anion), and R44(16) {Ni1-N1-H1C…O5-B4-O4-B1-O3…H1A-O11…H7-O7-B2-O1…H4A-N4-} (5) (two pentaborate(1-) anions and one interstitial H2O with O11). The R22(8) interaction for 3 is illustrated in Figure 7. Full details of all these interactions (and associated atomic numbering systems) are available in the Supplementary Materials.
Compound 6 is a heptaborate(2-) salt with numerous opportunities for interionic H-bonding interactions with the each anion possessing 14 potential acceptor sites and five donor sites and the two cations potentially having 20 donor sites and four acceptor sites; there are also four interstitial H2O molecules. Potentially templating cation–anion interactions include the R22(8) {Ni1-N12-H12A…O10-B4-O3…H11A-N11-}, the R33(8) {Ni2-N12-H12D…O23-H23B…O11…H11D-N11-}, and the R22(10) {Ni1-O21-H21B…O14-B1-O1-B2-O6…H2B-N2-}. The familiar R22(8) anion–anion interaction is present in {O12-H12…O9′-B7′-O13′-H13′…O7-B6-} and the reciprocal {O11-H11…O5′-B5′-O11′-H11′…O5-B5-} and there is also a R22(12) interaction, {O10-H10…O8′-B2′-O2′-B4′-O10′-H10′…O8-B2-O2-B4-}. The fifth heptaborate hydroxy group (containing O14) does not form a donor bond but O14 has a R22(10) acceptor interaction with a cation, as described above.
Intermolecular H-bonding, in addition to the intramolecular interaction found in 7 (Section 2.2.1) can be found in 7 and 8. Such interactions for 7 include R22(6) {Ni1-O8-H8…O2…H1AA-N1-} and {Ni1-O9-H9…O3…H2BD-N2-}, R44(8) {O21-H21B…O10H10…O21′-H21B’…O10′-H10′…} and a reciprocal R22(12) {O12-H12…O6′-B3′-O5′-B5′-O12′-H12′…O6-B3-O5-B5-}. There are five interstitial H2O’s per Ni(II) centre in 8 and their main roles are to act as ‘spacers’ [28] by forming H-bond bridges between neutral Ni(II) oxidoborate complexes. Templating interactions occur when the oxidoborate ligand of one complex enters the second coordination sphere of another. This occurs in reciprocal oxidoborate ligand R22(12) interactions, {O11-H11…O10′-B6′-O7′-B1′-O11′-H11′…O10-B6-O7-B1-} and {O12-H12…O9′-B5′-O4′-B2′-O12′-H12′…O9-B5-O4-B2-}; these are that similar to one observed in 7 and in another heptaborate(2-) system [52]. A R22(8) {O8-H8…O2′-B4′-O8′-H8′…O2-B4-} interaction also links oxidoborate sections of neighbouring complexes.
2.3. Magnetic, Spectroscopic, Thermal and p-XRD Characterization of Compounds 1–8
Magnetic susceptibility measurements on 1–8 confirmed that all, excepting 5, are paramagnetic with χm values ranging from 2500 × 10−6 to 3600 × 10−6 cm3·mol−1 (μeff of 2.6–3.0 BM) corresponding to two unpaired electrons per formula unit. Compound 5 is diamagnetic (χm = −170 × 10−6 cm3·mol−1), and this value is in accord with the Ni(II) centre being square-planar. The χm values of 1–4, 7 and 8 are typical of octahedral Ni(II) complexes [61] and have similar values to those obtained for the starting octahedral Ni(II) complexes. Compound 6 is unusual in that it has a μeff corresponding to two unpaired electrons but there are two Ni(II) centres in the formula unit; the XRD study confirmed that one Ni(II) centre is square planar and the other is octahedral.
Compound 5, in D2O solvent, gave 1H and 13C NMR signals for the AEN ligand at similar intensities and chemical shifts to those obtained for [Ni(AEN)]Cl.H2O and other reported data [62]. 1H and 13C NMR signals for the organic ligands (in D2O) were not observable for the other Ni(II) oxidoborates despite their apparent solubility, due to their paramagnetic properties. Despite the possible lability of the organic ligands the complexes the organic ligands are not sufficiently labile to be remote from the Ni(II) centres during the lifetime of the NMR experiment. 11B NMR spectra for all D2O solutions arising from the Ni(II) oxidoborates were observed and a probable explanation for this being two-fold: (i) compounds with insular oxidoborate anions (1–6) are not coordinated to the Ni(II) centres and hence are not in close proximity and unaffected by the paramagnetic centres, and (ii) the coordinated hexaborate(2-) ligands are significantly more labile than the N-donor ligands in 7 and 8. The 11B spectra were not particularly characteristic of the specific oxidopolyborate anions in 1–8 since it is known that dissolution of oxidopolyborate anions in aqueous solution yields solutions containing equilibrium mixtures of various oxidopolyborate anions whose concentrations are pH are boron concentration dependent [19,20]. However, pentaborate(1-) salts often afford spectra in a characteristic pattern with three signals at ca. +17, +13 and +1 ppm which have been assigned to B(OH)3/[B(OH)4]−, [B3O3(OH)4]− and [B5O6(OH)4]− species [5,27,28]; compounds 2–4 all showed this characteristic pattern. Dilute solutions often give just one signal, assigned to B(OH)3/[B(OH)4]−, at a chemical shift which is variable [19] but can be calculated assuming fast exchange of B(OH)3/[B(OH)4]− according to their relative proportions present in the oxidopolyborate anion originally present [27]. Unsurprisingly, therefore, compounds 1 and 5–8 all gave single signals at +11.1, +16.3, +15.8, +17.7 and +15.8 ppm respectively. The observed chemical shifts for the tetraborate(2-) (1) and the pentaborate(-) (5) salts are as would be expected from this calculation (+11.0 and +16.1, respectively) but chemical shifts for the hexaborate(2-) (7 and 8) and the heptaborate(2-) (6) salts are ca. 2 ppm more downfield than expected (13.8 and 14.6 ppm calc., respectively), indicating that more B(OH)3 might be present than expected. Similar behaviour has been observed before for hexaborate [34,35,36,37,38] and heptaborate [33] complexes.
