Physi-Sorption of H2 on Pure and Boron–Doped Graphene Monolayers: A Dispersion–Corrected DFT Study
by
 and
and
Iffat Nayyar
1, 
Bojana Ginovska
1,*,
Abhijeet Karkamkar
1,
Thomas Gennett
2,3 and
Thomas Autrey
1 1
Physical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
2
Department of Chemistry, Colorado School of Mines, Golden, CO 80401, USA
3
National Renewable Energy Laboratory, Golden, CO 80401, USA
*
Author to whom correspondence should be addressed.
C 2020, 6(1), 15; https://0-doi-org.brum.beds.ac.uk/10.3390/c6010015
Submission received: 28 February 2020
/
Revised: 17 March 2020
/
Accepted: 18 March 2020
/
Published: 22 March 2020
(This article belongs to the Collection Carbon-Based Materials for Hydrogen Production, Storage and Conversion)
Abstract
:High-surface-area carbons are of interest as potential candidates to store H2 for fuel–cell power applications. Earlier work has been ambiguous and inconclusive on the effect of boron doping on H2 binding energy. Here, we describe a systematic dispersion–corrected density functional theory study to evaluate the effect of boron doping. We observe some enhancement in H2 binding, due to the presence of a defect, such as terminal hydrogen or distortion from planarity, introduced by the inclusion of boron into a graphene ring, which creates hydrogen adsorption sites with slightly increased binding energy. The increase is from −5 kJ/mol H2 for the pure carbon matrix to −7 kJ/mol H2 for the boron–doped system with the boron content of ~7%. The H2 binding sites have little direct interaction with boron. However, the largest enhancement in physi-sorption energy is seen for systems, where H2 is confined between layers at a distance of about 7 Å, where the H2 binding nearly doubles to −11 kJ/mol H2. These findings suggest that interplanar nanoconfinement might be more effective in enhancing H2 binding. Smaller coronene model is shown to be beneficial for understanding the dependence of interaction energy on the structural configurations and preferential H2 binding sites.
1. Introduction
Among hydrogen storage materials, activated carbons, graphene, carbon nanotubes, and metal-organic frameworks are of great interest due to a combination of large surface area, low mass density, and high structural stability [1]. However, the typical H2 binding energy of these high-surface-area carbon materials is weak (–5.1 kJ/mol H2) [2]. The weak binding energy is attributed to physi-sorption dominated by van der Waals attraction, such that adsorbed molecules of H2 do not remain attached at ambient temperatures and require liquid nitrogen temperatures to achieve adsorption [3,4]. For practical applications, such as H2 fuel–cell electric vehicles, the operating temperatures are much higher, ranging from −40 to 85 °C, and require binding energies on the order of 15 to 25 kJ/mol for pressures ranging from 5 to 100 bar [5,6]. Unsaturated metal centers (e.g., Mg and Ni in metal–organic frameworks) provide an effective approach for enhancing the strength of binding between H2 and nanostructured porous materials [7,8]. Other approaches have involved templating porous carbons that can provide impressive H2 sorption properties and gravimetric and volumetric H2 storage capacities of approximately 5 wt% and 35 g/L, respectively, at 77 K and 20 bar [9]. Confinement effects have further been investigated in H2 binding in potassium-graphite-intercalation compound KC24, in which H2 is found to adsorb with an enthalpy of 8.4 kJ/mol H2 between layers of graphite with an interplanar spacing of 3.35 to 5.4 Å [10]. Similarly, graphite nanofibers have been studied as potential materials with enhanced storage capacity due to their ability to capture H2 molecules between layers, being estimated to be ~3.4 Å apart. Ahn et al. [11] reported that the performance of graphite nanofibers is not superior to other activated carbon materials, even though other reports have given ranges of storage between ~3 wt% and up to 13 wt% [12,13,14]. Calculations of H2 adsorption on slit pore models of graphite nanofibers by Wang et al. [15,16] also suggest that although the pores show increased adsorption relative to modeled single-wall carbon nanotubes, slit pores with a distance of 9 Å had the highest adsorption capacity. In addition, doping carbons with electron–deficient boron (B) has been investigated because of the lower mass of B compared to metal. Substituting B for a metal provides a potential approach to meeting gravimetric density targets for storing hydrogen onboard a fuel–cell electric vehicle [17]. In a pioneering computational study [18], doping fullerene C36 with B was predicted to lead to a significant increase in the hydrogen adsorption energy to −19.2 kJ/mol H2. The authors of that study suggested that the increase in binding energy is due to a partial charge transfer from the sigma-bond of an H2 molecule to the free localized pz–orbital of a B atom. However, a notable feature is the intrinsic non-planarity of the fullerene that induces distortion of the B center, allowing for better matching of the orbitals for enhanced interactions. In contrast, Zhou et al. [19] employed periodic one-dimensional calculations on carbon nanotubes using density functional theory (DFT) with a generalized gradient approximation (GGA) functional and supercells that range from 64 to 80 atoms and reported H2 binding and pristine carbon material binding energies that ranged between −1.8 and −10.3 kJ/mol H2, depending on the position of the H2. Furthermore, they observed that B doping reduces the binding energy by between 0.7 and 3.6 kJ/mol H2 and suggested that doping with electron–deficient B disrupts the conjugated six–membered ring electronic system. They also suggested that the polarity induced in the nanotubes by doping might play a role in the adsorption of the H2 molecule, resulting in decreased binding energy. More recently, a theoretical study on the adsorption of various gas molecules on graphene that was doped with B [20] reported energies of adsorption of −1.35 kJ/mol H2 using GGA, acknowledging that, for non-covalent interactions, the consequent neglect of long–range van der Waals (vdW) interactions, does not accurately describe the physi-sorption of H2. Computational work by Sha et al. on H2 storage in bulk BC3 material shows significant overestimation of the binding energies and spontaneous dissociation of H2 when local density approximation (LDA) functionals were used [21]. It has been established that LDA and GGA functionals are insufficient for capturing the vdW interactions of H2 with doped carbon materials [19,20]. These shortcomings have led to the development of the vdW–DF correlation functionals (vdW–DF1 [22,23] and vdW–DF2 [24]), which are parameterized to capture the vdW forces without any empirical corrections to the nonlocal dispersion interactions for a variety of systems, including graphene along with other carbon–based polymeric materials. Here, the nonlocal correlations are taken into account through the density–density interaction term [22]. Klimes et al. [25] showed that the accuracy of these functionals can be dramatically improved in dispersion bonded complexes. Benchmark studies of the vdW–DF2 functional with revPBE [26] exchange on H2 adsorption on coronene and graphene surfaces show physi-sorption energies for H2 on the hollow site of coronene (–5.7 kJ/mol H2) and graphene surfaces (–5.9 kJ/mol H2) to be in good agreement with experimental values [27]. A benchmark investigation of the physi-sorption of H2 on pure benzene and graphene shows that the revPBE based vdW–DF2 method with nonlocal dispersion is in good agreement with high–level coupled cluster (CCSD(T)) calculations and experimental values for both benzene and graphene [28].
