4.1. Influence of the Sulphur Source on the Structuration of AgCdS Photocatalysts
The physicochemical characterization of the CdS-X and AgCdS-X photocatalysts has revealed the strong impact of the sulphur source over the structure and photoactivity of the photocatalysts. It is well-known that the formation of CdS nanostructures under solvothermal conditions follows the solvent coordination molecular template (SCMT) model [
38,
39,
44] in which the ethylenediamine acts as solvent and structure director agent. According to this model the changes in the morphology and structure of the CdS particles are related with the metal (Cd
2+ and Ag
+) complexation [
67,
68,
69,
70] and the S
2−generation rate because both factors determine the nucleation and crystallization of the final nanostructures of CdS [
43,
51,
71]. In the solvothermal synthesis using ethylenediamine as solvent, the complexation of Cd and Ag take place in the first step according to its different complex stability constants (see
Table S2):
The lower stability of the [Ag(EDA)
2]
+ complex (K = 10
4.7) respect to that of [Cd(EDA)
2]
2+ (K = 10
10.1) and the low solubility of Ag
2S (Ksp = 6.3 × 10
−50) respect to CdS (Ksp = 8 × 10
−27) leads to the fractionated precipitation of the sulphides whose contact and growth depend on the sulphide and metal concentrations. The first precipitation of Ag
2S particles could act as a nucleating platform to form the first lamellar CdS structures without excluding some insertion of Ag
+ ions into the CdS during the nucleation step, taking into account the high mobility of Ag
+ in the Ag
2S lattice [
61]. In the last steps of the solvothermal mechanism, the lamellar CdS structures transform into nanobars by means of the rolling mechanism [
35] and subsequently, they can growth to nanobars by aggregation following an Ostwald mechanism [
72]. In such a way that the sulphur source used in the solvothermal synthesis produces important changes in the crystallinity and contacts between the CdS and Ag
2S as consequence of the changes in the metal (Cd
2+ and Ag
+) complexation and the S
2− generation rate that modifies the nucleation and kinetics of crystallization and growth. The S
2− generation from the elemental sulphur is produced by the nucleophilic attack of ethylenediamine to the Sulphur, leading to sulphide ions and polianions [
73]. The generation of S
2− is slow and it results in the growth of Ag
2S and CdS nuclei of small size. The precipitation of Ag
2S particles could act as nucleating platforms to form the first lamellar CdS structures without excluding some insertion of Ag
+ ions into the CdS during its nucleation [
61]. The slow generation of S
2− allows the evolution of the lamellar CdS structures into nanobars by means of the rolling mechanism [
35] and subsequently they can grow to nanobars by aggregation following the Ostwald mechanism [
72]. The use of L-cysteine can modify both the nucleation and growth steps of CdS because of the dual nature of L-cysteine acting as a complexing agent and sulphur source. In this case, the Cd
2+ and Ag
+ ions could complex with ethylenediamine and L-cysteine taking into account the relative stability of the complexes [
67]. As the temperature increases, the L-cysteine complexes lose their stability and Cd(Ag)-S bonds are obtained forming the initial sulphide nuclei:
Once the L-cysteine complexes decompose, the Cd-EDA complexes contribute to the growth of the small particles Ag-CdS L-cysteine:
The steric effects produced by the L-cysteine adsorbed on the CdS nuclei impedes the evolution of Ag-CdS into nanobars by means of the rolling mechanism [
35], obtaining the observed globular morphology. The use of thiourea can also modify both the nucleation and growth steps of CdS because the thiourea could act as both complexing agent and sulphur source. In this case, the thiourea forms stable complexes with the Ag
+ ions while the ethylenediamine is coordinated with the Cd
2+ ions according to the relative stability of the complexes [
67]. As the temperature increases, the hydrolysis of thiourea takes place to quickly produce the S
2− anions. At the same time, the Ag-Tu complex is decomposed and this factor, together with the high availability of S
2− ions formed from thiourea, leads to the initial fast nucleation and growth of the Ag
2S precipitates. The nucleation of CdS from Cd-EDA complexes takes place subsequently forming a high number of small CdS nuclei in contact with Ag
2S particles without excluding some insertion of Ag
+ ions in the CdS during this nucleation step. The subsequent growth step of CdS after the nucleation to form bidimensional structures (by Ostwald ripening) and the formation of nanorods by rolling mechanism are kinetically limited because of the low availability of S
2− after the quick hydrolysis of thiourea that results in low crystalline CdS nanostructures.
4.2. Nanostructuration of AgCdS and Its Relationship with Photoactivity
The differences in the photoactivity of the CdS-X and AgCdS-X samples must be discussed according to their different photophysical properties. Such properties have been strongly influenced by the nature of the sulphur source, affecting the photocatalytic stages from the light absorption to generate electron and holes, their transport and/or recombination and the redox reactions promoted by those electrons able to reach the surface of the photocatalyst.