Vibration spectroscopy is often used to help characterize oxidopolyborate species since strong, and often diagnostic, B-O stretches can be observed by IR spectroscopy between 1600 and 600 cm−1 [63]. In particular, diagnostic bands have been reported for pentaborate(1-) [5,27,28,64], hexaborate (2-) [35,37,63], and heptaborate(2-) [28,33,52] anions. Compounds 1–8 all displayed such diagnostic bands with the diagnostic band for the pentaborate(1-) anion (ca. 1925 cm−1) clearly present in 1- 5. Likewise, the diagnostic bands for hexaborate(2-) anion (ca. 960 and 810 cm−1) and the heptaborate(2-) anion (ca. 860 cm−1) were observed in IR spectra of 6 and 7, and 8, respectively.
Selected Ni(II) oxidoborates (2, 4–7) were thermally decomposed air and insights into these decompositions were obtained through TGA/DSC studies. The Ni(II) oxidoborates all yielded green glassy residues by 700 °C with masses consistent with anhydrous Ni(II) oxidoborates being produced with Ni:B stochiometries in the elemental ratios of their precursors. Thus, the pentaborates 2 and 4 were decomposed to NiB10O16 whilst 5 gave Ni2B10O17, the heptaborate (6) gave Ni2B14O23 and the hexaborate (7) yielded NiB6O10. These decomposition reactions are generally two-step processes with the first steps being a lower temperature (<280 °C) endothermic dehydration (loss of interstitial H2O and condensation reactions of the hydrated oxidoborates) followed by higher temperature (280–700 °C) exothermic oxidations of the organic ligands. Details for individual compounds (2, 4–7) are given in the experimental section. This type of decomposition behaviour has been observed previously for other hydrated Ni(II) oxidoborates [33,42,43,44,45,46,47,48] and more generally applies to the thermolysis of many hydrated transition-metal oxidopolyborates [30,33,35,37]. Powder XRD data were also obtained for this selection of Ni(II) oxidoborates (viz. 2, 4–7). The data obtained were good matches with data calculated from the single-crystal XRD data indicating that these samples were crystalline and homogeneous.
3. Experimental Methods
3.1. General
The Ni(II) complexes s-fac-[Ni(dien)2]Cl2.H2O [65] [Ni(dmen)2]Cl2.4H2O [66], [Ni(HEen)2]Cl2 [67], [Ni(AEN)]Cl.H2O [62], trans-[Ni(dach)2(H2O)2]Cl2 [68] were prepared by modified literature procedures. FTIR spectra were obtained as KBr pellets on a Perkin–Elmer 100FTIR spectrometer (Perkin–Elmer, Seer Green, UK). 11B NMR spectra were obtained on a Bruker Avance-400 spectrometer (Bruker, Coventry, UK) on samples dissolved in D2O at 128 MHz. TGA and DSC were performed on an SDT Q600 instrument (TA Instruments, New Castle DE, USA) using Al2O3 crucibles with a ramp rate of 10 °C per minute (RT to 1000 °C in air). X-ray crystallography was performed at the EPSRC national crystallography service centre at Southampton University. Magnetic susceptibility measurements were performed on a Johnson–Matthey magnetic susceptibility balance. (Johnson–Matthey, UK). CHN analyses were obtained from OEA Laboratories (Callingham, Cornwall).
3.2. X-ray Crystallography
Crystallographic data for 1–8 are given in the experimental section. The crystallographic data collection of compounds were performed on a Rigaku FR-E+ diffractometer (1, 3–8) equipped with an AFC12 goniometer, an HG Saturn 724+ detector and either HF varimax confocal mirrors (1, 3, 4, 6–8) or VHF varimax confocal mirrors (5). Data for 2 were obtained on DLS synchronous beamtime 119 equipped with a crystallographic goniometer and a Rigaku HG Saturn 724 detector. Data for all compounds were obtained at 100(2)K. Cell determinations and data collections were carried out using either CrysAlisPro [69] (1) or CrystalClear [70] (2–8). Data reduction, cell refinement and absorption correction were carried out using either CrysAlisPro [69] (1–3, 5–8) or CrystalClear [70] (4). Using Olex2 [71] the structures were solved using SHELXT [72] and models or refined with SHELXL [73]. Structure refinement details are given in the Supplementary Material. CCDC 2101911-2101918 (1–8 respectively), contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from CCDC, 12 Union Road, Cambridge, CB2 1EZ. Fax: +44 1223 336033; E-mail: [email protected])
3.3. Preparation of Trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O (1)
NiCl2.6H2O (1.00 g, 4.0 mmol) dissolved in ethanol (25 mL) and gave a clear green solution. Aqueous NH3 solution (2.2 mL, 25%, 24 mmol) was added dropwise to the green solution with stirring and then was left to stir for a further 5 min to give a dark blue solution. Ag2O (0.926 g, 4 mmol) was rapidly added and stirred at room temperature for 5 min and the precipitate which had formed (AgCl) was removed by filtration. B(OH)3 (1.23 g, 20 mmol) was added to the dark blue filtrate and left to stir for a further 10 min. The reaction mixture was filtered into vials and left to allow for slow evaporation of the solvent. After 2 weeks a light blue solid (1) had formed in the bottom of a vial and this was collected by filtration. (0.6 g, 40%). M.p. ≥ 300 °C. χm = 2500×10−6 cm3·mol−1. H22B4N4NiO12. Anal. Calc.: H = 6.0%, N = 15.1%. Found: H = 5.7%, N = 15.3. 11B/ppm: 11.1. IR (KBr/cm−1): 3375(s), 3245(s), 1634(m), 1450(s), 1402(s), 1345(s), 1230(m), 1152(m), 1059(m), 1002(s), 936(m), 828(m). sc-XRD crystal data: Mr = 372.16, monoclinic, P21/c (No. 14), a = 8.27730(10) Å, b = 20.5277(3) Å, c = 8.01330(10) Å, β = 91.2830(10)°, α = γ = 90°, V = 1361.23(3) Å3, T = 100(2) K, Z = 4, Z’ = 1, μ(MoKα) = 1.492 mm−1, 15,477 reflections measured, 3128 unique (Rint = 0.0329) which were used in all calculations. The final wR2 was 0.0528 (all data) and R1 was 0.0216 (I > 2σ(I)).