Being encouraged by theoretical predictions of enhanced binding of H2 to B-doped carbon materials, several experimental studies have been undertaken to dope these materials with boron. Some of these experimental approaches targeted the increased heteroatom substitution, which is the nature of the dopant in frameworks with high surface area and tunable porosity. Wang et al. [29] used the substitution reactions to prepare B-doped microporous carbons. They reported that B doping led to an increase in the surface area and the isosteric heat of adsorption [Qst] for H2 compared to the undoped carbon (i.e., an increase from ca. −6 kJ/mol to −10 kJ/mol at higher coverage). Chung et al. [30] developed a new class of polymeric B-containing precursors that were thermally transformed to microporous B/C materials with a boron content of 7.2% and surface area of 780 m2/g. The substitutional p–type B dopant was shown to polarize the C surface, resulting in higher Qst of −10.8 kJ/mol for 0.62 wt % at temperatures of 77 to 87 K and a pressure of 1.2 bar for H2 binding. Tour and coworkers [31] reported a bottom-up, solution–phase technique for the preparation of pristine and heteroatom-substituted carbon scaffolds that show high surface areas and enhanced H2 physi-sorption capacities relative to the pure carbon scaffolds. This approach involved heating chorine-containing small organic molecules (precursors) with metallic sodium at reflux in high-boiling solvents, being subsequently followed by the addition of the heterotopic electrophiles to the mixture for dopant incorporation. The substituted carbon scaffolds enriched with 7.3% boron were observed to have higher surface area of around 900 m2/g and enhanced reversible hydrogen physi-sorption capacity (–8.6 kJ/mol at zero coverage). Jeong et al. [32] developed a method for preparing a broad range of porous B-doped carbon materials by using B precursors containing inorganic additives. They reported the chemical structure of the doped sheets changed from a disordered (less π–conjugated) state with a B-puckered configuration at 600 to 800 °C to an ordered (highly π–conjugated) state with a planar configuration at 1500 °C. The planar graphitic layers were reported to accommodate <3% of B content, while the amorphous, BCx-like materials exhibited much higher surface areas (500 to 800 m2/g) and 12% B content with electron–deficient B moieties, resulting in enhanced H2 binding energies (–12 to −20 kJ/mol at higher coverage). In a recent joint experimental and computational effort [33], a site–specific selective incorporation of B into the graphene sublattices was demonstrated to form a tunable band gap controlled by the dopant concentration. Shcherban et al. [34] reported a nanocasting approach to prepare B-doped CMK-3 using SBA-15 as an exo-template. They reported no increase in H2 binding energy as a result of incorporating B and a decrease in the overall capacity of H2 adsorption. In 2012, Bult et al. [35] reported enhanced hydrogen binding energy for a high-surface-area, B-doped carbon material made by chemical vapor deposition of a B-doped carbon (BCx) layer onto CM-Tec activated carbon. The enhancement was attributed to increased interlayer spacing of the material (~4 Å) and higher concentration of B sites. Table 1 provides a summary of the previously measured and calculated binding energies.
Experimental reports of B-containing carbon materials for H2 storage are relatively limited when compared to computational studies. The highly oxophilic nature of boron makes synthesis of these materials with precise control and purity very challenging. In addition, experimental data for H2 capacities and binding energy show significant discrepancies, potentially due to different measurement protocols and a lack of a standard method of measurement, as well as wide range of the materials and the impurities they contain. Thus, reliable theoretical methods for predicting H2 physi-sorption energies can be helpful in rationally designing these storage materials and achieving improved structure–activity relationships.
In this study, we report a detailed investigation of H2 adsorption on a coronene model and a periodic graphene monolayer and bilayer, as well as their boron–doped variations. We also investigate the effect of B-doping on graphene at different substitutional sites and the effect of interlayer spacing on the binding properties of pure and doped graphene surface. We found that explicit inclusion of the vdW dispersion correction through nonlocal correlation parameterized by density and its gradient is key to capture the physi-sorption of H2. The accuracy of these binding energies is quite sensitive to the optimal density gradient correction in the vdW kernel. Using improved DFT exchange with the dispersion corrected correlation functional provides excellent agreement with experiment [2]. The coronene model was found to adequately describe the binding of H2 and it can be used both for exploratory and detailed electronic structure calculations of the interacting sites in lieu of the much more expensive periodic calculations. The adsorption properties of the doped coronene are also analyzed using quantum molecular descriptors, such as charge and bond order analyses, as a function of the position of B-atom substitution and the H2 adsorption site.
2. Methods
Quantum mechanical calculations for the coronene (C24H12) and the doped coronene were done with the NWChem computational package [37] while using DFT with the hybrid B3LYP (a = 20) functional [38,39]. The results also were compared against the GGA functionals PBE0 [40] (a = 25) and PBE [41] (a = 0). Here, parameter a is the fraction of Hartree–Fock (HF) exchange in the Exchange–Correlation (XC) functional, given as:
For all calculations, we used the 6-311G++** basis set [42], and dispersion interaction was included within the framework developed by Grimme (D3) with the semiempirical correction [43]. Vibrational analysis was done to confirm the minima at the potential energy surface, at the rigid rotor-harmonic oscillator approximation, as well as to provide zero–point correction to the electronic energies. As we did not observe any significant change in the energies with the vibrational corrections, we only report electronic energies in this study for consistency with the solid state calculations. All the calculations were done in the gas phase, at 0 K temperature.
Three-dimensional periodic system calculations were done using the VASP code [44]. To model the H2 adsorption into the graphene surface, we selected a rectangular supercell structure with the cell size of 12.3 × 12.3 Å separated by the vacuum layer with thickness of 15 Å and 7 Å between two monolayers along the z–axis. Vacuum layer with thickness of 15 Å behaves as a two-dimensional material, with the presence of the two monolayers having no impact on the energetics. The unit cell contained 50 carbon atoms. We optimized the structure using the DFT PBE [41] level of theory with projected augmented wave potential [45], with a cutoff of 875 eV. The total energy of the system was minimized with respect to the internal coordinates. The energies of self-consistent calculations were converged to the 10−6 eV/cell. A Monkhorst–Pack grid of 36 k–points (4 × 4 × 4 mesh) and 52 k–points was used (4 × 4 × 6 mesh) to sample the Brillouin zone. Both of the meshes yielded similar results. The dispersion corrections were added using the vdW–DF2 [24] nonlocal correlation functional with its semilocal PW86 exchange [41] replaced with that of GGA PBE [41], revPBE [26], optPBE [25], and RPBE [46] functionals as implemented in VASP. The performance of vdW–DF2 functional at PBE [41] exchange level was compared to the erlier vdW–DF1 [23] version and other DFT–D3 methods (with zero and Becke-Johnson damping) [43,47] with semiempirical dispersion corrections. The vdW–DF2 functional uses a gradient correction in its parameterized vdW kernel in contrast to the older vdW–DF1 version [24]. The effect of the interplanar spacing between two graphene monolayers was tested using both the vdW–DF1 [23] and vdW–DF2 [24] correlation functionals with different DFT exchange contributions [25,41,46].
3. Results
3.1. Coronene Model Calculations
Coronene is often used as a model system for a graphene surface. It is a polycyclic aromatic hydrocarbon that contains six benzene rings (Figure 1). The smaller size of coronene, allows for detailed calculations and the rapid evaluation of variety of doped structures, while maintaining the periodic aromaticity of graphene. The approach taken in this study was to use the smaller model for evaluation of the doping effects on the H2 binding and subsequently refine the calculations for the most promising systems with periodic calculations.
The binding energy () was calculated as the difference between the electronic energy of the molecule with and without H2; that is
where n is the number of hydrogen molecules adsorbed. We used the electronic energies to evaluate the binding energies of the materials, for consistency with the solid–state calculations. We do not expect significant thermal contributions since H2 physi-sorption occurs at low temperatures.
3.1.1. Benchmarking Level of Theory for H2 Binding to Coronene
Validation of the calculations on coronene as a model system show that adding the semiempirical Grimme dispersion correction to the DFT electronic energies significantly affects the energies for H2 physi-sorption.