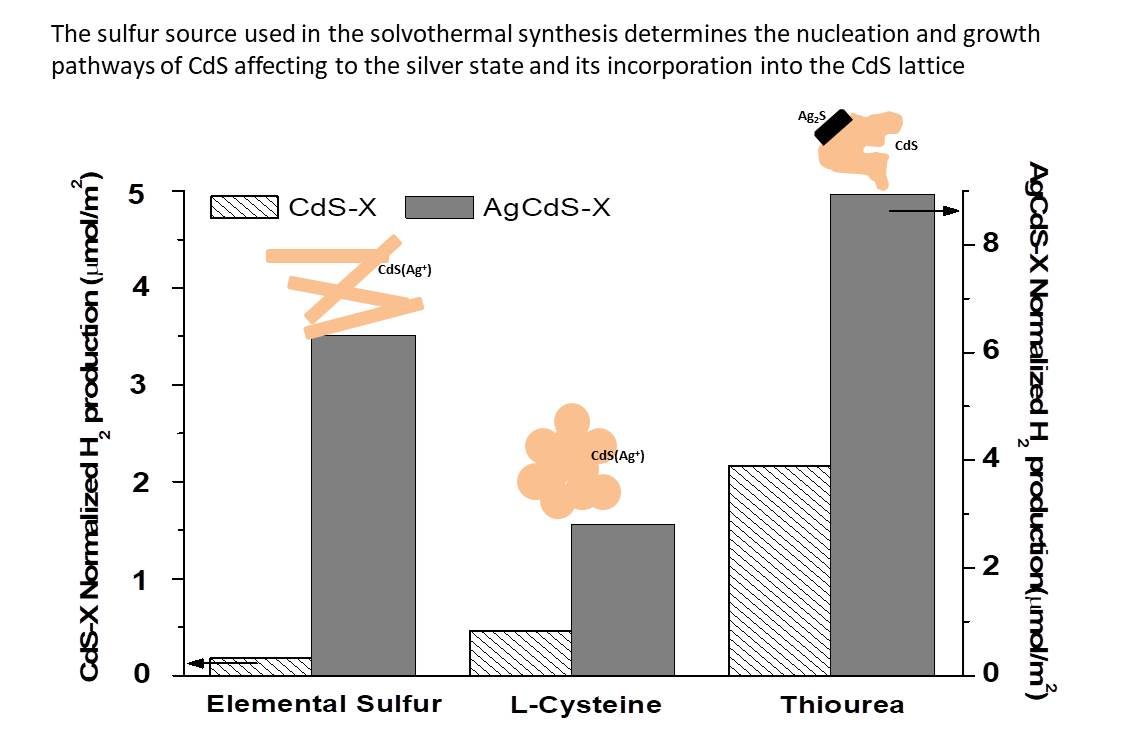
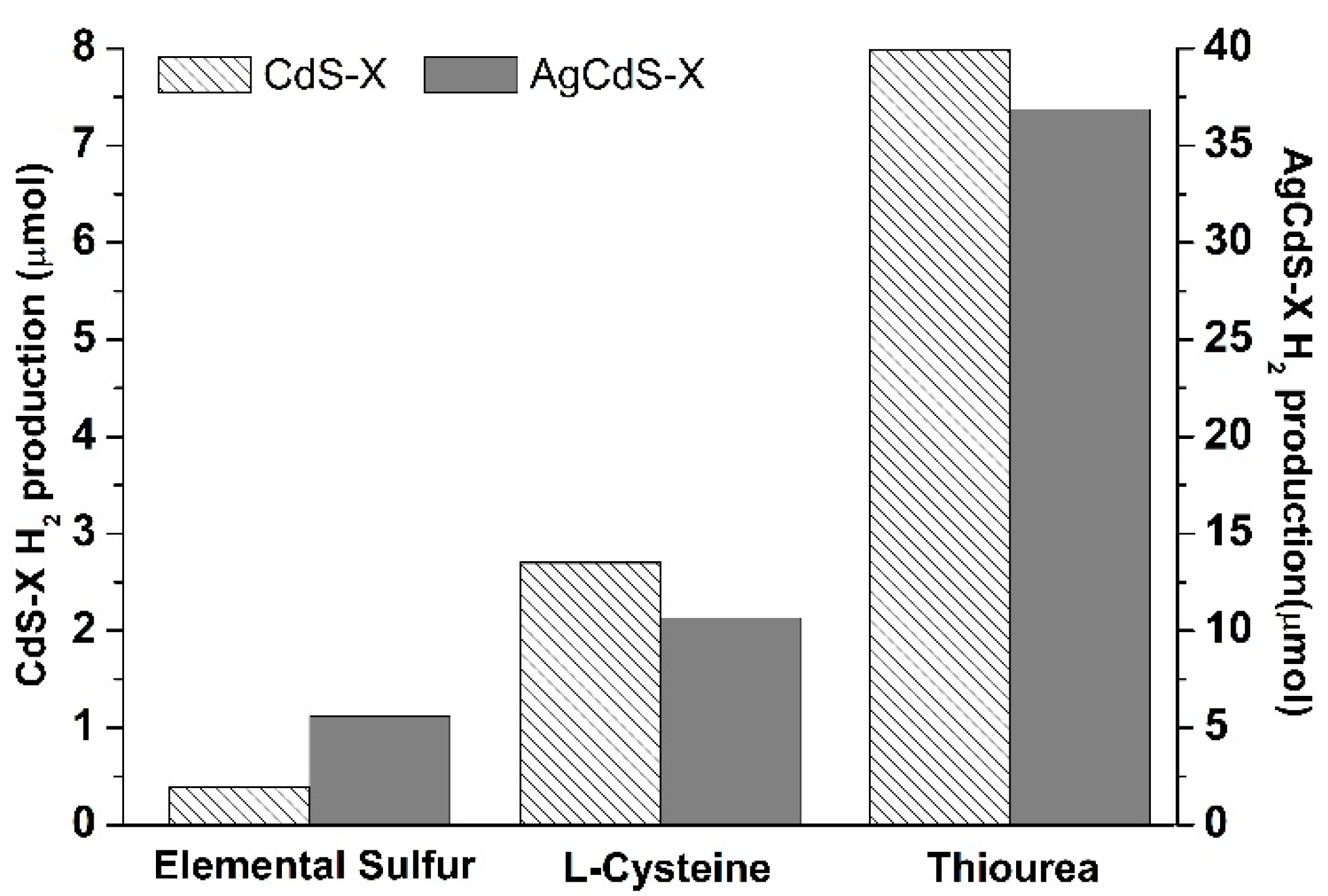
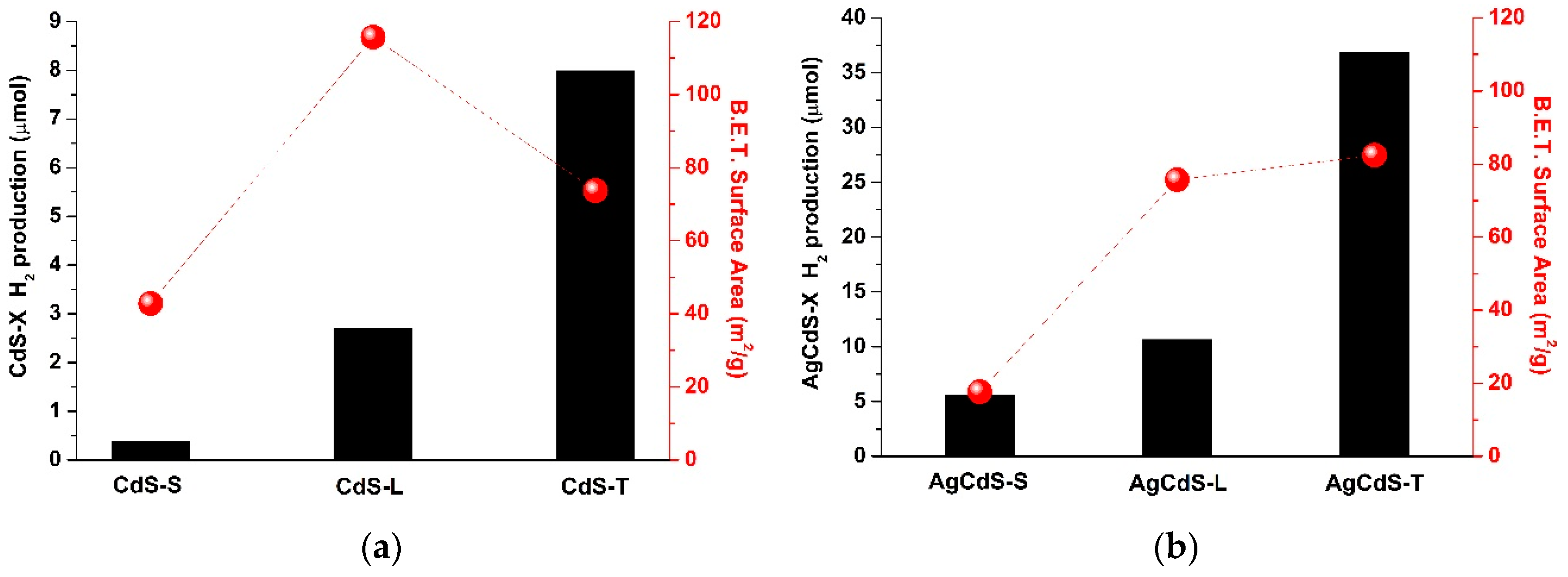
One of the most important parameters involved in the light absorption ability of the photocatalysts is related with the surface area exposed to irradiation. The
Figure 10 shows the evolution of the hydrogen production (µmol) respect to the surface area in the CdS-X and AgCdS-X series. For both photocatalyst series it was found that the photoactivity does not evolve in parallel with the surface area (
Figure 10), which indicates that there are additional factors to the surface area to justify the differences in the photoactivity of the CdS-X and AgCdS samples.
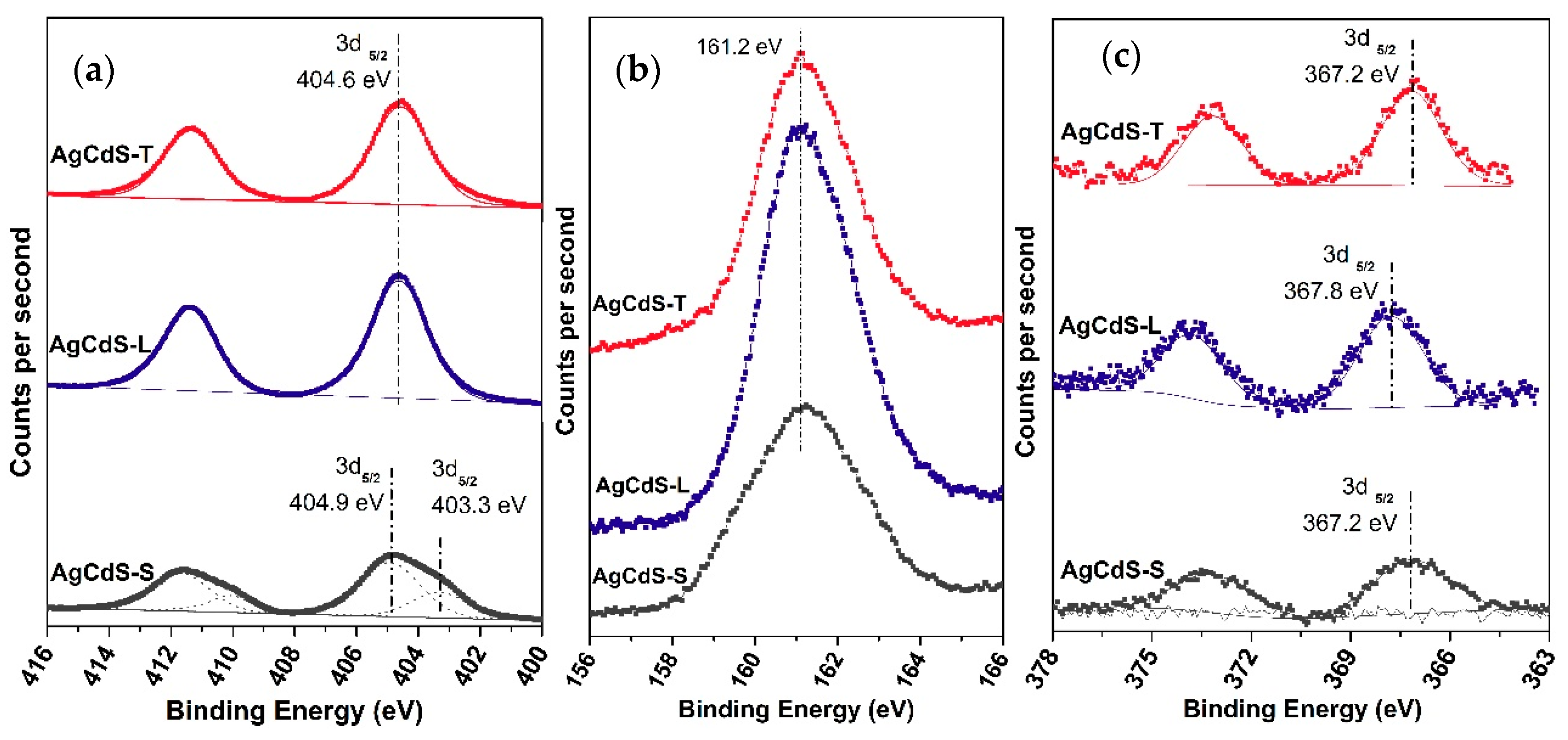