3.4. Preparation of s-fac-[Ni(dien)2][B5O6(OH)4]2 (2)
s-fac-[Ni(dien)2]Cl2.H2O (1.05 g, 3.0 mmol) was dissolved in H2O (10 mL) and added to a suspension of excess DOWEX® monosphere 550A OH− activated anion exchange resin (Dupont, Wilmington, DE, USA) (40 g) in H2O (40 mL). The mixture was left to stir for 30 h at RT and then filtered to remove the resin. The filtrate, containing s-fac-[N i(dien)2](OH)2, was reduced in volume to 15 mL and MeOH (15 mL) was added. B(OH)3 (1.83 g, 29.7 mmol) was added to the H2O/MeOH solution which was then gently warmed with stirring for 3 h. The solution was concentrated to 5 mL under reduced pressure and the concentrated solution was left for a few days for crystallization. Purple crystals of 2 were carefully recovered by filtration and dried in air (0.70 g, 34%). M.p. = 287–289 °C (dec.). χm = 3516 × 10−6 cm3 mol−1. C8H34B10N6NiO20. Anal. Calc.: C = 13.7%, H = 4.9%, N = 12.0%. Found: C = 14.0%, H = 5.4%, N = 12.1%. 11B/ppm: 1.5 (1%), 13.3 (13%), 17.4 (86%). IR (KBr/cm−1): 3362(s), 3333(s), 3297(s), 3263(s), 1412(s), 1330(s), 1303(s), 1135(m), 1059(m), 1042(m), 1028(m), 964(m), 931(m), 915(s), 774(s), 772(m), 707(s). TGA: 100–200 ∘C, condensation of polyborate which loss of four H2O 14.9% (10.2% calc.); 200–650 ∘C, oxidation of organic content 40.3% (39.7% calc.); residual NiB8O13 59.7% (60.3% calc.). p-XRD d-spacing/Å (% rel. int.): 6.22 (24), 5.88 (100), 5.57 (94), 4.27 (15), 3.79 (19), 3.71 (28), 2.94 (17). sc-XRD crystal data: Mr = 701.22, triclinic, P-1 (No. 2), a = 8.5763(5) Å, b = 9.2902(6) Å, c = 9.3493(6) Å, α = 78.098(5)°, β = 89.108(5)°, γ = 89.110(5)°, V = 728.75(8) Å3, T = 100(2) K, Z = 1, Z’ = 0.5, μ(Synchrotron0.6889Å) = 0.702 mm−1, 6342 reflections measured, 3152 unique (Rint = 0.0476) which were used in all calculations. The final wR2 was 0.2109 (all data) and R1 was 0.0767 (I > 2σ(I)).
3.5. Preparation of Trans-[Ni(dmen)2(H2O)2][B5O6(OH)4]2.2H2O (3)
[Ni(dmen)2]Cl2.4H2O (0.97 g, 3.0 mmol), and Ag2O (0.75 g, 3.2 mmol) were rapidly stirred in H2O (30 mL) at room temperature for 40 min and the precipitate which had formed (AgCl) was removed by filtration. B(OH)3 (1.85 g, 30 mmol) was added to the dark blue filtrate and left to stir for a further 40 min. The reaction mixture was filtered into vials and then left to allow for slow evaporation of solvent. After 3 weeks a light blue solid (3) had formed in the bottom of a vial and this was collected by filtration. (0.9 g, 40%). M.p. ≥ 300 °C. χm = 3600 × 10−6 cm3·mol−1. C8H40B10N4NiO24. Anal. Calc.: C = 12.9%, H = 5.4%, N = 7.5%. Found: C = 13.0%, H = 5.5%, N = 7.5. 11B/ppm 16.6 (85%), 13.9 (14%), 1.2 (1%). IR (KBr/cm−1): 3510 (m), 3348(s), 3285(s), 1608(m), 1415(s), 1310(s), 1129(m), 1043(m), 1015(m), 920(s), 780(s), 707(m), 486(m). sc-XRD crystal data: Mr = 743.25, triclinic, P-1 (No. 2), a = 8.6570(3) Å, b = 9.7443(3) Å, c = 10.2101(3) Å, α = 107.087(3)°, β = 102.747(3)°, γ = 95.002(3)°, V = 792.23(5) Å3, T = 100(2) K, Z = 1, Z’ = 0.5, μ(MoKα) = 0.708 mm−1, 12,978 reflections measured, 3628 unique (Rint = 0.0163) which were used in all calculations. The final wR2 was 0.0731 (all data) and R1 was 0.0270 (I > 2σ(I)).
3.6. Preparation of [Ni(HEen)2][B5O6(OH)4]2 (4)
The preparation of [Ni(HEen)2](OH)2 was carried out as described previously for 2 from [Ni(HEen)2]Cl2 (1.00 g, 2.95 mmol) and Dowex (40 g). B(OH)3 (1.83 g, 29.6 mmol) was added with stirring. The reaction mixture was stirred for a further 2 h at room temperature. The solvent was then evaporated to 5 mL using a rotary evaporator, then the resulting solution was left for 10 d in several NMR tubes for crystallization to yield purple crystals of 4 (0.76 g, 37%). M.p. = 288–289 °C (dec.). χm = 3430 × 10−6 cm3 mol−1 C8H32B10N4NiO22. Anal. Calc.: C = 13.7%, H = 4.6%, N = 8.0%. Found: C = 13.7%, H = 4.7%, N = 8.0%. 11B/ppm: 1.2 (3%), 13.4 (23%), 18.0 (74%). IR (KBr/cm−1): 3375(s), 3288(s), 3322(s), 1410(s), 1321(s), 1196(m), 1141(s), 1022(s), 919(s), 774(s), 706(s). TGA: 230–290 ∘C, condensation of polyborate which loss of four H2O 10.3% (10.2% calc.); 290–700 ∘C, oxidation of organic content 37.9% (39.9% calc.); residual NiB10O16 62.1% (60.1 calc.). p-XRD d-spacing/Å (% rel. int.): 6.18 (40), 5.88 (75), 5.50 (100), 3.94 (26), 3.78 (38), 3.66 (25). sc-XRD crystal data: Mr = 703.18, Monoclinic, P21/c, a = 8.3891(2) Å, b = 11.6666(3) Å, c = 14.3092(4) Å, α = γ = 90°, β = 90.035(2)°, V =1400.47(6) Å3, T = 100(2) K, Z = 2, Z’ = 0.5, μ(MoKα) = 0.791 mm−1, 6239 reflections measured, 6239 unique (Rint = 0) which were used in all calculations. The final wR2 was 0.1124 (all data) and R1 was 0.0499 (I > 2σ(I)).