Table 2 presents the binding energies for coronene with and without the DFT–D3 dispersion correction for PBE, PBE0, and B3LYP XC functionals. The lack of dispersion correction in all cases grossly underestimates the electronic binding energies, with B3LYP predicting an endothermic reaction. The addition of the Grimme D3 dispersion results in the increase in the binding of about 8 to 9 kJ/mol H2, depending on the functional used, predicting binding energies in the range of −6.2 to −9.8 kJ/mol H2. The binding energies calculated at B3LYP level with D3 semiempirical correction is in excellent agreement with the calculated [27] H2 binding on coronene sheets. For all additional studies on the coronene model, we used the D3 dispersion–corrected B3LYP functional. All of the structures and absolute energies for the molecular models are provided in the Supplementary Material (Pages p8–p14).
3.1.2. Effect of Boron Substitution on H2 Binding
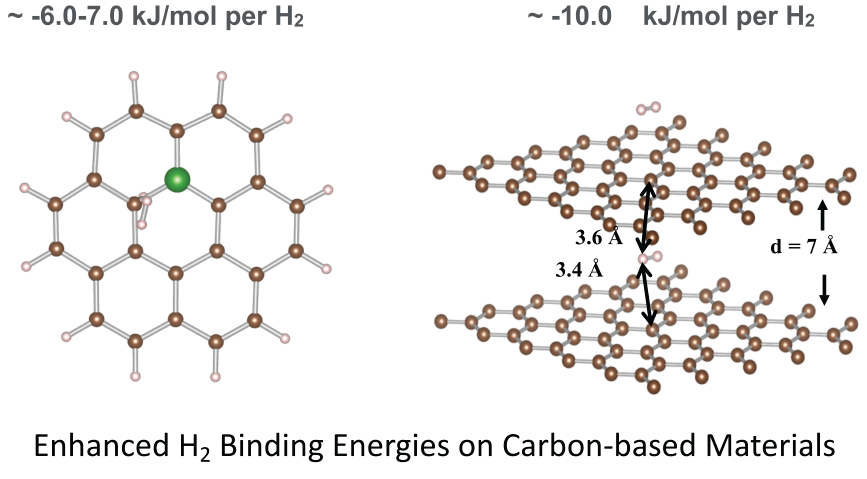
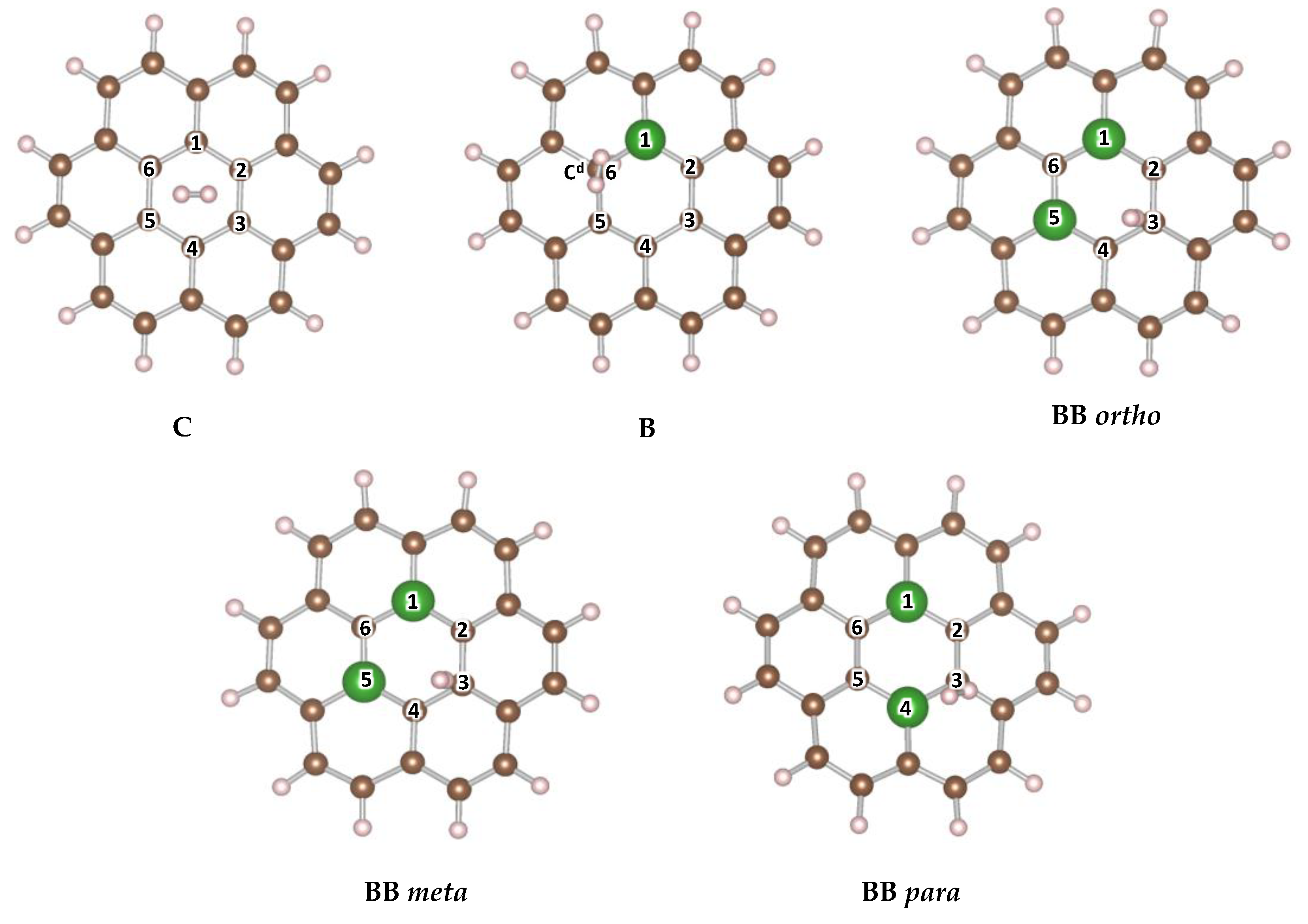
We considered several plausible configurations, as indicated in Figure 1, for boron atoms occupying sites on the central ring of the coronene model. All structures were doped with either one or two B atoms. In the systems where only one B atom was introduced, the charge was preserved by terminating an adjacent carbon atom with hydrogen. The nomenclature of the systems refers to the position of the dopant on the central ring of the coronene. For the ortho position, the two dopant atoms are adjacent to each other. In the meta position, there is one C atom between the two dopants, while para means that there are two C atoms between the two dopant atoms.
Doping with one B atom enhances H2 physi-sorption by 1.4 kJ/mol H2 to −7.6 kJ/mol H2, whereas the addition of a second B molecule to the central ring results in a decrease, with the strongest binding energy being only 0.4 kJ/mol H2 higher than pristine coronene, as observed for ortho substitution. The meta and para positions slightly reduce the energy of physi-sorption in comparison to the undoped system. Table 3 summarizes these results and the trends are discussed in detail in the following two sections. The dependence of site of physi-sorption and strength of binding energies as a function of B–doping is explained in Section 3.1.3 and Section 3.1.4, respectively.