The capacity to generate electrons and holes is another important parameter related with the photoactivity. This fact is governed by the energy of the bandgap, because it determines the effective photons that are able to excite electrons from the valence band to the conduction band. The lower bandgap, the higher range of photons can be harnessed to generate electrons and holes. The energy gap of the AgCdS-X photocatalysts decreases respect to the CdS-X samples in the case of the samples prepared with elemental sulphur and L-cysteine (AgCdS-S and AgCdS-L). For these samples, the Ag
+ insertion into the CdS lattice, demonstrated by XRD and TEM, generated new electronic states near the valence band of the CdS, decreasing its band gap respect the CdS-X samples (
Table 6).
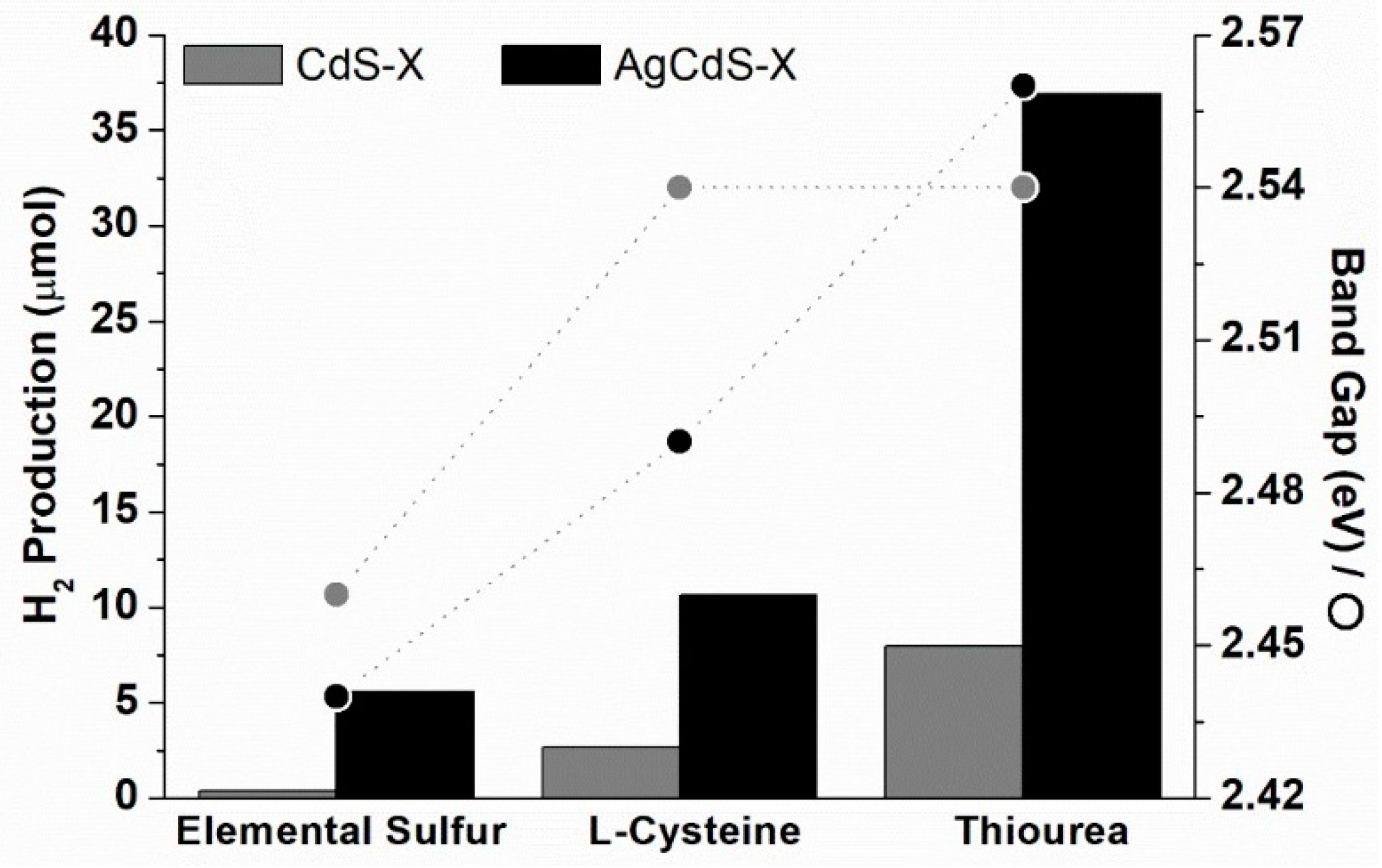