3.7. Preparation of [Ni(AEN)][B5O6(OH)4].H2O (5)
The preparation of [Ni(AEN)](OH) was carried out as described previously for 2 from [Ni(AEN)]Cl.H2O (2.10 g, 7.0 mmol) and Dowex (25 g). B(OH)3 (2.22 g, 36 mmol) was added to the aqueous solution which was gently warmed with stirring for 3 h, and then its volume was reduced to 5 mL under reduced pressure. The solution was then distributed in several NMR tubes for crystallization and left for 10 days to yield dark red crystals of 5 (1.30 g, 38%). M.p. = 279–280 °C (dec.). χm = −170 × 10−6 cm3 mol−1. C9H25B5N4NiO11. Anal. Calc.: C = 22.6%, H = 5.3%, N = 11.7%. Found: C = 22.7%, H = 5.4%, N = 11.7%. 1H/ppm: 2.0 (s, 6H, CH3), 2.5 (t, 4H, J = 6 Hz, CH2 of en), 3.2 (t, 4H, J = 6 Hz, CH2 of en), 4.8 (s, 11H, NH2, H2O, OH, CH). 13C/ppm: 19.9, 43.0, 53.6, 160.5. 11B/ppm: 16.3. IR (KBr/cm−1): 3377(s), 3326(s), 3288(s), 3246(s), 3169(s), 1615(m), 1566(m), 1536(m), 1476(s), 1442(s), 1416(s), 1393(s), 1333(s), 1295(s), 1129(m), 1094(s), 1072(m), 1019(m), 924(s), 778(m). TGA: 100-180 ∘C, loss of one interstitial H2O 2.5% (3.7% calc.); 180–280 ∘C, condensation of polyborate which loss of two further H2O 10.0% (11.3% calc.); 280–700 °C, oxidation of organic content 46.1% (48.0% calc.); residual Ni2B10O17 53.9% (52.0% calc.). p-XRD d-spacing/Å (% rel. int.): 5.65 (69), 5.38 (100), 4.14 (46), 3.89 (33), 3.81 (53), 3.65 (58), 2.03 (41), 1.43 (33). sc-XRD crystal data: Mr = 478.09, Triclinic, P-1, a = 8.5386(2) Å, b = 11.3328(3) Å, c = 11.8717(3) Å, α = 115.914(3)°, β = 101.626(2)°, γ = 99.670(2)°, V = 968.39(5) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(MoKα) = 1.065 mm−1, 16,639 reflections measured, 4433 unique (Rint = 0.0216) which were used in all calculations. The final wR2 was 0.0765 (all data) and R1 was 0.0278 (I > 2σ(I)).
3.8. Preparation of Trans-[Ni(dach)2(H2O)2] [Ni(dach)2] [B7O9(OH)5]2.4H2O (6)
The preparation of [Ni(dach)2(H2O)2](OH)2 was carried out as previously described for 2 from trans-[Ni(dach)2(H2O)2]Cl2 (1.00 g, 2.53 mmol) and Dowex (40 g). B(OH)3 (1.56 g, 25.2 mmol) was added to the solution which was then gently warmed with stirring for 3 h. The solution volume was reduced to 5 mL under reduced pressure. The product (6) was isolated by filtration, and then allowed to dry in an oven at 40 °C for 3 h to afford orange prismatic crystals (0.78 g, 47%). M.p. = 270–272 °C (dec.). χm = 3030 × 10−6 cm3 mol−1. C24H78B14N8Ni2O34. Anal. Calc.: C = 22.3%, H = 6.1%, N = 8.7%. Found: C = 21.8%, H = 6.3%, N = 8.4%. 11B/ppm: 15.8. IR (KBr/cm−1): 3663(s), 3271(s), 2929(m), 2863(m), 1468(m), 1453(s), 1350(s), 1180(s), 1125(m), 1066(s), 986(w), 854(m), 807(m). TGA: 100–190 ∘C, loss of four interstitial and two coordinated H2O 7.5% (8.4% calc.); 190–270 ∘C, condensation of polyborate which loss of five further H2O 15.0% (15.3% calc.); 270–720 ∘C, oxidation of organic content 48.2% (50.7% calc.); residual Ni2B14O23 51.8% (49.3% calc.). p-XRD d-spacing/Å (% rel. int.): 10.67 (100), 9.81 (89), 6.25 (58), 5.35 (70), 4.26 (48), 3.74 (55), 2.03(59). sc-XRD crystal data: Mr = 645.85, Monoclinic, C2/c, a = 22.3539(4) Å, b = 11.0192(2) Å, c = 22.8834(4) Å, α = γ = 90°, β = 107.630(2)°, V = 5371.94(18) Å3, T = 100(2) K, Z = 8, Z’ = 1, μ(MoKα) = 0.806 mm−1, 33628 reflections measured, 6154 unique (Rint = 0.0281) which were used in all calculations. The final wR2 was 0.1087 (all data) and R1 was 0.0384 (I > 2σ(I)).
3.9. Preparation of Trans-[Ni(en)(H2O)2{B6O7(OH)6}].H2O (7)
An aqueous solution of en (2.30 g, 70%, 26.79 mmol) was added to an aqueous solution (10 mL) of NiSO4.6H2O (2.12 g, 8.06 mmol). The reaction mixture was stirred for 5 min, and then an aqueous solution (10 mL) of Ba(OH)2.8H2O (2.54 g, 8.06 mmol) was added. The reaction mixture was stirred for 30 min and then filtered to remove the BaSO4. A solution of B(OH)3 (4.98 g, 80.6 mmol) in H2O (10 mL) was added to the filtrate with stirring. The reaction mixture was stirred at room temperature for 2 h. The solution was then distributed in several vials and left for 10 d to yield faint blue crystals of 7 (1.20 g, 33%). M.p. = 245–247 °C (dec.). χm = 2588 × 10−6 cm3 mol−1. C2H20B6N2NiO16. Anal. Calc.: C = 5.3%, H = 4.5%, N = 6.2%. found: C = 5.4%, H = 4.5%, N = 6.2%. 11B/ppm: 17.7. IR (KBr/cm−1): 3400(s), 3350(s), 2924(w), 1420(m), 1380(s), 1360(s), 1133(s), 1095(s), 1044(s), 955(m), 908(m), 809(s). TGA: 100–180 ∘C, loss of one interstitial H2O and two coordinated water molecules 0.8% (12% calc.); 180–280 ∘C, condensation of polyborate which loss of three further H2O 24.3% (23.9% calc.); 280–700 ∘C, oxidation of organic content 38.6% (37.2% calc.); residual NiB6O10 61.4% (62.8% calc.). p-XRD d-spacing/Å (% rel. int.): 9.78 (100), 8.26 (24), 6.77 (53), 4.88 (48), 3.99 (34), 3.38 (30), 2.74 (47). sc-XRD crystal data: Mr = 451.77, triclinic, P-1 (No. 2), a = 8.5116(7) Å, b = 9.7946(5) Å, c = 9.8073(8) Å, α = 89.873(5)°, β = 82.901(7)°, γ = 74.372(6)°, V = 780.94(10) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(MoKα) = 1.333 mm−1, 5883 reflections measured, 5883 unique (Rint = 0) which were used in all calculations. The final wR2 was 0.1902 (all data) and R1 was 0.0682 (I > 2σ(I)).