3.1.3. Effect of Boron Doping on the Charge Distribution
We consider the distribution of charges in the central ring of the model to better understand the sites of H2 adsorption in different models and variations in the binding energies. We report the Mulliken charges for all atoms in Table 4. There is no separation of charges in the central ring due to the symmetry of the carbon atoms in the coronene ring, resulting in H2 interaction with the full ring, positioning in the middle of the ring, known as the ‘hollow’ site, parallel to the plane of the surface.
Single and double substitutions of boron result in either positive charge distribution on the boron centers (BB meta and BB para) or nearly neutral (B and BB ortho), and negative charge on the adjacent carbon atoms. In the singly doped B system, the puckered C atom (Cd) adjacent to boron, which has terminal H, becomes the site for H2 physi-sorption. In the systems that were doped with two boron atoms, the H2 molecule physi-sorbs over the BB bond (‘bridge’ site in the BB ortho) even though this site has no deficit of electron density, or in the case of the BB meta and BB para on the most neutral C atom (‘top’ site on C3) on the central ring. This suggests that H2 prefers to interact with the coronene with sites that are neutral when no geometric distortions are present; however, the H2 prefers to interact with the distorted site if present in spite of the excess charge that is developed at this site.
3.1.4. Effect of Boron Doping on the Bond Order
In coronene, the electrons on the carbon centers are delocalized over the central ring, thus resulting in all C–C bonds attaining partial double bond character (1.5 ≥ bond order ≥ 1.2). B–doping changes the electronic structure, thus affecting the bond order. We report the bond orders for the pure and doped coronene systems under study in Table 5.
The analysis of the bond order in the doped coronene suggests that a singly substituted boron system maintains a bond order ~1 for the B–C bonds, which represents a single bond character. The addition of the terminating hydrogen results in all single C–C bonds for the tetra–coordinate carbon (C6). This observation is consistent with the excess electron density being accumulated in the tetrahedral carbon, which results in negative charge (see Table 4). The complete aromaticity (six partial double bonds) within the ring that was observed for the C system is partially perturbed with the formation of four single and two partial double bonds. We emphasize the charge of the B atom for this system, being less electropositive, which is attributed to the electron redistribution within the ring (see Table 4). On the other hand, the coronene system doped with two borons at ortho substitution results in an alternately single and partial double bond arrangement of bond orders. To our surprise, the B5–B6 bond in this system attains a slight partial character, suggesting that the presence of electron–rich boron centers for this system is a result of charge delocalization, being reflected as partial double bond orders. In the BB meta model, all bonds adopt nearly single bond character, consistent with accumulation of electron density on all the carbons of the central ring bound to the borons, with borons having +0.5 charge (see Table 4). However, the para substitution of the borons results in a ring with two double bonds between the C–C atoms. The H2 binding enhancement correlates with doping that less drastically disturbs the bonding distribution (i.e., singly doped B and BB ortho), whereas a more significant redistribution of bond order and charges within the ring results in reduced binding strength (i.e., BB meta and BB para). This suggests that a complete disruption of the aromaticity in the central ring and creation of electron–deficient or electron–rich density sites leads to decreased binding energy. The binding energy is enhanced for the B-doped variations where boron is electro neutral as a result of the charge delocalization within the ring. H2 adsorbs on the most electro neutral site in the ring when no geometrical distortions are present in the planar structure.
3.1.5. Other Possibilities for H2 Binding
We also explored the binding of a second H2 molecule and the possibilities of H2 adsorption at the external rings of the coronene models, corresponding to a model where B–doping is at the edge of a graphitic monolayer. When only one B-doped site on the central ring is considered, the binding of the second H2 is slightly weaker (by 0.3 to 0.6 kJ/mol H2, depending on the position of the second binding event (Figure S1 in Supplementary Materials). No significant enhancement was found for H2 binding when B-doping occurs at the boundary of the model system (Figure S2 in Supplementary Materials).
3.2. Periodic Two–Dimensional and Three–Dimensional Doped Graphene
We selected few promising models that were used to calculate the H2 binding to a periodic two-dimensional graphene surface based on the initial screening of doped coronene model. We also considered the effects of increased boron concentration and the effects of confining the H2 between two surfaces with an interplanar distance of 15 Å and 7 Å. Table S2 in Supplementary Materials illustrates the absolute energies for all layered structures.
3.2.1. Benchmarking Level of Theory for H2 Binding on Graphene
The experimental binding energy for H2 on graphene is measured at −5.1 kJ/mol H2 [2]. The benchmarking of the level of theory used was done on pristine graphene sheets. The initial relaxation of the surface was done using the PBE XC functional, for a periodic surface with 12.3 × 12.3 × 15 Å cell size. Single point energies were calculated for this structure with different dispersion correction methods used in this work (Table 6, top four entries). The H2 binding energy was significantly underestimated with PBE (–0.8 kJ/mol H2) for all entries, except those using vdW–DF1 and vdW–DF2. Whereas vdW–DF1 overestimates the binding (Table 6, and Table S1 in Supplementary Materials), vdW–DF2 provides the best agreement with experiment [2] for graphene at −6.0 kJ/mol H2. We also investigated the sensitivity of the binding energy on the DFT exchange, using vdW–DF2 with three other functionals: revPBE, optPBE, and RPBE (Table 6, bottom four entries). RPBE exchange provides even closer agreement with experiment [2] at −5.2 kJ/mol H2 with vdW–DF2. Slightly weaker binding energy was calculated with revPBE and optPBE.
3.2.2. Effect of Boron Doping Concentration on H2 Binding
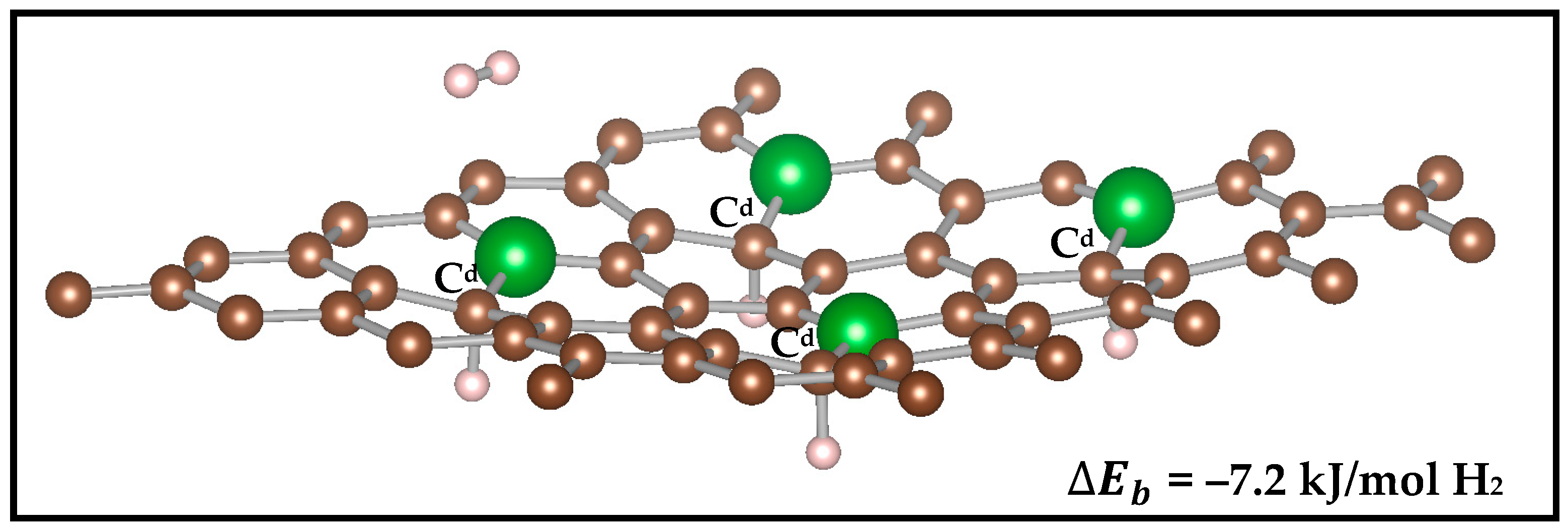
We substituted a single carbon atom with boron for a C–to–B ratio of 49:1 (2% doping) and four carbon atoms for C–to–B ratio of 46:4 (7.4% doping) because the coronene model shows largest enhancement of binding energy for the singly doped B site in the central benzene ring (see Table 3). We find that for ~2% B–doping, the enhancement in H2 binding energy increases to −6.6 kJ/mol H2 from that of −5.2 kJ/mol H2 for pure graphene (Figure 2, top and middle). From the several plausible configurations for H2 physi-sorption sites that ranged from the ‘top’ site on puckered C (Cd) atom to the ‘bridge’ B–C and ‘hollow’ sites (Figure S3 in the Supplementary Materials), most favorable interaction was found with the Cd atom (‘top’ Cd and ‘bridge’ B–Cd sites), as also observed with the coronene model (Figure 1). At the hollow site, the binding energy is slightly lower at −5.8 kJ/mol H2.
