A Protein Assembly Hypothesis for Population-Specific Decrease in Dementia with Time



Department of Biochemistry and Structural Biology, The University of Texas Health Center at San Antonio, San Antonio, TX 78229-3900, USA
*
Author to whom correspondence should be addressed.
Biophysica 2021, 1(1), 15-21; https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1010002
Submission received: 16 December 2020
/
Revised: 13 January 2021
/
Accepted: 18 January 2021
/
Published: 7 February 2021
{kind=link}
{kind=link}
{kind=link}
Abstract
:A recent report in the journal, Neurology, documents age-normalized, nation-specific (e.g., United States and Western Europe), progressive decrease of dementia, beginning about 25 years ago. This observation has, thus far, not had explanation. We begin our proposed explanation with the following previous disease construct. (1) Some dementia is caused by innate immune over-response to infections. (2) The innate immune over-response occurs via excessive conversion of amyloid protein to α-sheet conformation. (3) This conversion evolved to inhibit invading microbes by binding microbe-associated α-sheet, e.g., in hyper-expanded capsid intermediates of some viruses. The rarity of human α-sheet makes this inhibition specific for microbial invaders. As foundation, here we observe directly, for the first time, extreme, sheet-like outer shell thinness in a hyper-expanded capsid of phage T3. Based on phage/herpesvirus homology, we propose the following. The above decrease in dementia is caused by varicella-zoster virus (VZV) vaccination, USFDA-approved about 25 years ago; VZV is a herpesvirus and causes chickenpox and shingles. In China and Japan, a cotemporaneous non-decrease is explained by lower anti-VZV vaccination. Co-assembly extension of α-sheet is relatively independent of amino acid sequence. Thus, we project that additional dementia is suppressible by vaccination against other viruses, including other herpesviruses.
1. Introduction
In a previous study [1] of 49,202 individuals in the United States and Europe, the finding was made that (1) incidence of dementia dramatically increased with age, as previously reviewed in [2], and (2) in any given age range, the incidence has been significantly decreasing for the last ~25 years. The decreases per 10 years were 24% in men and 8% in women. Improved health care was suggested as a reason for the decrease. However, further details were not proposed [1].
The individuals of this study were from the United States (2596), France (2960), Netherlands (10,235), Sweden (1168), United Kingdom (6441 + 11,788), France (8250) and Iceland (5764) [1]. These countries have relatively advanced health care, supporting the hypothesis that improvement in health care is a reason for the decrease.
A hypothesis with further details should provide explanation for the above gender-specificity. It should also provide explanation for the observation, quoted in [1], that such decrease was not observed in either China [3] or Japan [4], both with advanced heath care systems. Developing such an explanatory hypothesis, within the strategic objective of preventing dementia, is the aim of the current communication.
1.1. Foundation for an Explanation: Basics
In search of an explanation, we note that evidence has been accumulating that Alzheimer’s disease-caused dementia is correlated with previous virus and bacterial infection. The infection-based studies have focused on herpes simplex viruses (HSVs), especially HSV-1 and HSV-2 (cold sores and genital infections). These viruses and varicella zoster (VZV; chickenpox), also a herpesvirus, are neurotropic and sometimes re-activate after release from nerves (shingles in the case of VZV). However, the onset of neurodegenerative disease typically occurs after, not during, infection [5,6,7,8].
Thus, correlation with virus infection is best explained by a long-term host reaction initiated by the virus. Other data have suggested that this host reaction is a primitive, acellular, innate immune reaction. These data were obtained either via homology [9] or via studies of the effects of virus infection on either mice or cell cultures [10]. If the virus-generated trigger for the innate immune response has structural features in common among several different viruses, then vaccination against all of these viruses would be an obvious way to reduce the frequency of Alzheimer’s disease. This raises the question of what, structurally, is the source of this innate immune function. The answer is likely to be a protein structure that is intrinsically complementary to itself so that it is extensible by amyloid proteins with the same basic structure.