3.10. Preparation of [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (8)
The preparation of [Ni(dmen)2](OH)2 was carried out as described for 2 from [Ni(dmen)2]Cl2.4H2O (1.00 g, 2.6 mmol) and Dowex (30 g). B(OH)3 (1.57 g, 25 mmol) was added to the filtrate and left to stir for 30 min. The turquoise solution was filtered into vials and left to allow for slow evaporation of the solvents. After 2 weeks, a blue-green solid (8) had formed in the bottom of a vial and this was collected by filtration. Yield: (0.5 g, 36%). M.p. ≥ 300 °C. χm = 3050 × 10−6 cm3 mol−1. C4H30B6N2NiO19. Anal. Calc.: C = 9.0%, H = 5.7%, N = 5.2%. Found: C = 9.3%, H = 5.5%, N = 5.5. 11B/ppm: 15.8. IR (KBr/cm−1): 3399(s), 1642(m), 1416(s), 1355(s),1247(s), 1120(s), 1052(m), 1009(m), 962(m), 859(m), 813(s), 700(m). sc-XRD crystal data: Mr = 533.87, triclinic, P-1 (No. 2), a = 9.1508(4) Å, b = 9.8599(4) Å, c = 13.5971(4) Å, α = 88.525(3)°, β = 70.627(3)°, γ = 65.245(4)°, V = 1041.79(8) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(MoKα) = 1.023 mm−1, 15,537 reflections measured, 4713 unique (Rint = 0.0461) which were used in all calculations. The final wR2 was 0.1048 (all data) and R1 was 0.0403 (I > 2σ(I)).
4. Conclusions
Eight new oxidoborate Ni(II) compounds were prepared by self-assembly crystallization processes from aqueous solution starting from B(OH)3 and selected Ni(II) complexes. Six of these compounds were salts containing insular oxidoborate anions, but two products were complexes containing coordinated oxidoborate(2-) anions, with energetically favourable Ni-O bonds. The two products that contained coordinated hexaborate(2-) ligands were neutral monomeric complexes, rather than the 1-D coordination polymers that we have occasionally observed in related Cu(II) and Zn(II) chemistry. The new compounds were templated in all cases by numerous strong structure-directing inter/intramolecular H-bonding interactions involving the oxidoborate ligands and many novel H-bonding motifs are noted and described. This work further demonstrates that in general the inclusion of labile metal complexes into aqueous solutions containing B(OH)3 can lead to the self-assembly of novel species including those with coordinated oxidoborate ligands and that this synthetic strategy could be applied successfully to other systems.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/inorganics9090068/s1: single-crystal XRD data, including H-bond interactions.
Author Contributions
M.A.B. conceived the experiments; M.A.A. synthesized and characterized the complexes and grew the single-crystals; P.N.H. and S.J.C. solved the crystal structures; M.A.B. wrote the paper with contributions from all co-authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Crystallographic data available from CCDC, UK, see Section 3.2 for details.
Acknowledgments
We thank the EPSRC for the use of the X-ray Crystallographic Service (NCS, Southampton, UK).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beckett, M.A.; Brellocks, B.; Chizhevsky, I.T.; Damhus, T.; Hellwich, K.-H.; Kennedy, J.D.; Laitinen, R.; Powell, W.H.; Rabinovich, D.; Vinas, C.; et al. Nomenclature for boranes and related species (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 355–381. [Google Scholar] [CrossRef] [Green Version]
- Heller, G. A survey of structural types of borates and polyborates. Top. Curr. Chem. 1986, 131, 39–98. [Google Scholar]
- Grice, J.D.; Burns, P.C.; Hawthorne, F.C. Borate minerals II. A hierarchy of structures based upon the borate fundamental building block. Canad. Min. 1999, 37, 731–762. [Google Scholar]
- Christ, C.L.; Clark, J.R. A crystal-chemical classification of borate structures with emphasis on hydrated borates. Phys. Chem. Miner. 1977, 2, 59–87. [Google Scholar] [CrossRef]
- Beckett, M.A. Recent Advances in crystalline hydrated borates with non-metal or transition-metal complex cations. Coord. Chem. Rev. 2016, 323, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.M. Borates in industrial use. Struct. Bond. 2003, 105, 1–40. [Google Scholar]
- Schubert, D.M. Boron oxide, boric acid, and borates. In Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed.; John and Wiley and Sons: Hoboken, NJ, USA, 2011; pp. 1–68. [Google Scholar]
- Schubert, D.M. Hydrated zinc borates and their industrial use. Molecules 2019, 24, 2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, L.-Q.; Huang, H.-S.; Liu, Z.-H. Solvotheramal synthesis and crystal structures of two novel borates [(CH3)3NH][B5O6(OH)4] and Na2[H2TMED][B7O9(OH)5]2. J. Clust. Sci. 2014, 25, 893–903. [Google Scholar] [CrossRef]
- Liang, J.; Wang, Y.-G.; Wang, Y.-X.; Liao, F.-H.; Lin, J.-H. Supramolecular assembly of borate with quaternary ammonium: Crystal structure and tunable luminescent properties. J. Solid State Chem. 2013, 200, 99–104. [Google Scholar] [CrossRef]
- Jemai, N.; Rzaigui, M.; Akriche, S. Stabilization of hexaborate nets with mixed Co(II) metal and organic cations: Synthesis, rationale, characterization, comparative study and enhancement of bioactivity. J. Clus. Sci. 2015, 26, 2051–2064. [Google Scholar] [CrossRef]
- Becker, P. Borate materials in nonlinear optics. Adv. Mater. 1998, 10, 979–992. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, S. Recent developments of metal borate halides: Crystal chemistry and application in second-order NLO materials. Coord. Chem. Rev. 2016, 323, 15–35. [Google Scholar] [CrossRef]