Previous experimental study [31] on carbon scaffolds has revealed an increase up to −8.6 kJ/mol H2 in the physi-sorption energy with about 7.3% of boron substitution in the C network. Because the coronene model predicts largest increase when one boron is substituted per carbon ring, in the graphene monolayer with 7.4% doping we substitute one boron atom per alternate ring (Figure 2, bottom). The binding energy in this case is −7.2 kJ/mol H2, which suggests that increasing the B concentration from 2% to 7.4% slightly increases the H2 binding.
Because higher concentrations are not likely to be experimentally achieved with good dispersion of B sites, and due to the low enhancement in binding energy, we don’t anticipate significant benefit from exploring higher boron doping concentrations.
3.2.3. Effect of Interlayer Distance Between Graphene Monolayers on the H2 Binding
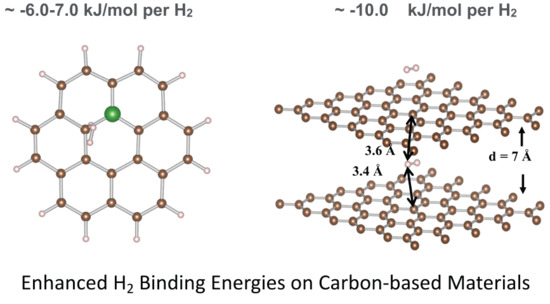
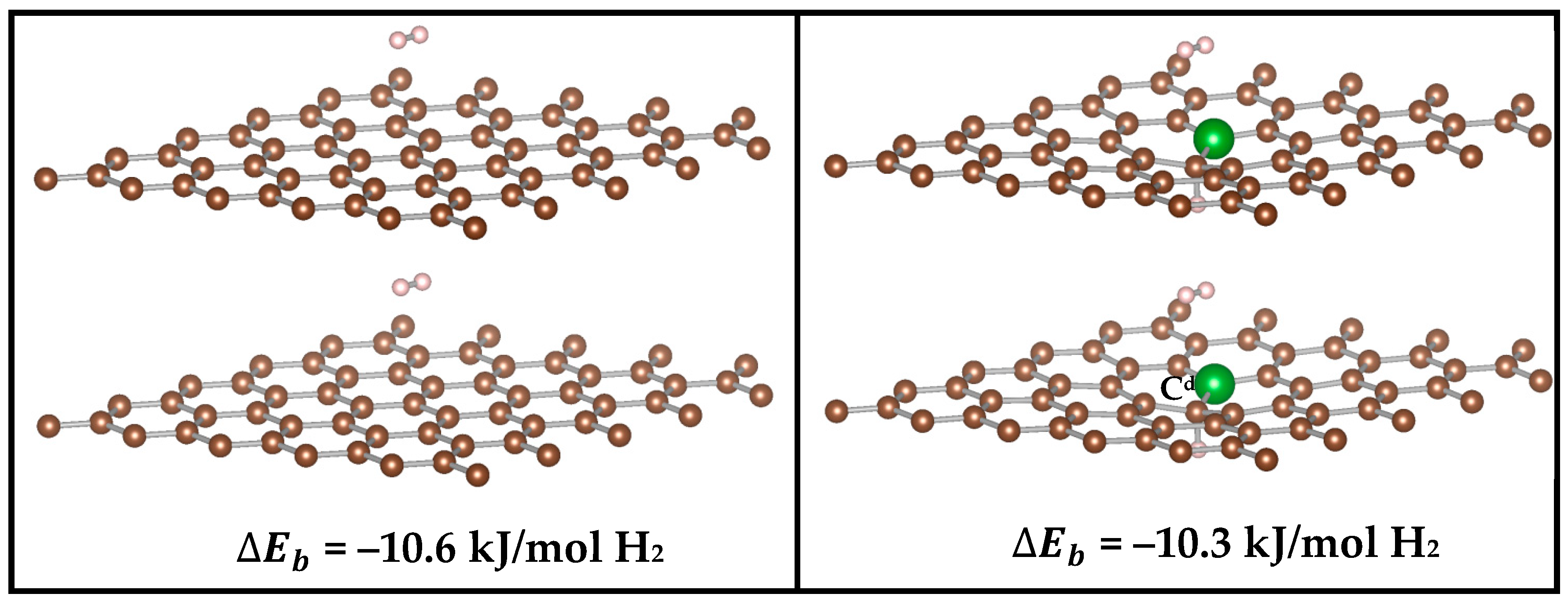
The addition of periodicity in the direction perpendicular to the plane of the graphene sheets, allowed us to evaluate the effects of multi–layered structure on the H2 binding. Interestingly, the position and orientation of H2 does not change when the spacing between the surfaces is reduced from 15 Å to 7 Å, implicating long–range interactions with the second surface (Figure 3). For graphene, a bilayer separated by 7 Å, results in a significant increase of the binding energy to −10.6 kJ/mol H2. This finding is consistent with previous calculations by Patchkovskii et al. [48], which utilize post–HF methods, giving confidence in the DFT methodology employed here.
Interestingly, a bilayer of B-doped graphene with doping concentration of 2% (C–to–B ratio of 49:1) and a separation of 7Å gave similar binding energy of (–10.3 kJ/mol H2.) as the bilayer of undoped graphene. This observation suggests that, even though some enhancement might be gained from doping, or distortion of the surface, a much larger effect is achieved when H2 is confined between two surfaces.
4. Discussion and Conclusion
Systematic DFT calculations for both the coronene and the two-dimensional periodic sheets of graphene systems demonstrated the importance of explicitly considering the effects of long–range van der Waals forces to the binding energies by including semiempirical (DFT–D3) and nonempirical (vdW–DF) dispersion correction, respectively. We find that using the vdW–DF2 method with the corrected density gradient term is necessary in order to accurately evaluate the H2 physi-sorption energy for graphene. We also find that judicious selection of the underlying exchange of the vdW–DF2 functional is important, with the alternative revision of PBE functional, RPBE, showing significant improvement over the previously recommended revPBE. We benchmarked the binding energy calculations at this level of theory for graphene surface, and find that RPBE/vdW–DF2 gives the best agreement.