1.2. Foundation for an Explanation: Details
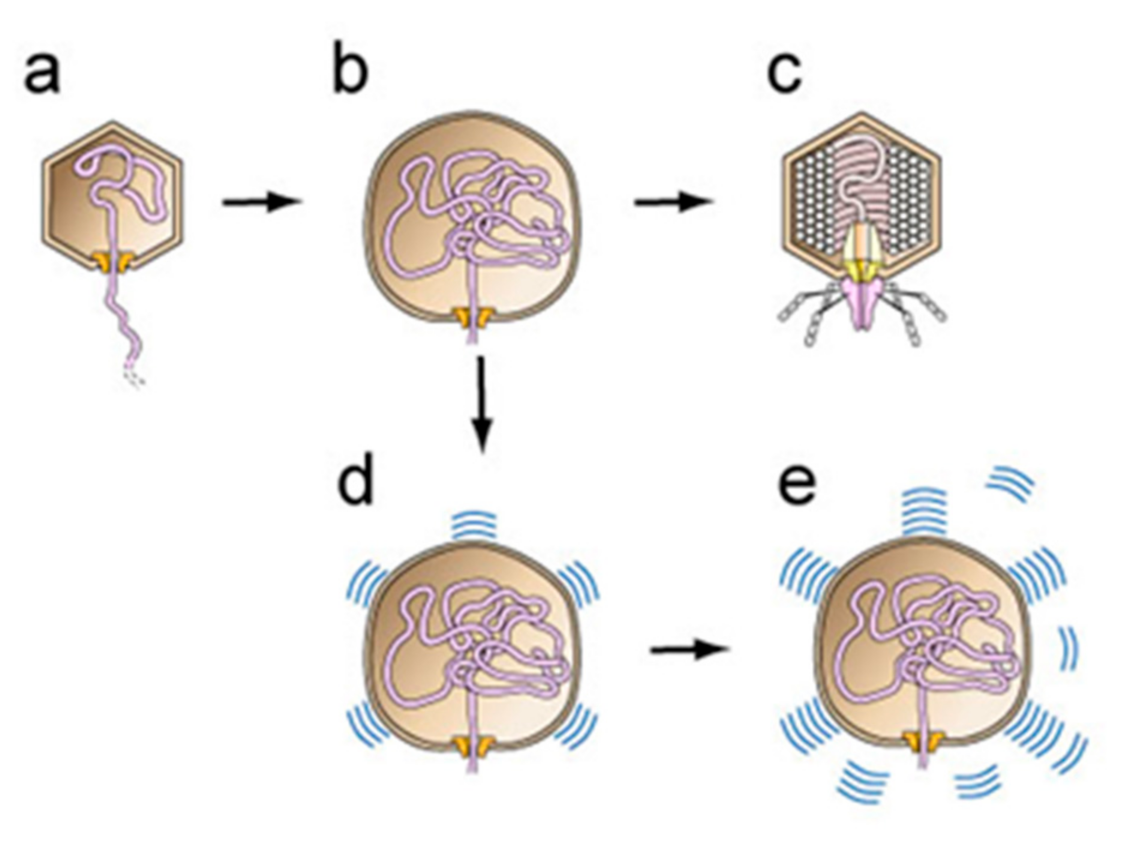
More recently, yet additional data suggest that the details of this innate immune reaction are the following [11,12]. (1) The structural basis is a (self-complementary) sheet-like protein structure, originally discovered by Pauling and Corey [13], and later called α-sheet [14,15,16]. Moreover, α-sheet structure can be visualized by starting with β-sheet and rotating every other amino acid by ~180° [13,14]. This rotation places all α-carboxyl groups (negatively charged at neutral pH) on one edge of the sheet and all α-amino groups (positively charged at neutral pH) on the opposite edge. Based on observation and characterization of DNA packaging-generated hyper-expanded capsids (details, below), α-sheet is proposed to be a major conformation of virus capsid proteins during virus assembly [11,12]. However, in healthy human cells, α-sheet is present only in stretches of 4–6 amino acids [14,15]. (2) This aspect makes the intracellular presence of more extended α-sheet a signal of the presence of a hostile invader, if the invader produces particles with α-sheet structure. (3) Amyloid protein performs innate immunity function by converting to α-sheet structure and then extending viral assembly-produced α-sheet. (4) A result is that normal virus assembly (Figure 1a–c, an abbreviated version of a DNA packaging pathway proposed for phage T3 [17]) is blocked (Figure 1d). (5) Neurodegenerative disease is caused by over-activity of this innate immunity protein, thereby producing toxic levels of α-sheet structured amyloid protein (Figure 1e). (6) Cellular efforts at detoxification subsequently convert most α-sheet to the β-sheet found [18,19] in amyloid plaques.
The innate immunity proteins include amyloid beta (Aβ) that accumulates to form extracellular plaques in the case of Alzheimer’s disease. Anti-herpesvirus activity of polymerized Aβ has been shown in both mice and cell culture ([10] and included references).
Herpesviruses also package a double-stranded DNA genome in a pre-formed capsid. This aspect is analogous to what occurs with both phage T3, and all other studied double-stranded DNA phages. Each of the major herpesvirus proteins involved is homologous to the phage counterpart [12,20,21,22,23,24]. For developing a hypothesis, this is a pivotal point because phage assembly generated particles are generally more accessible than those of eukaryotic virus counterparts. This advantage increases as particles become more difficult to detect, isolate and characterize.