- Becker, P.; Held, P.; Bohaty, L. Crystal growth and optical properties of the polar hydrated pentaborates Rb[B5O6(OH)4].2H2O and NH4[B5O6(OH)4].2H2O and structure redetermination of the ammonium compound. Cryst. Res. Technol. 2000, 35, 1251–1262. [Google Scholar] [CrossRef]
- Liu, H.-X.; Liang, Y.-X.; Jiang, X. Synthesis, crystal structure and NLO properties of a nonmetal pentaborate [C6H13N2][B5O6(OH)4]. J. Solid State Chem. 2008, 181, 3243–3247. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Jones, C.L.; Krueger, K. Synthesis, XRD studies and NLO properties of [p-H2NC6H4CH2NH3][B5O6(OH)4].2H2O and NLO properties of some related salts. J. Clus. Sci. 2017, 28, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, G.; Tian, S.; Liao, F.; Lin, J. Synthesis and structure of [C2H10N2][B5O8(OH)]: A nonmetal pentaborate with nonlinear optical properties. J. Cryst. Growth Des. 2007, 7, 1246–1250. [Google Scholar] [CrossRef]
- Paul, A.K.; Sachidananda, K.; Natarajan, S. [B4O9H2] cyclic borates as the building unit in a family of zinc borate structures. Cryst. Growth Des. 2010, 10, 456–464. [Google Scholar] [CrossRef]
- Salentine, G. High-field 11B NMR of alkali borate. Aqueous polyborate equilibria. Inorg. Chem. 1983, 22, 3920–3924. [Google Scholar] [CrossRef]
- Anderson, J.L.; Eyring, E.M.; Whittaker, M.P. Temperature jump rate studies of polyborate formation in aqueous boric acid. J. Phys. Chem. 1964, 68, 1128–1132. [Google Scholar] [CrossRef]
- Corbett, P.T.; Leclaire, J.; Vial, L.; West, K.R.; Wietor, J.-L.; Sanders, J.K.M.; Otto, S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. [Google Scholar] [CrossRef]
- Sola, J.; Lafuente, M.; Atcher, J.; Alfonso, I. Constitutional self-selection from dynamic combinatorial libraries in aqueous solution through supramolecular interactions. Chem. Commun. 2014, 50, 4564–4566. [Google Scholar] [CrossRef] [Green Version]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Wiebcke, M.; Freyhardt, C.C.; Felsche, J.; Engelhardt, G. Clathrates with three-dimensional host structures of hydrogen bonded pentaborate [B5O6(OH)4]− ions: Pentaborates with the cations NMe4+, NEt4+, NPhMe3+ and pipH+ (pipH+ = piperidinium). Z. Naturforsch. 1993, 48, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Visi, M.Z.; Knobler, C.B.; Owen, J.J.; Khan, M.I.; Schubert, D.M. Structures of self-assembled nonmetal borates derived from α,ω-diaminoalkanes. Cryst. Growth Des. 2006, 6, 538–545. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Davies, R.A.; Horton, P.N.; Jones, C.L. Pentaborate(1-) salts templated by substituted pyrrolidinium cations: Synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations. Dalton Trans. 2015, 44, 7032–7040. [Google Scholar] [CrossRef] [Green Version]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Jones, C.L. Polyborate anions partnered with large nonmetal cations: Triborate(1-) pentaborate(1-) and heptaborate(2-) salts. Eur. J. Inorg. Chem. 2017, 4510–4518. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. A new polyborate anion [B7O9(OH)6]3-: Self-assembly, XRD and thermal properties of s-fac-[Co(en)3][B7O9(OH)6].9H2O. Inorg. Chem. Commun. 2015, 59, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. A new decaoxidooctaborate(2-) anion, [B8O10(OH)6]2-: Synthesis and characterization of [Co(en)3][B5O6(OH)4][B8O10(OH)6].5H2O (en = 1,2-diaminoethane). Inorg. Chem. 2015, 54, 412–414. [Google Scholar] [CrossRef]
- Taube, H. Rates and mechanisms of substitutions in inorganic complexes in aqueous solution. Chem. Rev. 1952, 50, 69–126. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [H3NCH2CH2NH2)Zn{k3O,O’,O’’-B12O18(OH)6-k1O’’’} Zn(en)(NH2CH2CH2NH3)].8H2O: A neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6-) anion. Pure Appl. Chem. 2018, 90, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Oxidopolyborate anions templated by transition-metal complexes: Self-assembled synthesis and structural studies (XRD) of [Co(NH3)6]2[B4O5(OH)4]3.11H2O, [Ni(phen)3][B7O9(OH)5].9.5H2O and [Zn(dac)2(H2O)2][B7O9(OH)5].H2O. J. Mol. Struct. 2020, 1200, 127071. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Two 1-D Coordination Polymers containing Zinc(II) Hexaborates: [Zn(en){B6O7(OH)6}].2H2O (en = 1,2-diaminoethane) and [Zn(pn){B6O7(OH)6}].1.5H2O (pn = (+/−) 1,2-diaminopropane). Crystals 2018, 8, 470. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Hexaborate(2-) and dodecaborate(12-) anions as ligands to zinc(II) centres: Self-assembly and single crystal XRD characterization of [Zn{k3O-B6O7(OH)6}(k3-N-dien)]0.5H2O (dien = NH(CH2CH2NH2)2), (NH4)2[Zn{k2-O-B6O7(OH)6} (H2O)2].2H2O and (1,3-pn)3[({k1-NH3{CH2}3NH2)Zn{k3-O-B12O18(OH)6}]2.14H2O. Inorganics 2019, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Oxidoborate chemistry: The self-assembled, templated, synthesis, and an XRD study of a 1-D coordination polymer [Cu(en){B6O7(OH)6}].3H2O (en = 1,2-diaminoethane). Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 952–956. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Synthesis and characterization of polyborates templated by cationic copper(II) complexes: Structural (XRD), spectroscopic, thermal (TGA/DSC) and magnetic properties. Polyhedron 2017, 135, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Copper(2+) complexes of hydroxyoxidoborates. Synthesis and characterization of two clusters containing the hexaborate(2-) ligand: [Cu(NH2CH2CH2NEt2){B6O7(OH)6}].5H2O and [Cu(NH3)2{B6O7(OH)6}].2H2O. J. Cluster Sci. 2019, 30, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Synthesis and characterization by a single-crystal XRD study of [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}].13H2O: An unusual bis(hydroxytrioxidodiborate) tri-metallic chain supported by a [{Cu4O}{B20O32(OH)8]6- cluster. J. Cluster Sci. 2018, 29, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Copper(2+) complexes of hydroxyoxido polyborates: Synthesis and characterization of [Cu(MeNHCH2CH2NMeH)2(H2O)2][B5O6(OH)4]2 2B(OH)3. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 948–951. [Google Scholar] [CrossRef]