Our calculations show that only marginal improvements in binding energy are observed when the doping does not induce any distortion to the surface, even though earlier calculations have suggested possible enhancement to the binding energy by doping graphene with boron atoms. This is consistent with the lack of experimental evidence of increased binding that would confirm the earlier work. More significant changes to the binding energy (up to 2 kJ/mol H2) were found in cases when the doping induces a defect, such as distortion from planarity, i.e. puckered carbon atom adjacent to boron, or hydrogenation of an unsaturated bond. This has also been observed in the H2 interaction with BX3:NH3 (X = H,F,Cl) frustrated Lewis acids [49]. These types of defects may provide another avenue for exploring irregular graphene–based materials, provided they can be introduced in a controlled manner. When a defect is present, the H2 molecule tends to adsorb at the defect site. Although the effect of surface defects can be somewhat different in the bulk material, they are still likely to affect adsorption energies and should be considered for future experiment design. In addition, the coronene calculations provide an excellent model for understanding the governing criteria for the site selection and strength for H2 physi-sorption. Analysis of the coronene calculations show that change in the charge distribution (delocalization) and the small disturbance of the aromaticity correlate with the enhancement of binding.
The graphite monolayer models allowed for us to investigate the possible enhancement of the binding energy by increasing the boron concentration. Increasing the concentration from 2% to 7%, results in enhancement of only 0.6 kJ/mol H2.
A notable enhancement, resulting in binding energy of −11 kJ/mol H2 is accomplished by confinement of an H2 molecule between two layers of graphene, with the layers being separated by a distance of 7 Å. This models a scenario in which the H2 molecule interacts with both surfaces, resulting in a largely additive effect. Increasing the distance between the layers to 15 Å no longer benefits from this additive effect and no substantial increase in binding is observed. The addition of boron sites in the case of bilayer models shows no enhancement in the binding energies. This observation suggests that another option to increase the H2 binding might be accomplished by understanding the effects of the confinement on the H2 adsorption, consistent with previous findings [9,10,15,16].
Direct comparison of calculations with experiment is still difficult since it is difficult to quantify the distribution of boron in the carbon lattice in experiments [31,33], however, this work provides useful insight into both the atomistic details that may guide the preferential binding sites for H2 in these materials, as well as the possibility of enhancing this binding by the intentional design of defect sites and confinement in the material. These approaches may be more effective in developing materials that can reach the U.S. Department of Energy targets for onboard H2 storage.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2311-5629/6/1/15/s1, Figure S1. Binding of second H2 molecule on the boron–doped model; Figure S2. H2 binding of systems where boron doping is at the edge; Table S1. H2 binding energy for different DFT exchange methods using vdW–DF1; Figure S3. Interactions sites for H2 physi-sorption on 2% boron–doped graphene; Figure S4. Top and side view of H2 interactions on graphene with different doping; Table S2. Absolute electronic energies for extended systems; Geometries and electronic energy for the molecular models.
Author Contributions
Conceptualization, B.G., T.A. and A.K.; methodology, I.N. and B.G; calculations, formal analysis, and visualization, I.N. and B.G.; writing—original draft preparation, I.N., B.G and T.A.; writing—review and editing, All authors; project administration, T.A and T.G.; funding acquisition T.A and T.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Hydrogen Materials–Advanced Research Consortium, established as part of the Energy Materials Network under the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel–Cell Technologies Office.
Acknowledgments
Computational resources were provided by Pacific Northwest National Laboratory Research Computing. PNNL is operated by Battelle for DOE.
Conflicts of Interest
The authors declare no conflict of interest.
References
- He, T.; Pachfule, P.; Wu, H.; Xu, Q.; Chen, P. Hydrogen carriers. Nat. Rev. Mater. 2016, 1, 1–7. [Google Scholar] [CrossRef]
- Panella, B.; Hirscher, M.; Roth, S. Hydrogen adsorption in different carbon nanostructures. Carbon 2005, 43, 2209–2214. [Google Scholar] [CrossRef]
- Candelaria, S.L.; Shao, Y.Y.; Zhou, W.; Li, X.L.; Xiao, J.; Zhang, J.G.; Wang, Y.; Liu, J.; Li, J.H.; Cao, G.Z. Nanostructured carbon for energy storage and conversion. Nano Energy 2012, 1, 195–220. [Google Scholar] [CrossRef]
- Yang, J.; Sudik, A.; Wolverton, C.; Siegel, D.J. High capacity hydrogen storage materials: Attributes for automotive applications and techniques for materials discovery. Chem. Soc. Rev. 2010, 39, 656–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Du, Y.; Sun, L.X. Ca-decorated novel boron sheet: A potential hydrogen storage medium. Int J Hydrogen Energy 2016, 41, 5276–5283. [Google Scholar] [CrossRef]
- Cha, M.H.; Nguyen, M.C.; Lee, Y.L.; Im, J.; Ihm, J. Iron-Decorated, Functionalized Metal Organic Framework for High-Capacity Hydrogen Storage: First-Principles Calculations. J. Phys. Chem. C 2010, 114, 14276–14280. [Google Scholar] [CrossRef]
- Kapelewski, M.T.; Runcevski, T.; Tarver, J.D.; Jiang HZ, H.; Hurst, K.E.; Parilla, P.A.; Ayala, A.; Gennett, T.; FitzGerald, S.A.; Brown, C.M.; et al. Record High Hydrogen Storage Capacity in the Metal-Organic Framework Ni-2(m-dobdc) at Near-Ambient Temperatures. Chem. Mater. 2018, 30, 8179–8189. [Google Scholar] [CrossRef]
- Runcevski, T.; Kapelewski, M.T.; Torres-Gavosto, R.M.; Tarver, J.D.; Brown, C.M.; Long, J.R. Adsorption of two gas molecules at a single metal site in a metal-organic framework. Chem. Commun. 2016, 52, 8251–8254. [Google Scholar] [CrossRef]
- McNicholas, T.P.; Wang, A.M.; O’Neill, K.; Anderson, R.J.; Stadie, N.P.; Kleinhammes, A.; Parilla, P.; Simpson, L.; Ahn, C.C.; Wang, Y.Q.; et al. H-2 Storage in Microporous Carbons from PEEK Precursors. J. Phys. Chem. C 2010, 114, 13902–13908. [Google Scholar] [CrossRef] [Green Version]
- Purewal, J.J.; Keith, J.B.; Ahn, C.C.; Fultz, B.; Brown, C.M.; Tyagi, M. Adsorption and melting of hydrogen in potassium-intercalated graphite. Phys. Rev. B 2009, 79, 054305. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.C.; Ye, Y.; Ratnakumar, B.V.; Witham, C.; Bowman, R.C.; Fultz, B. Hydrogen desorption and adsorption measurements on graphite nanofibers. Appl. Phys. Lett. 1998, 73, 3378–3380. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Liao, B.; Liu, M.; Wei, Y.L.; Lu, M.Q.; Cheng, H.M. Hydrogen uptake in vapor-grown carbon nanofibers. Carbon 1999, 37, 1649–1652. [Google Scholar] [CrossRef]
- Huang, C.W.; Wu, H.C.; Li, Y.Y. Hydrogen storage in platelet graphite nanofibers. Sep. Purif. Technol. 2007, 58, 219–223. [Google Scholar] [CrossRef]
- Browning, D.J.; Gerrard, M.L.; Lakeman, J.B.; Mellor, I.M.; Mortimer, R.J.; Turpin, M.C. Studies into the storage of hydrogen in carbon nanofibers: Proposal of a possible reaction mechanism. Nano Lett. 2002, 2, 201–205. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Johnson, J.K. Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores. J. Chem. Phys. 1999, 110, 577–586. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Johnson, J.K. Computer simulations of hydrogen adsorption on graphite nanofibers. J. Phys. Chem. B 1999, 103, 277–281. [Google Scholar] [CrossRef]
- U.S. DRIVE Partnership. Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles. 2017. Available online: https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_targets_onboard_hydro_storage_explanation.pdf (accessed on 10 January 2020).