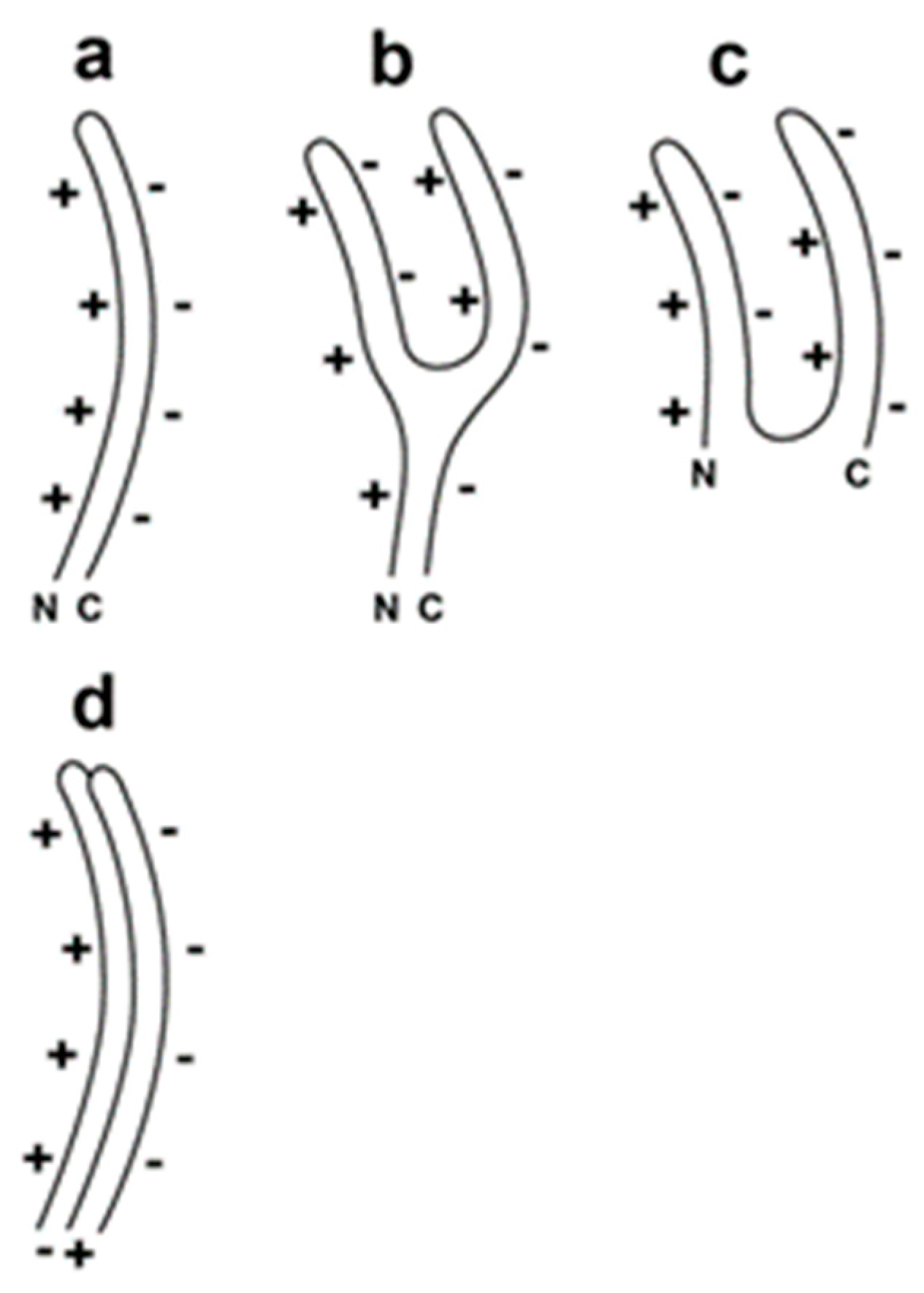
For the double-stranded DNA phage, T3, we have observed the following evidence of a DNA packaging-generated capsid with α-sheet structured subunits. Some DNA packaging-generated capsids are hyper-expanded, as illustrated in Figure 1b. The hyper-expansion constrains these capsids to have an outer shell so thin that the protein subunits of the capsid’s outer shell must have a sheet structure [11,12]. This structure is likely to be α-sheet for the following reasons. (1) The outer shell has a relatively high negative electrical surface charge density, which would be a characteristic of a capsid with α-sheet structured subunits and the α-carboxyl edge at the capsid exterior [11]. (2) Moreover, α-sheet structure explains the flexibility of the outer shell of the capsid (Figure 2 [11]). However, the thinness of the shell was not previously observed directly by electron microscopy. The reason was impermeability of the capsid’s shell to negative stains.
Here, we find an example of a hyper-expanded T3 capsid with outer shell thinning directly observed by electron microscopy of a negatively stained specimen. We transfer the observations with T3 to herpesviruses and develop an explanation of the dementia frequency observations of [1]. We use this explanation to suggest a practical means for decreasing the incidence of dementia.
2. Materials and Methods
Heads of phage T3 were obtained and purified by the use of procedures previously described. The final purification step was buoyant density centrifugation in a cesium chloride density gradient [25]. This purification step separated heads (tail-free) from phage particles (with the relatively short T3 tail), although the bands formed were close enough for low level cross-contamination. Otherwise, the heads were over 95% pure by protein composition.