- Lan, S.M.; Di, W.-J.; Shao, Z.-D.; Liang, Y.-X. Two new transition metal inorganic–organic hybrid borates: [tris(2-aminoethoxy)trihydroxyhexaborato]cobalt(II) and its nickel(II) analogue. Acta Cryst. 2011, C67, m338–m341. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, J.; Liu, Z. Synthesis, crystal structures and thermal behavior of Co(en)3[B4O5(OH)4]Cl.3H2O and [Ni(en)3][B5O6(OH)4]2.2H2O. J. Chin. Chem. 2009, 27, 494–500. [Google Scholar] [CrossRef]
- Pan, C.-Y.; Hu, S.; Li, D.-G.; Ouyang, P.; Zhao, F.-H.; Zheng, Y.-Y. The first ferroelectric templated borate: [Ni(en)2pip][B5O6(OH)4]2. Dalton Trans. 2010, 39, 5772–5777. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Zhang, J.-J.; Zhang, W.-J. Synthesis, crystal structure and vibrational spectroscopy of a novel mixed ligands Ni(II) pentaborate [Ni(C4H10N2)(C2H8N2)2][B5O6(OH)4]2. Inorg. Chim. Acta 2006, 359, 519–524. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, G.M.; He, H.; Yang, B.-F.; Yang, G.-Y. Syntheses and Crystal Structures of Two New Pentaborates Templated by Transition-Metal Complexes. J. Clust. Sci. 2014, 25, 1295–1305. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Malinina, E.A.; Vologzhanina, A.V.; Sivaev, I.B.; Kuznetsov, N.T. Formation of oxidopolyborates in destruction of the [B11H14]− anion promoted by transition metals. Inorg. Chim. Acta 2020, 509, 119693. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, F.; Yin, X.H. Hydrothermal synthesis and crystal structure of organic-hybrid borate [Ni(dien)2][B5O6(OH)4]. Chem. Res. Appl. 2010, 22, 1587–1591. [Google Scholar]
- Kose, D.A.; Yurdakul, O.; Sahin, O.; Ozturk, Z. The new metal complex templated polyoxoborate(s) (POB(s)) structures. Synthesis, structural characterization, and hydrogen storage capacities. J. Mol. Struct. 2017, 1134, 806–813. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Hodgson, P.G. Copper-ligand bond-lengths in axial complexes of the copper(II) ion. J. Inorg. Nucl. Chem. 1973, 35, 4071–4081. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Li, L.-Q.; Zhang, W.-J. Two new borates containing the first examples of large isolated polyborate anions: Chain [B7O9(OH)5]2− and Ring [B14O20(OH)6]4−. Inorg. Chem. 2006, 45, 1430–1432. [Google Scholar] [CrossRef]
- Schubert, D.M.; Visi, M.Z.; Khan, S.; Knobler, C.B. Synthesis and structure of a new heptaborate oxoanion isomer: B7O9(OH)52-. Inorg. Chem. 2008, 47, 4740–4745. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L.; Varma, K.S. Templated heptaborate and pentaborate salts of cyclo-alkylammonium cations: Structural and thermal properties. Dalton. Trans. 2012, 41, 4396–4403. [Google Scholar] [CrossRef]
- Sacconi, L.; Mani, F.; Bencini, A. Nickel. In Comprehensive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Books Ltd.: Oxford, UK, 1987; Volume 5, Chapter 50; pp. 1–347. [Google Scholar]
- Ihara, Y.; Satake, Y.; Fujimolo, Y.; Senda, H.; Suzuki, M.; Uehara, A. X-ray structures of octahedral diaqua(N,N)dialkylethelenediamine)nickel(II) complexes possessing elongated nickel(II)-nitrogen bonds along axial direction. Bull. Chem. Soc. Jpn. 1991, 64, 2349–2352. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yin, X.-H.; Zhang, F. Two novel thioindate-thioantimonate compounds with [Ni(dien)2]In2Sb4S11 and [Ni(dien)2](In3Sb2S9)2.2H2O with transition metal complexes. Inorg. Chem. 2010, 49, 9671–9676. [Google Scholar] [CrossRef]
- Natarajan, S.; Klein, W.; Panthoefer, M.; Wuellen, L.V.; Jansen, M. Solution mediated synthesis and structure of the first anionic bis(hexaborato)-zincate prepared in the presence of organic base. Z. Anorg. Allg. Chem. 2003, 629, 959–962. [Google Scholar] [CrossRef]
- Jemai, N.; Rzaigui, S.; Akriche, S. Piperazine-1,4-diium bis(hexahydroxidoheptaoxidohexaborato-k3O,O’,O’’)cobaltate(II) hexahydrate. Acta Cryst. 2014, 70, m167–m169. [Google Scholar] [CrossRef]
- Xin, S.-S.; Zhou, M.-H.; Beckett, M.A.; Pan, C.-Y. Recent advances in crystalline oxidoborate complexes of d-block or p-block metals: Structural aspects, syntheses and physical properties. Molecules 2021, 26, 3815. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic chemistry. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Zolatarev, P.N.; Arshad, M.N.; Asiri, A.M.; Al-amshany, Z.M.; Blatov, V.A. A possible route toward expert systems in supramolecular chemistry: 2-periodic H-bonded patterns in molecular crystals. Cryst. Growth Des. 2014, 14, 1938–1949. [Google Scholar] [CrossRef]
- Figgis, B.N.; Lewis, J. The magnetic properties of transition metal complexes. Prog. Inorg. Chem. 1964, 6, 37–239. [Google Scholar] [CrossRef]