- Kim, Y.H.; Zhao, Y.F.; Williamson, A.; Heben, M.J.; Zhang, S.B. Nondissociative adsorption of H-2 molecules in light-element-doped fullerenes. Phys. Rev. Lett. 2006, 96, 016102. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gao, X.P.; Yan, J.; Song, D.Y. Doping effects of B and N on hydrogen adsorption in single-walled carbon nanotubes through density functional calculations. Carbon 2006, 44, 939–947. [Google Scholar] [CrossRef]
- Dai, J.Y.; Yuan, J.M.; Giannozzi, P. Gas adsorption on graphene doped with B, N, Al, and S: A theoretical study. Appl. Phys. Lett. 2009, 95, 232105. [Google Scholar] [CrossRef]
- Sha, X.W.; Cooper, A.C.; Bailey, W.H.; Cheng, H.S. Revisiting Hydrogen Storage in Bulk BC3. J. Phys. Chem. C 2010, 114, 3260–3264. [Google Scholar] [CrossRef]
- Vanin, M.; Mortensen, J.J.; Kelkkanen, A.K.; Garcia-Lastra, J.M.; Thygesen, K.S.; Jacobsen, K.W. Graphene on metals: A van der Waals density functional study. Phys. Rev. B 2010, 81, 081408. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.; Rydberg, H.; Schroder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Murray, E.D.; Kong, L.Z.; Lundqvist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef] [Green Version]
- Klimes, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J Phys. Condens. Mat. 2010, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Yang, W.T. Comment on “Generalized gradient approximation made simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Varenius, E. Hydrogen Adsorption on Graphene and Coronene A van der Waals Density Functional Study; Chalmers University of Technology: Gothenburg, Sweden, 2011. [Google Scholar]
- Cabria, I.; Lopez, M.J.; Alonso, J.A. Searching for DFT-based methods that include dispersion interactions to calculate the physisorption of H-2 on benzene and graphene. J. Chem. Phys. 2017, 146, 214104. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Yang, F.H.; Yang, R.T. Hydrogen Storage Properties of B- and N-Doped Microporous Carbon. AIChE J. 2009, 55, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.M.; Jeong, Y.; Chen, Q.; Kleinhammes, A.; Wu, Y. Synthesis of microporous boron-substituted carbon (B/C) materials using polymeric precursors for hydrogen physisorption. J. Am. Chem. Soc. 2008, 130, 6668–6669. [Google Scholar] [CrossRef]
- Jin, Z.; Sun, Z.Z.; Simpson, L.J.; O’Neill, K.J.; Parilla, P.A.; Li, Y.; Stadie, N.P.; Ahn, C.C.; Kittrell, C.; Tour, J.M. Solution-Phase Synthesis of Heteroatom-Substituted Carbon Scaffolds for Hydrogen Storage. J. Am. Chem. Soc. 2010, 132, 15246–15251. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Chung, T.M. The synthesis and characterization of a super-activated carbon containing substitutional boron (BCX) and its applications in hydrogen storage. Carbon 2010, 48, 2526–2537. [Google Scholar] [CrossRef]
- Usachov, D.Y.; Fedorov, A.V.; Vilkov, O.Y.; Petukhov, A.E.; Rybkin, A.G.; Ernst, A.; Otrokov, M.M.; Chulkov, E.V.; Ogorodnikov, I.I.; Kuznetsov, M.V.; et al. Large-Scale Sublattice Asymmetry in Pure and Boron-Doped Graphene. Nano Lett. 2016, 16, 4535–4543. [Google Scholar] [CrossRef] [PubMed]
- Shcherban, N.; Filonenko, S.; Yaremov, P.; Dyadyun, V.; Bezverkhyy, I.; Ilyin, V. Boron-doped nanoporous carbons as promising materials for supercapacitors and hydrogen storage. J. Mater. Sci. 2017, 52, 1523–1533. [Google Scholar] [CrossRef]
- Bult, J.B.; Lee, J.; O’Neill, K.; Engtrakul, C.; Hurst, K.E.; Zhao, Y.F.; Simpson, L.J.; Parilla, P.A.; Gennett, T.; Blackburn, J.L. Manipulation of Hydrogen Binding Energy and Desorption Kinetics by Boron Doping of High Surface Area Carbon. J. Phys. Chem. C 2012, 116, 26138–26143. [Google Scholar] [CrossRef]
- Ariharan, A.; Viswanathan, B.; Nandhakumar, V. Hydrogen storage on boron substituted carbon materials. Int. J. Hydrogen Energy 2016, 41, 3527–3536. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam HJ, J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods.20. Basis Set for Correlated Wave-Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Norskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patchkovskii, S.; Tse, J.S.; Yurchenko, S.N.; Zhechkov, L.; Heine, T.; Seifert, G. Graphene nanostructures as tunable storage media for molecular hydrogen. Proc. Natl. Acad. Sci. USA 2005, 102, 10439–10444. [Google Scholar] [CrossRef] [Green Version]
- Camaioni, D.M.; Ginovska-Pangovska, B.; Schenter, G.K.; Kathmann, S.M.; Autrey, T. Analysis of the Activation and Heterolytic Dissociation of H-2 by Frustrated Lewis Pairs: NH3/BX3 (X = H, F, and Cl). J. Phys. Chem. A 2012, 116, 7228–7237. [Google Scholar] [CrossRef]
Figure 1.
Coronene (C24H12) and boron–doped coronene structural models with H2 adsorbed. The atoms on the central ring are numbered 1 through 6 for ease of discussion in the text and in the data reported in Tables 3 and 4. For the system B, a terminal H is added on the neighboring C atom (C6) denoted as Cd.
Figure 1.
Coronene (C24H12) and boron–doped coronene structural models with H2 adsorbed. The atoms on the central ring are numbered 1 through 6 for ease of discussion in the text and in the data reported in Tables 3 and 4. For the system B, a terminal H is added on the neighboring C atom (C6) denoted as Cd.

Figure 2.
Binding energy ( of H2 on periodic graphene and different boron–doped concentrations with interlayer spacing of 15 Å along z–axis. Both 2% and 7% B-doping (middle and bottom panel, respectively) show the H2 interacting near the Cd atom. The second monolayer at 15 Å along z–axis is not shown here for simplicity. The side and top views are shown in Figure S4 in the Supplementary Materials.
Figure 2.
Binding energy ( of H2 on periodic graphene and different boron–doped concentrations with interlayer spacing of 15 Å along z–axis. Both 2% and 7% B-doping (middle and bottom panel, respectively) show the H2 interacting near the Cd atom. The second monolayer at 15 Å along z–axis is not shown here for simplicity. The side and top views are shown in Figure S4 in the Supplementary Materials.


Figure 3.
Position of the H2 molecule between two monolayers of graphene (left) and 2% boron–doped graphene (right) at a distance of 7 Å along z–axis. The position and orientation of the H2 does not change when the spacing between the surfaces is reduced from 15 Å to 7 Å (15 Å not shown), implicating long–range interactions with the second surface. The H2 is physi-sorbed by both the top and bottom monolayers at a distance of 3.6 and 3.4 Å, respectively.