For electron microscopy, the heads were dialyzed against 0.2 M NaCl, 0.01 M Tris, pH 7.4, 0.001 M MgCl2 and stored at 4 °C. The dialyzed heads of Figure 3 were prepared for electron microscopy 1.5 years after the dialysis. The procedure of preparation was negative staining with 1.0% sodium phosphotungstate, pH 8.4 [26]. The procedure includes a carbon support film designed to increased particle adherence without glow discharge [26]. A 1.5 μL droplet of specimen was placed on the carbon support film and left for 2.0 min at room temperature (25 ± 3 °C). The grid was then washed with 3 drops of Milli-Q-purified water and then 2 drops of 1.0% sodium phosphotungstate, pH 8.4. Excess stain was withdrawn with Whatman #1 filter paper. The image presented here had particles that were on the support film, not over holes in the film.
Specimens were observed in a JEOL100CX electron microscope in The Department of Pathology at The University of Texas Health Science Center at San Antonio.
3. Results and Discussion
3.1. Outer Shell-Thinning of Hyper-Expanded T3 Capsids
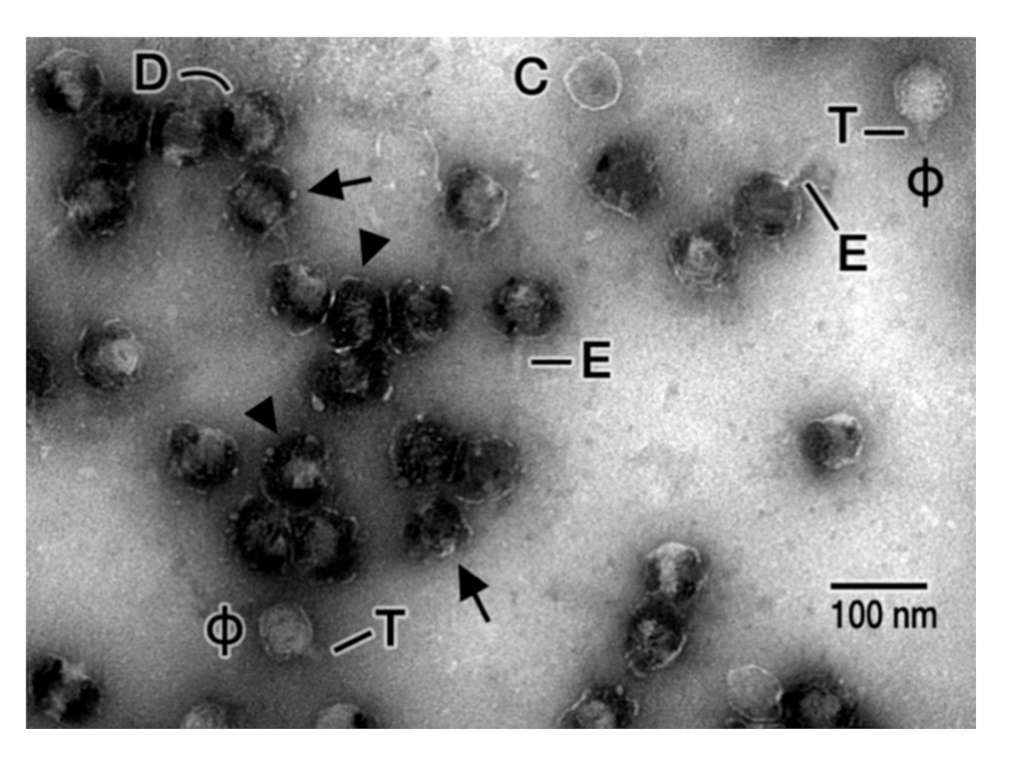
Given the possibility that hyper-expanded T3 capsids are prototypes for viral triggers of Alzheimer’s disease, we searched for and found a more electron microscopy-analyzable, hyper-expanded T3 capsid. The hyper-expansion occurred after, rather than during, DNA packaging. The particles were (DNA-filled) capsids that had not yet added a tail (heads). The heads had been isolated [25] from lysates of cells infected by a T3 mutant selected to propagate at relatively high sodium chloride concentration (0.9 M) [25]. This preparation was highly purified, except that some phages were not removed from the “head fraction” during buoyant density centrifugation in a cesium chloride density gradient (see Figure 3 in [25]).
After negative staining with 1.0% sodium phosphotungstate, pH 8.4, heads were penetrated by the negative stain. Most of these heads had the following two linked features, as seen for the particles indicated by arrowheads in Figure 3: (1) outer shells with thinner-than typical regions (1 nm or less) encompassing over 50% of their length, sometimes to the point that part of a shell was difficult to see and (2) 1.2–1.6× increased diameter. Size references for both shell-thinning and shell-diameter increase were (1) the particles indicated with a “ϕ” in Figure 3, which were unexpanded versions of (contaminating) phage particles (shell thickness: ~2 nm) and (2) the particle indicated with a “C” in Figure 3, which was an unexpanded, empty (no DNA) capsid (shell thickness: ~2 nm). This particle presumably was a head that lost its DNA. The linkage of the difference in shell diameter to the difference in shell thickness was universal in this sample. This linkage made unlikely the possibility that increased diameter was caused only by particle flattening.