The comparison between the bandgap energy of the CdS-X and AgCdS-X samples and its photoactivity is shown in
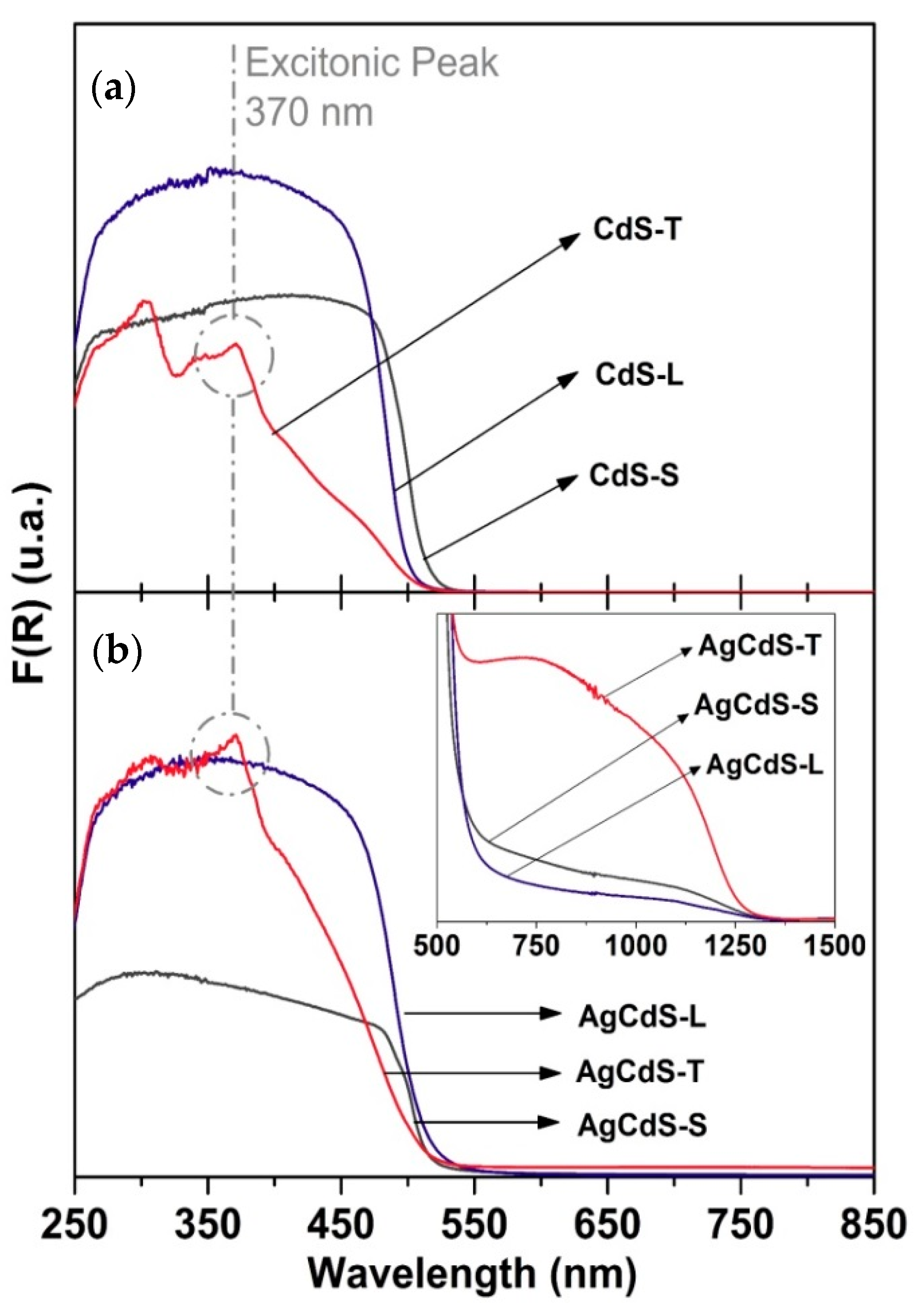
Figure 11. It is observed an improvement in the photoactivity of the AgCdS-X respect to bare CdS-X counterparts only for the samples prepared with elemental sulphur and L-cysteine (AgCdS-S and AgCdS-L). This improvement is parallel to the band gap narrowing observed for the AgCdS-S and AgCdS-L samples. However, for the samples prepared with thiourea (CdS-T and AgCdS-T), a different trend was observed, showing the AgCdS-T higher photoactivity in spite of the increase in the energy gap observed for this sample respect to those prepared with elemental sulphur and L-cysteine. The better photoactivity of the AgCdS-T photocatalyst in spite of its higher energy gap could be related with additional factors related, with the presence of small crystalline structures (quantum dots), as the UV-vis spectra showed (
Figure 7). The origin of these quantum dots is associated to the fast nucleation of sulphides derived from the use of thiourea. Due to the extremely fast nucleation stage, there exist a large number of nuclei, which aggregation and growth through the Ostwald mechanism was hindered, remaining as small crystalline domains. These small nanostructures generate quantum confinement effects that have been reported to strongly improve the photoactivity of CdS because they are able to absorb solar light, enhancing the surface amplitude of electrons and holes facilitating their interfacial reactions [
39].