Figure 3.
Position of the H2 molecule between two monolayers of graphene (left) and 2% boron–doped graphene (right) at a distance of 7 Å along z–axis. The position and orientation of the H2 does not change when the spacing between the surfaces is reduced from 15 Å to 7 Å (15 Å not shown), implicating long–range interactions with the second surface. The H2 is physi-sorbed by both the top and bottom monolayers at a distance of 3.6 and 3.4 Å, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Binding energy of H2 (kJ/mol H2) on carbon and boron–doped carbon materials. The * denotes calculated values.
Table 1.
Binding energy of H2 (kJ/mol H2) on carbon and boron–doped carbon materials. The * denotes calculated values.
| System | Binding Energy of H2 | Reference |
|---|---|---|
| Pure Graphene | −5.1 kJ/mol H2 | [2,3] |
| Microporous B/C | −10.8 kJ/mol (0.62 wt %) | [30] |
| B doped Carbon Scaffolds | −8.6 kJ/mol (zero coverage) | [31] |
| Porous BCx | −12 to −20 kJ/mol (high coverage) | [32] |
| B doped CMK-3 | −5 kJ/mol H2 | [34] |
| B doped C | NA | [36] (increased capacity to 5 wt% at 298 K) |
| C36 Fullerenes | −19.2 kJ/mol H2 | [18] * |
| C–Nanotubes | Reduced from −10.3 kJ/mol H2 in pristine | [19] * |
| B doped Graphene | −1.35 kJ/mol H2 | [20] * |
| Pure Coronene | −5.7 kJ/mol H2 | [27] * |
| Pure Graphene | −5.9 kJ/mol H2 | [27] * |
| Pure Graphene | −5.5 kJ/mol H2, −4.6 kJ/mol H2 | [28] * |
Table 2.
Binding energy ( in kJ/mol H2) for H2 physi-sorption on coronene using different XC functionals.
Table 2.
Binding energy ( in kJ/mol H2) for H2 physi-sorption on coronene using different XC functionals.
| XC Functional | (D3 Corrected) | |
|---|---|---|
| PBE | −0.3 | −9.8 |
| PBE0 | −0.7 | −7.3 |
| B3LYP | 3.1 | −6.2 |
| vdW–DF2 (solid state) [27] | −5.7 | |
Table 3.
Binding energy ( in kJ/mol H2) of adsorbed H2 in coronene and boron–doped models. The nomenclature introduced here will be used for shorthand notation of the systems hereon.
Table 3.
Binding energy ( in kJ/mol H2) of adsorbed H2 in coronene and boron–doped models. The nomenclature introduced here will be used for shorthand notation of the systems hereon.
| System | Nomenclature | |
|---|---|---|
| C24 H12 | C | −6.2 |
| B C23 H13 | B | −7.6 |
| B2 C22 H12 | BB ortho | −6.6 |
| BB meta | −5.9 | |
| BB para | −5.6 |
Table 4.
Mulliken charges for the atoms on the central ring in the coronene models. The numbering of atoms and naming of system is as shown on Figure 1 and Table 3. The substituted boron atoms are identified by a superscript ‘b’. The distorted Cd atom is identified by a superscript ‘Cd’.
| Atom # | C | B | BB ortho | BB meta | BB para |
|---|---|---|---|---|---|
| 1 | 0.10 | 0.01 b | −0.22 | 0.51 b | 0.56 b |
| 2 | 0.06 | −0.28 | 0.13 | −0.22 | −0.21 |
| 3 | 0.06 | 0.11 | 0.13 | 0.16 | −0.12 |
| 4 | 0.11 | 0.09 | −0.27 | −0.22 | 0.37 b |
| 5 | 0.04 | 0.14 | −0.03 b | 0.50 b | −0.19 |
| 6 | 0.05 | −0.54 Cd | −0.03 b | −0.56 | −0.20 |
Table 5.
Bond order analysis for the bonds between the atoms on the central ring in the coronene models. The numbering of atoms and naming of system is as shown in Figure 1 and Table 3. Single asterisk (*) denotes the B–C bonds, while double asterisk (**) represents the B–B bond. The bonds containing Cd atom are identified by a superscript ‘Cd’.
Table 5.
Bond order analysis for the bonds between the atoms on the central ring in the coronene models. The numbering of atoms and naming of system is as shown in Figure 1 and Table 3. Single asterisk (*) denotes the B–C bonds, while double asterisk (**) represents the B–B bond. The bonds containing Cd atom are identified by a superscript ‘Cd’.
| Bond Order | C | B | BB ortho | BB meta | BB para |
|---|---|---|---|---|---|
| 1–2 | 1.25 | 1.12 * | 1.24 | 1.01 * | 0.95 * |
| 2–3 | 1.21 | 1.30 | 1.06 | 1.16 | 1.43 |
| 3–4 | 1.25 | 1.14 | 1.25 | 1.17 | 1.00 * |
| 4–5 | 1.26 | 1.30 | 1.04 * | 1.01 * | 1.01 * |
| 5–6 | 1.23 | 1.06 Cd | 1.24 ** | 1.13 * | 1.42 |
| 6–1 | 1.26 | 1.00 *Cd | 1.02 * | 1.13 * | 0.96 * |
Table 6.
Comparison of H2 binding energy ( in kJ/mol H2) for different dispersion correction and density functional theory (DFT) exchange methods for interlayer spacing of 15 Å between periodic graphene sheets. The calculated values are in good agreement when the vdW–DF2 dispersion correction is used at different DFT exchange levels, with the RPBE providing the best agreement with experiment.
Table 6.
Comparison of H2 binding energy ( in kJ/mol H2) for different dispersion correction and density functional theory (DFT) exchange methods for interlayer spacing of 15 Å between periodic graphene sheets. The calculated values are in good agreement when the vdW–DF2 dispersion correction is used at different DFT exchange levels, with the RPBE providing the best agreement with experiment.
| DFT Exchange | Dispersion Method | |
|---|---|---|
| PBE | no dispersion | −0.75 |
| PBE | D3—no damping | −0.8 |
| PBE | D3—Becke-Johnson damping | −0.75 |
| PBE | vdW–DF1 | −9.5 |
| PBE | vdW–DF2 | −6.0 |
| revPBE | vdW–DF2 | −3.8 |
| optPBE | vdW–DF2 | −4.6 |
| RPBE | vdW–DF2 | −5.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nayyar, I.; Ginovska, B.; Karkamkar, A.; Gennett, T.; Autrey, T. Physi-Sorption of H2 on Pure and Boron–Doped Graphene Monolayers: A Dispersion–Corrected DFT Study. C 2020, 6, 15. https://0-doi-org.brum.beds.ac.uk/10.3390/c6010015
AMA Style
Nayyar I, Ginovska B, Karkamkar A, Gennett T, Autrey T. Physi-Sorption of H2 on Pure and Boron–Doped Graphene Monolayers: A Dispersion–Corrected DFT Study. C. 2020; 6(1):15. https://0-doi-org.brum.beds.ac.uk/10.3390/c6010015
Chicago/Turabian StyleNayyar, Iffat, Bojana Ginovska, Abhijeet Karkamkar, Thomas Gennett, and Thomas Autrey. 2020. "Physi-Sorption of H2 on Pure and Boron–Doped Graphene Monolayers: A Dispersion–Corrected DFT Study" C 6, no. 1: 15. https://0-doi-org.brum.beds.ac.uk/10.3390/c6010015
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.