3.2. Other Features of Hyper-Expanded T3 Capsids
In Figure 3, packaged DNA was seen in most hyper-expanded heads and no longer filled the capsid interior, presumably because of hyper-expansion. The observed shape of the DNA periphery varied. Rod-shaped, sometimes bent, DNA was often observed (indicated by the letter, D, for one particle in Figure 3). Studies are ongoing to determine the role of shell hyper-expansion/thinning in DNA packaging.
Furthermore, variable folding of shell subunits was indicated by limited shell regions that were thickened; arrows point at two of these regions in Figure 3. Such folding is explained by an α-sheet conformation, with the folding driven, at least in part, by charge neutralization (Figure 2).
Finally, an additional feature was the presence of a rod-shaped external extension on some particles. This extension (E in Figure 3) was distinguishable from the tail via both the tail’s trapezoidal shape and the tail’s lesser length. The extension was formed by externalization of proteins, originally internal, as previously seen for T3 phage particles [27].
3.3. The Hypothesis and Its Explanation of the Data on the Incidence of Alzheimer’s Disease
Thus, the complete hypothesis (including previously presented aspects) is the following. (1) A significant number of Alzheimer’s disease cases are triggered by loss of control of an innate immunity response to previous infection with viruses, including herpesviruses [12]. (2) The innate immunity response is based on the adoption of an α-sheet conformation by amyloid-forming proteins, followed by co-assembly with (and, therefore, inactivation of) the α-sheet of viral assembly intermediates [11,12]. The low amino acid sequence specificity of α-sheet co-assembly [13,14,15] implies that this innate immune response can be made by one protein for several viruses. (3) The post- ~1995 decrease with time of the incidence of Alzheimer’s disease is caused by vaccination against VZV. This vaccination lowered the number of, but did not eliminate, viral infections that trigger Alzheimer’s disease.
The linkage of Alzheimer’s disease to previous herpesvirus infection depends on the presence of the ApoE-ε4 allele [2,5,6,7,8]. As previously discussed [12], a possible reason is the effect of this allele on basement membranes, which are gels, and the subsequent stimulatory effect of basement membranes on the production of capsids homologous to the T3 capsids with α-sheet-structured subunits. The production of the latter is stimulated by propagation in cells embedded in an agarose gel [12].
The hypothesis provides the following explanation for the otherwise paradoxical observation, mentioned above, that the decrease in Alzheimer’s disease with time is not replicated in China [3] and Japan [4]. Until the last ~5 years, China [28] and Japan [29] did not sponsor routine VZV vaccination, unlike the United States (and presumably Western Europe), even though the vaccine strain of VZV was isolated in Japan [29]. The evidence for VZV-triggering of Alzheimer’s disease was especially strong in the case of untreated ocular shingles [6].
Finally, the above hypothesis provides an explanation for the higher time-dependent decrease of Alzheimer’s disease in women (above). The explanation is VZV vaccination and the observed [30] greater susceptibility of women to VZV-caused shingles. The above hypothesis assumes that some of the accompanying aspects of Alzheimer’s disease, such as decreased brain energy metabolism and increased inflammation [31], are either contributors to, or effects of, but not the main cause of, the basic disease process.
A corollary of this hypothesis is that the correlation with Alzheimer’s disease is with a step of reactivated herpesvirus infection (i.e., DNA packaging) that occurs before the completion of progeny virus particles. Thus, the hypothesis predicts a relatively weak correlation with either the DNA/capsid of mature virus particles or the induction of antibodies, as is often empirically found (review [32]).
The explanations of this section indicate that the above hypothesis is useful. However, while usefulness is a trademark of a correct hypothesis, it is not proof of the details of the hypothesis. In the future, these details can be tested by (1) tracking the sequence of events associated with the onset of Alzheimer’s disease and (2) determining the effects of specifically blocking each of these events in animal models. A prototype for doing this is the work done on the assembly of monomeric proteins to form the capsids of bacteriophages, e.g., the assembly of the tail of phage T4 [33,34]. The evidence presented here, although detailed, is circumstantial.
However, even without these details, the above hypothesis provides a lead that should be pursued as follows. Attempts to further lower the incidence of Alzheimer’s disease should be made by accelerating development of vaccines against the other herpesviruses, especially HSV-1 and HSV-2. Data suggest that vaccines for HSV-6A and HSV-7 should also be high on the priority list [7]. Vaccines are already in development for HSV-1 and HSV-2 infections (review [35]).