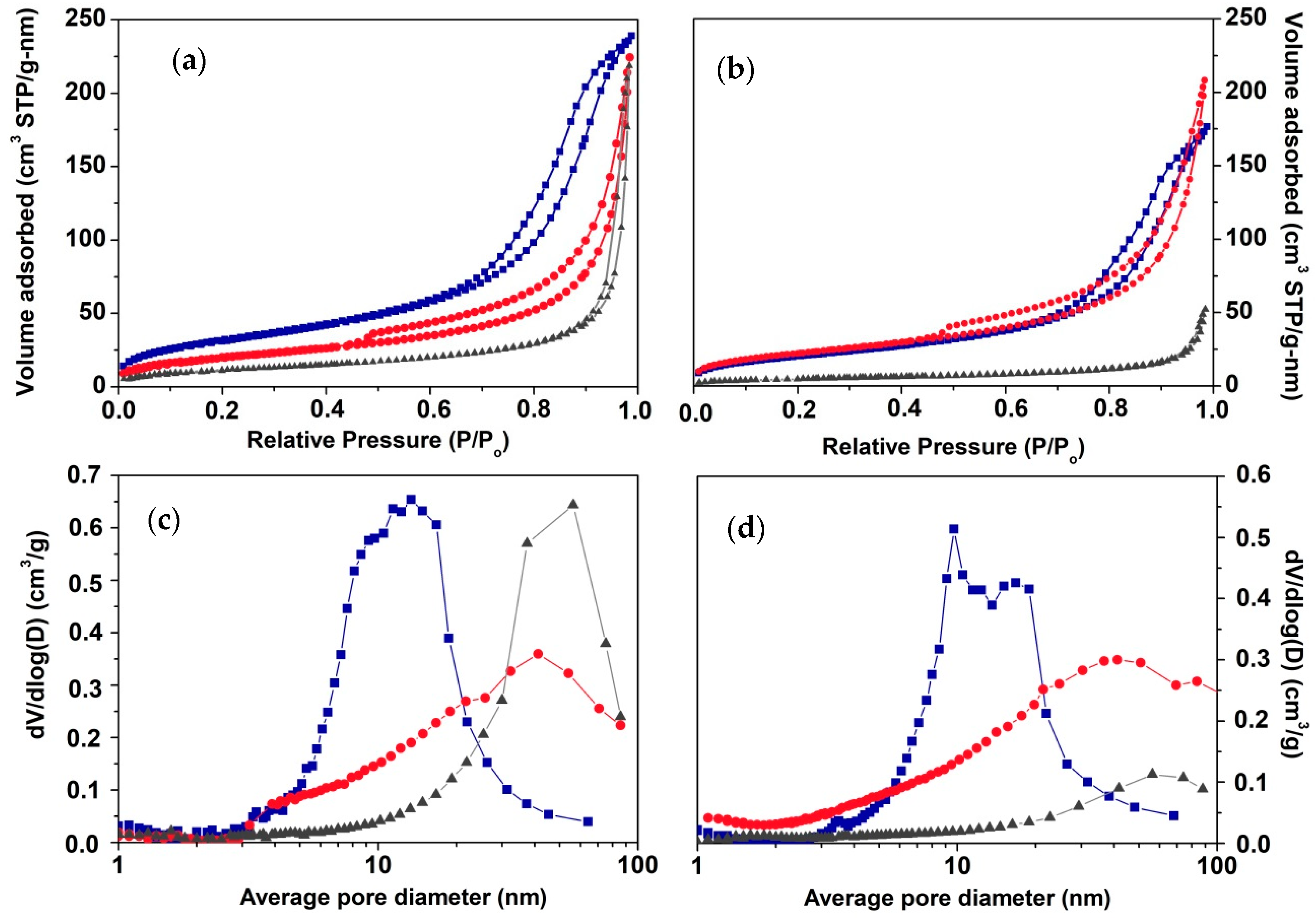
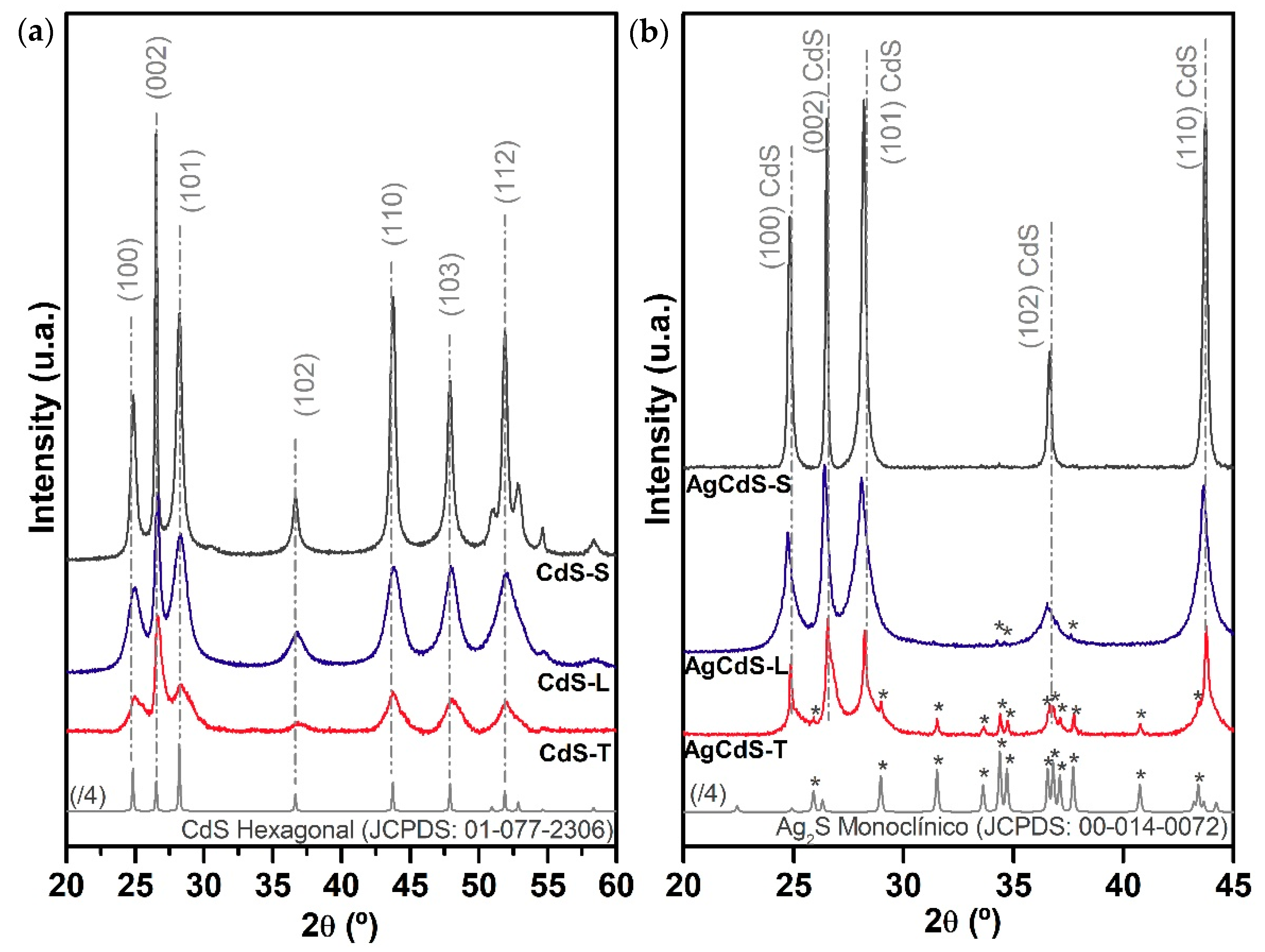
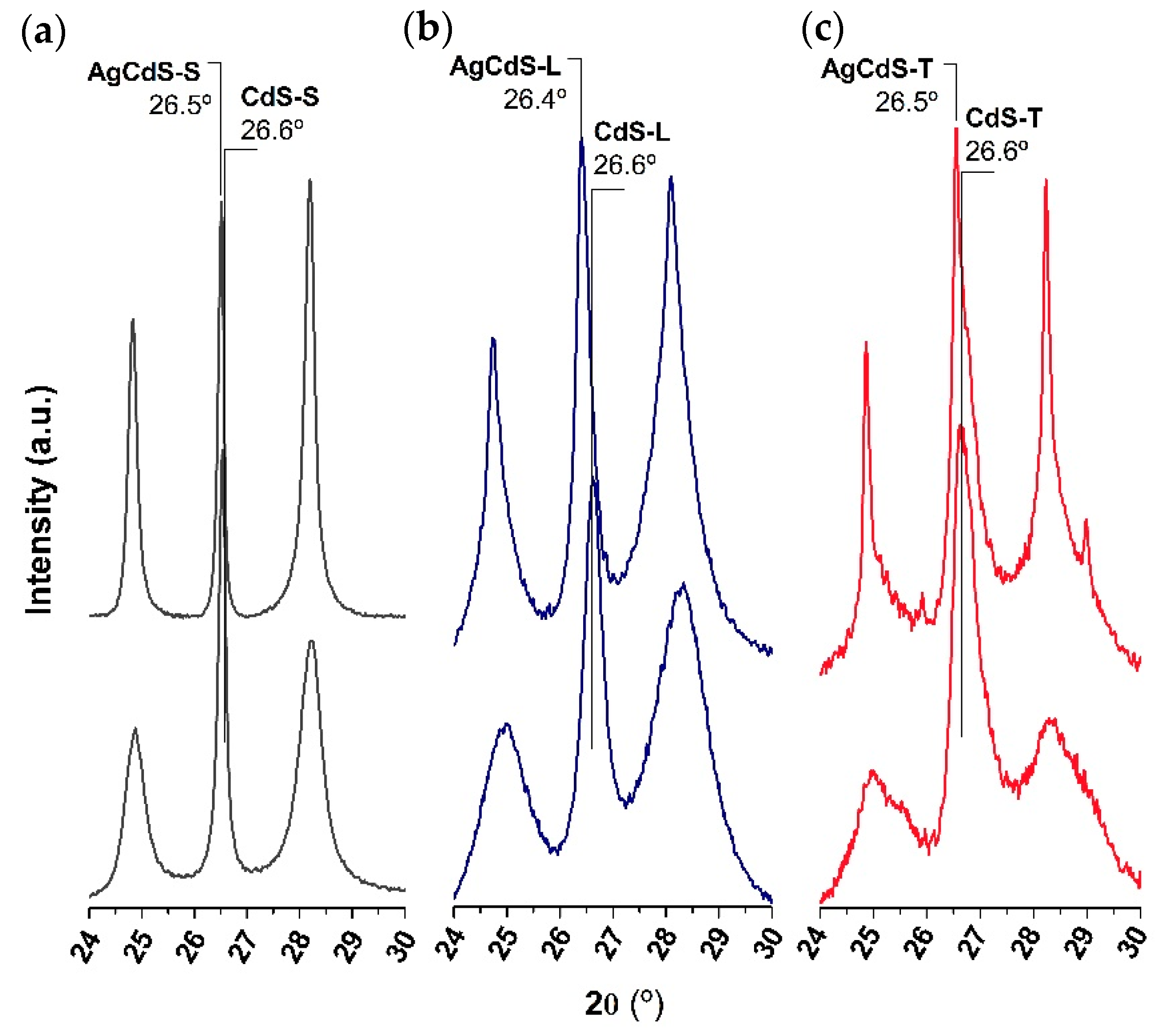
From the XRD analysis it was found differences in crystallinity and size of CdS-X and AgCdS-X photocatalysts, and thus, these parameters must be analysed because it is known that crystallinity and size are involved in the effectiveness of the mobility of electron and holes to achieve the photocatalytic surfaces. In this sense the effectiveness of the carrier mobility can be improved in CdS structures of low size and high crystallinity because they decrease the distance to reach the surface and the number of grain boundaries, which act as recombination sites.