- Elfring, W.H.; Rose, N.J. Studies of three nickel(II) containing different macrocyclic ligands derived from acetylacetone and triethlenetetramine. Inorg. Chem. 1975, 14, 2759–2768. [Google Scholar] [CrossRef]
- Li, J.; Xia, S.; Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta 1995, 51, 519–532. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Coles, S.J.; Kose, D.A.; Kreuziger, A.-M. Structural and thermal studies of non-metal cation pentaborate salts derived from 1,5-diazobicyclo[4.3.0]non-5-ene, 1,8-diazobicyclo[5.4.0]undec-7-ene and 1,8-bis(dimethylamino)naphthalene. Polyhedron 2012, 38, 157–161. [Google Scholar] [CrossRef]
- Breckenridge, J.G. Copper(II) and nickel(II) coordination compounds with diethyltriamine and hydroxyethylethylenediamine ligands. Can. J. Res. 1948, 6, 11–19. [Google Scholar] [CrossRef]
- Cui, A.; Chen, X.; Sun, J.; Wei, J.; Yang, J.; Kou, H. Preparation and thermochromic properties of copper(II)-N,N-diethylethylenediamine. J. Chem Educ. 2011, 88, 311–312. [Google Scholar] [CrossRef]
- Wendlandt, W.W. The thermal decomposition of metal complexes—VI. Some amine complexes of copper(II) sulphate. J. Inorg. Nucl. Chem. 1963, 25, 833–842. [Google Scholar] [CrossRef]
- Saito, R.; Kidani, Y. Preparative separation of cis- and trans-1,2-diaminocyclohexane by means of selective nickel(II) complex formation. Chem. Lett. 1976, 5, 123–126. [Google Scholar] [CrossRef] [Green Version]
- CrysAlis Pro Software System; Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2021.
- CrystalClear-SM; Expert 2.1 Software; Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2013.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement, and analysis programme. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelXT-intergrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Drawings of the organic ligand structures found in compounds 1–8 with abbreviations as follows: (a) en, 1,2-diaminoethane), (b) dien, N-(2-aminoethyl)-1,2-ethanediamine, (c) HEen, N-(2-hydroxyethyl)-1,2-diaminoethane, (d) dmen, N,N-dimethyl-1,2-diaminoethane, (e) dach, 1,2-diaminocyclohexane, (f) AEN, 1-(3-azapropyl)-2,4-dimethyl-1,5,8-triazaocta-2,4-dienato(1-).
Figure 1.
Drawings of the organic ligand structures found in compounds 1–8 with abbreviations as follows: (a) en, 1,2-diaminoethane), (b) dien, N-(2-aminoethyl)-1,2-ethanediamine, (c) HEen, N-(2-hydroxyethyl)-1,2-diaminoethane, (d) dmen, N,N-dimethyl-1,2-diaminoethane, (e) dach, 1,2-diaminocyclohexane, (f) AEN, 1-(3-azapropyl)-2,4-dimethyl-1,5,8-triazaocta-2,4-dienato(1-).

Scheme 1.
Reagents and stoichiometry of reactants involved in self-assembly of Ni(II) oxidoborates from cationic Ni(II) complexes with B(OH)3 in aqueous or methanolic aqueous solution.
Scheme 1.
Reagents and stoichiometry of reactants involved in self-assembly of Ni(II) oxidoborates from cationic Ni(II) complexes with B(OH)3 in aqueous or methanolic aqueous solution.

Figure 2.
Drawings of the oxidoborate structures found in (a) 1, (b) 2–5, (c) 7, 8 and (d) 6.

Figure 3.
Drawing of structure trans-[Ni(en)(H2O)2{B6O7(OH)6}].H2O (7), showing selected crystallographic numbering.
Figure 3.
Drawing of structure trans-[Ni(en)(H2O)2{B6O7(OH)6}].H2O (7), showing selected crystallographic numbering.

Figure 4.
Drawing of structure of [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (8), showing (selected) crystallographic numbering.
Figure 4.
Drawing of structure of [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (8), showing (selected) crystallographic numbering.

Figure 5.
Drawing of structure of trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (1), showing (selected) crystallographic numbering and the R33(8) ring system: {Ni1-O11A-H11A…O6-H6…O21…H11E-N11}.
Figure 5.
Drawing of structure of trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O [Ni(dmen)(H2O){B6O7(OH)6}].5H2O (1), showing (selected) crystallographic numbering and the R33(8) ring system: {Ni1-O11A-H11A…O6-H6…O21…H11E-N11}.

Figure 6.
Drawing of the R44(12) ring, {O6-H6…O21-H21A…O3′-B1′-O6′-H6′…O21′-H21A’…O3-B1-}, in trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O (1).
Figure 6.
Drawing of the R44(12) ring, {O6-H6…O21-H21A…O3′-B1′-O6′-H6′…O21′-H21A’…O3-B1-}, in trans-[Ni(NH3)4(H2O)2][B4O5(OH)4].H2O (1).

Figure 7.
Drawing of the R22(8) {Ni1-O11-H11B…O7-B2-O2…H12A-N12-} interaction involving the Ni(II) in trans-[Ni(dmen)2(H2O)2] [B5O6(OH)4]2.2H2O (3).
Figure 7.
Drawing of the R22(8) {Ni1-O11-H11B…O7-B2-O2…H12A-N12-} interaction involving the Ni(II) in trans-[Ni(dmen)2(H2O)2] [B5O6(OH)4]2.2H2O (3).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Oxidoborates Templated by Cationic Nickel(II) Complexes and Self-Assembled from B(OH)3. Inorganics 2021, 9, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090068
AMA Style
Altahan MA, Beckett MA, Coles SJ, Horton PN. Oxidoborates Templated by Cationic Nickel(II) Complexes and Self-Assembled from B(OH)3. Inorganics. 2021; 9(9):68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090068
Chicago/Turabian StyleAltahan, Mohammed A., Michael A. Beckett, Simon J. Coles, and Peter N. Horton. 2021. "Oxidoborates Templated by Cationic Nickel(II) Complexes and Self-Assembled from B(OH)3" Inorganics 9, no. 9: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090068
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.