Author Contributions
Conceptualization, P.S.; methodology, P.S. and E.T.W.; validation, P.S.; resources, P.S.; data curation, P.S. and E.T.W.; writing—original draft preparation, P.S.; writing—review and editing, P.S.; visualization, P.S.; supervision, P.S.; project administration, P.S.; funding acquisition, P.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Welch Foundation, grant number AQ-764, and the Morrison Trust.
Data Availability Statement
The data presented in this study are in Figure 3 and in co-taken electron micrographs stored by P.S.
Acknowledgments
We thank the Department of Pathology at our institution for providing facilities for electron microscopy.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Wolters, F.J.; Chibnik, L.B.; Waziry, R.; Anderson, R.; Berr, C.; Beiser, A.; Bis, J.C.; Blacker, D.; Bos, D.; Brayne, C.; et al. Twenty-seven-year time trends in dementia incidence in Europe and the United States: The Alzheimer Cohorts Consortium. Neurology 2020, 95, e519–e531. [Google Scholar] [CrossRef]
- Anonymous. Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Li, S.; Yan, F.; Li, G.; Chen, C.; Zhang, W.; Liu, J.; Jia, X.; Shen, Y. Is the dementia rate increasing in Beijing? Prevalence and incidence of dementia 10 years later in an urban elderly population. Acta Psychiatr. Scand. 2007, 115, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Hata, J.; Yoshida, D.; Mukai, N.; Nagata, M.; Iwaki, T.; Kitazono, T.; Kanba, S.; Kiyohara, Y.; Ninomiya, T. Trends in dementia prevalence, incidence, and survival rate in a Japanese community. Neurology 2017, 88, 1925–1932. [Google Scholar] [CrossRef]
- Dobson, C.B.; Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease. Neurobiol. Aging 1999, 20, 457–465. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat. Rev. Neurol. 2020, 16, 193–197. [Google Scholar] [CrossRef]
- Bandea, C.I. Aβ, tau, α-synuclein, Huntingtin, TDP-43, PrP and AA are Members of the Innate Immune System: A Unifying Hypothesis on the Etiology of AD, PD, HD, ALS, CJD and RSA as Innate Immunity Disorders. bioRxiv 2013. Available online: http://biorxiv.org/content/early/2013/11/18/000604 (accessed on 29 October 2020). [CrossRef] [Green Version]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018, 99, 56–63, Erratum in: Neuron 2018, 100, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P.; Wright, E.T. Nanomedicine and phage capsids. Viruses 2018, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P.; Hunter, B.; Wright, E.T. Electron microscopy of in-plaque phage T3 assembly: Proposed analogs of neurodegenerative disease triggers. Pharmaceuticals 2020, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Milner-White, E.J.; Russell, M.J. Predicting the conformations of peptides and proteins in early evolution. Biol. Direct 2008, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Milner-White, E.J.; Russell, M.J. Functional capabilities of the earliest peptides and the emergence of life. Genes 2011, 2, 671–688. [Google Scholar] [CrossRef] [Green Version]
- Armen, R.S.; DeMarco, M.L.; Alonso, D.O.; Daggett, V. Pauling and Corey’s alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA 2004, 101, 11622–11627. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P. Proposed ancestors of phage nucleic acid packaging motors (and cells). Viruses 2011, 3, 1249–1280. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Cao, Q.; Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Cascio, D.; Eisenberg, D.S. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nat. Commun. 2020, 11, 4090. [Google Scholar] [CrossRef]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [Green Version]
- Cardone, G.; Winkler, D.C.; Trus, B.L.; Cheng, N.; Heuser, J.E.; Newcomb, W.W.; Brown, J.C.; Steven, A.C. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 2007, 361, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElwee, M.; Vijayakrishnan, S.; Rixon, F.; Bhella, D. Structure of the herpes simplex virus portal-vertex. PLoS Biol. 2018, 16, e2006191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaljeh, L.F.; Rothschild, J.A.; Naderi, M.; Coghill, L.M.; Brown, J.M.; Brylinski, M. Hinge region in DNA packaging terminase pUL15 of herpes simplex virus: A potential allosteric target for antiviral drugs. Biomolecules 2019, 9, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, Q.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Wu, Y.; Zhao, X.; Zhang, S.; et al. Terminase large subunit provides a new drug target for herpesvirus treatment. Viruses 2019, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serwer, P.; Wright, E.T.; Liu, Z.; Jiang, W. Length quantization of DNA partially expelled from heads of a bacteriophage T3 mutant. Virology 2014, 456, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P. Internal proteins of bacteriophage T7. J. Mol. Biol. 1976, 107, 271–291. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; Demeler, B.; Jiang, W. States of phage T3/T7 capsids: Buoyant density centrifugation and cryo-EM. Biophys Rev. 2018, 10, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, L.; Sun, X.; Cao, Y.; Wang, Z.; Liu, L.; Xu, Y.; Zhou, M.; Liu, Y. Effectiveness and failure rate of the varicella vaccine in an outbreak in Jiangsu, China: A 1:2 matched case-control study. Hum. Vaccin. Immunother. 2020, 16, 506–512. [Google Scholar] [CrossRef]
- Ozaki, T.; Asano, Y. Development of varicella vaccine in Japan and future prospects. Vaccine 2016, 34, 3427–3433. [Google Scholar] [CrossRef]
- Fleming, D.M.; Cross, K.W.; Cobb, W.A.; Chapman, R.S. Gender difference in the incidence of shingles. Epidemiol. Infect. 2004, 132, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren-Gash, C.; Forbes, H.J.; Williamson, E.; Breuer, J.; Hayward, A.C.; Mavrodaris, A.; Ridha, B.H.; Rossor, M.N.; Thomas, S.L.; Smeeth, L. Human herpesvirus infections and dementia or mild cognitive impairment: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, W.B.; Eiserling, F.A.; Crowther, R.A. 1994. Long tail fibers: Genes, proteins, structure, and assembly. In Molecular Biology of Bacteriophage T4; Karam, J.D., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 282–290. [Google Scholar]
- Arisaka, F.; Yap, M.L.; Kanamaru, S.; Rossmann, M.G. Molecular assembly and structure of the bacteriophage T4 tail. Biophys. Rev. 2016, 8, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, K.P.; Hook, L.M.; Naughton, A.; Pardi, N.; Awasthi, S.; Cohen, G.H.; Weissman, D.; Friedman, H.M. An HSV-2 nucleoside-modified mRNA genital herpes vaccine containing glycoproteins gC, gD, and gE protects mice against HSV-1 genital lesions and latent infection. PLoS Pathog. 2020, 16, e1008795. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A phage T3-based hypothesis for virus-derived initiation of Alzheimer’s disease. DNA packaging is initiated by a DNA-free procapsid, as illustrated for phage T3 in [11,12]. Then, (a) DNA enters an expanded, more angular conversion product of the procapsid, (b) hyper-expansion, with subunit conversion to α-sheet structure, occurs to accelerate DNA packaging when packaging is slowed in (a) (proposed details [17]), (c) DNA packaging finishes and a tail is added to the capsid to form a mature phage particle. When this process occurs during herpesvirus infection of a human cell, (d) amyloid proteins block progression of the hyper-expanded capsids by converting to α-sheet structure and, then, extending the α-sheet of subunits of the capsid. Alzheimer’s disease is initiated by (e) production of toxic amounts of amyloid protein α-sheet. The latter is subsequently converted to the β-sheet amyloid protein of plaques.
Figure 1.
A phage T3-based hypothesis for virus-derived initiation of Alzheimer’s disease. DNA packaging is initiated by a DNA-free procapsid, as illustrated for phage T3 in [11,12]. Then, (a) DNA enters an expanded, more angular conversion product of the procapsid, (b) hyper-expansion, with subunit conversion to α-sheet structure, occurs to accelerate DNA packaging when packaging is slowed in (a) (proposed details [17]), (c) DNA packaging finishes and a tail is added to the capsid to form a mature phage particle. When this process occurs during herpesvirus infection of a human cell, (d) amyloid proteins block progression of the hyper-expanded capsids by converting to α-sheet structure and, then, extending the α-sheet of subunits of the capsid. Alzheimer’s disease is initiated by (e) production of toxic amounts of amyloid protein α-sheet. The latter is subsequently converted to the β-sheet amyloid protein of plaques.

Figure 2.
Proposed α-sheet-polypeptide backbone of the subunits of a capsid’s outer shell (Adapted from [11]). (a) hyper-expanded capsid, (b) intermediate capsid and (c) contracted capsid (N and C indicate the N- and C-terminals of the major protein of the capsid’s outer shell; the + symbols indicate the positive electrical charge of the α-amino edge; the − symbols indicate the negative electrical charge of the α-carboxyl edge), (d) proposed [11] staggering of subunits in the unperturbed outer shell to minimize charge-charge repulsion.
Figure 2.
Proposed α-sheet-polypeptide backbone of the subunits of a capsid’s outer shell (Adapted from [11]). (a) hyper-expanded capsid, (b) intermediate capsid and (c) contracted capsid (N and C indicate the N- and C-terminals of the major protein of the capsid’s outer shell; the + symbols indicate the positive electrical charge of the α-amino edge; the − symbols indicate the negative electrical charge of the α-carboxyl edge), (d) proposed [11] staggering of subunits in the unperturbed outer shell to minimize charge-charge repulsion.
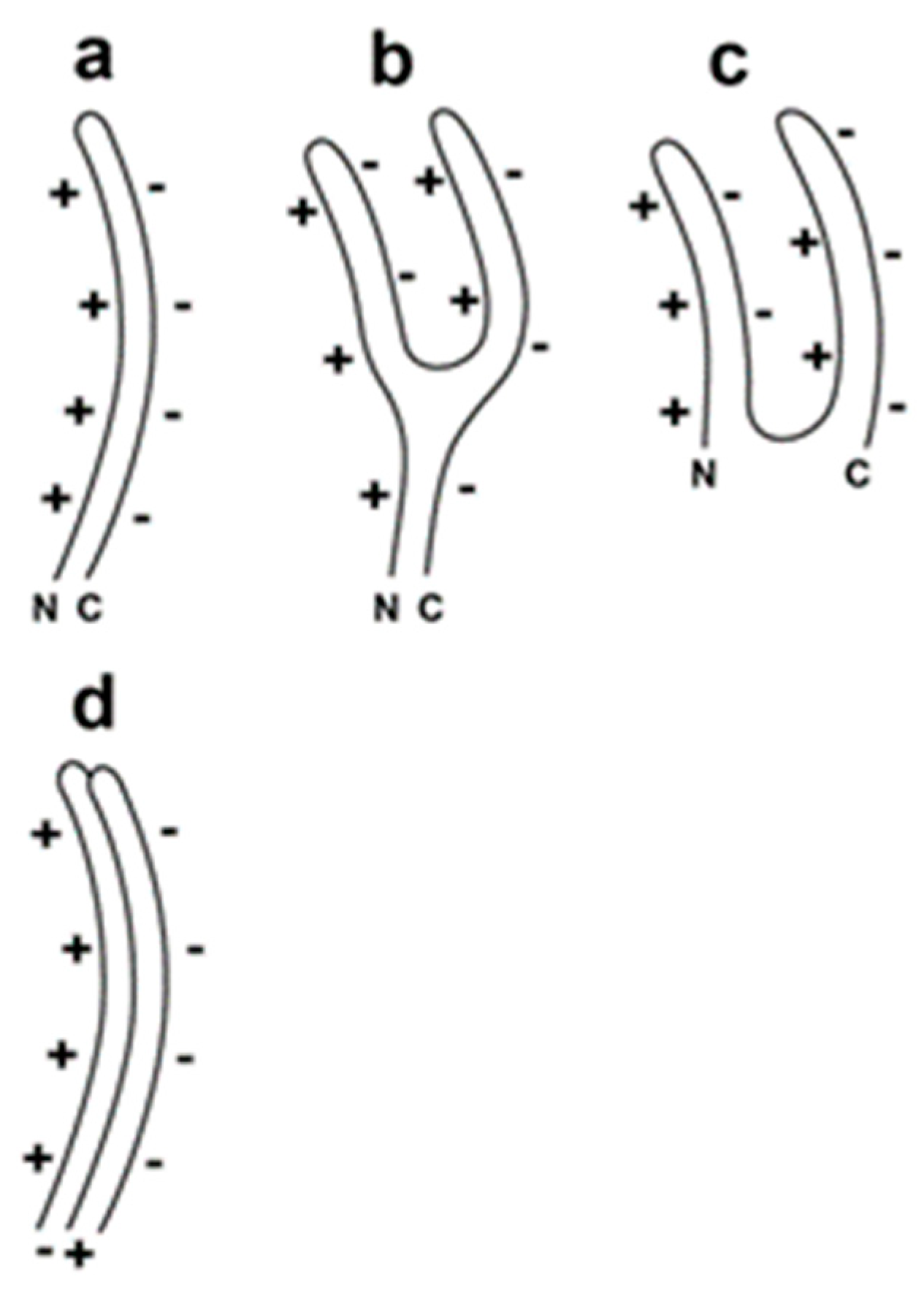
Figure 3.
Electron microscopy of negatively stained, hyper-expanded T3 heads. This image has several hyper-expanded heads, two indicated by arrowheads, two by arrows. The arrows point to shell regions that appear folded. Other aspects are C, empty, non-hyper-expanded capsid; D, DNA packaged in a head; E, external extension originating in the interior of heads; ϕ, phage contaminating from a neighboring fraction during purification; T, tail on phage particle.
Figure 3.
Electron microscopy of negatively stained, hyper-expanded T3 heads. This image has several hyper-expanded heads, two indicated by arrowheads, two by arrows. The arrows point to shell regions that appear folded. Other aspects are C, empty, non-hyper-expanded capsid; D, DNA packaged in a head; E, external extension originating in the interior of heads; ϕ, phage contaminating from a neighboring fraction during purification; T, tail on phage particle.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Serwer, P.; Wright, E.T. A Protein Assembly Hypothesis for Population-Specific Decrease in Dementia with Time. Biophysica 2021, 1, 15-21. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1010002
AMA Style
Serwer P, Wright ET. A Protein Assembly Hypothesis for Population-Specific Decrease in Dementia with Time. Biophysica. 2021; 1(1):15-21. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1010002
Chicago/Turabian StyleSerwer, Philip, and Elena T. Wright. 2021. "A Protein Assembly Hypothesis for Population-Specific Decrease in Dementia with Time" Biophysica 1, no. 1: 15-21. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1010002