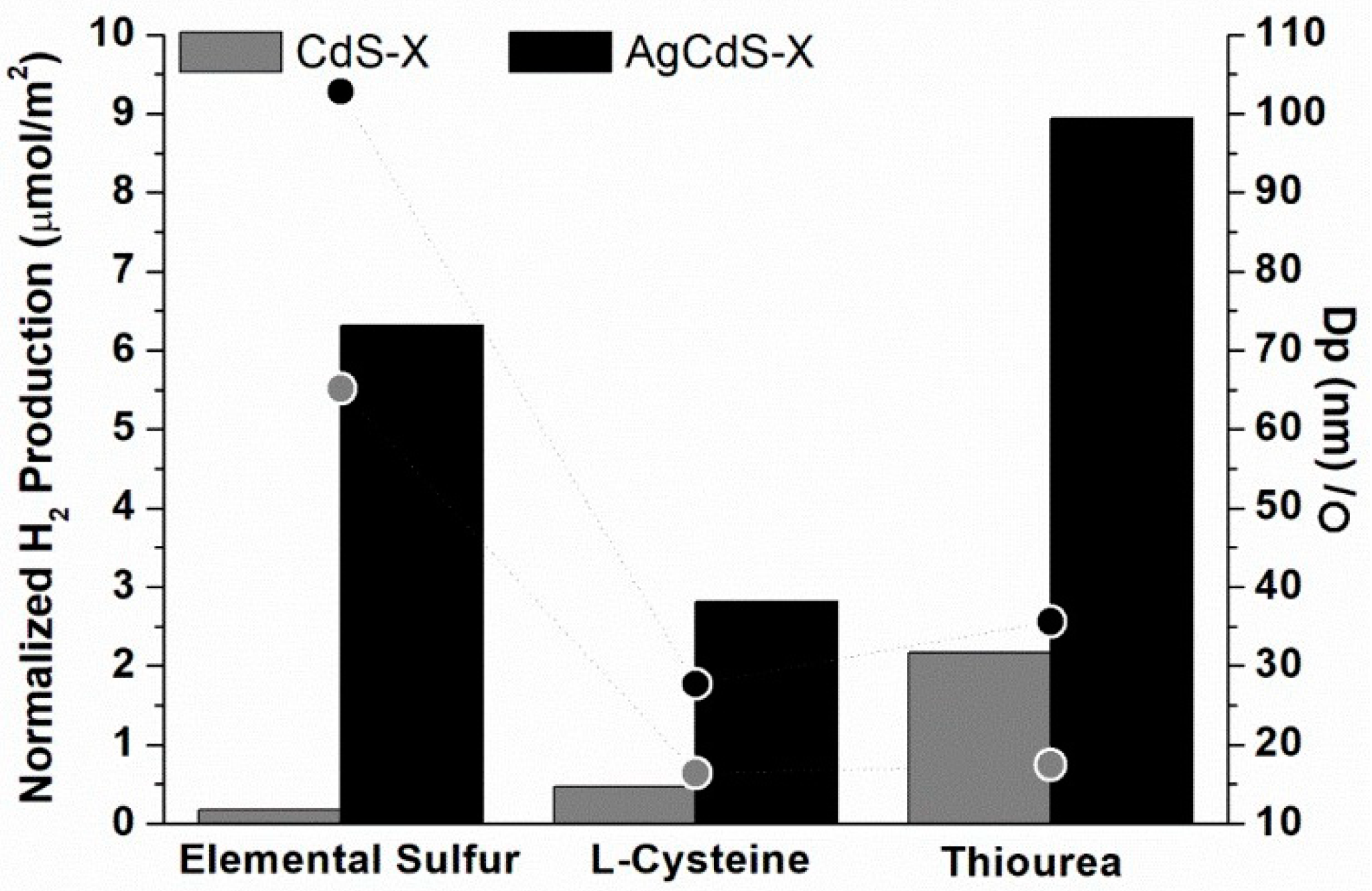
Figure 12 compares the intrinsic photoactivity (µmol/m
2) of the AgCdS-X and CdS-X samples respect to the crystallite size (Dp) calculated from XRD. As shown in
Figure 12 there is not a clear correlation between photocatalytic activity and crystallite size. The effectiveness of the transport of electron and holes to achieve the surface of the photocatalysts can also be determined by the presence of Ag
2S on the surface that can act as co-catalysts facilitating the transport of the energy carriers. According to this argument, the different intensities of the PL spectra (
Figure 8) indicate that Ag
2S species on surface play an active role to avoid the recombination processes.
The effect of residual nitrogen on surface of the samples prepared with thiourea could also be responsible of the high photoactivity observed on these samples because nitrogen linked to the surface Cd
2+ ions can create surface states that promotes the transference of the photogenerated electrons to the protons to produce hydrogen [
74].
The differences in the crystallite size, crystallinity and textural properties described above are translated into different morphologies depending on the sulphur source employed (SEM analysis,
Figure 4). However, none of these parameters showed a direct relationship with the photoactivity, and therefore it seems that the photoactivity of the AgCdS-X samples is linked to a combination of these factors together with the insertion of Ag
+ in the CdS or the segregation of Ag
2S on CdS.
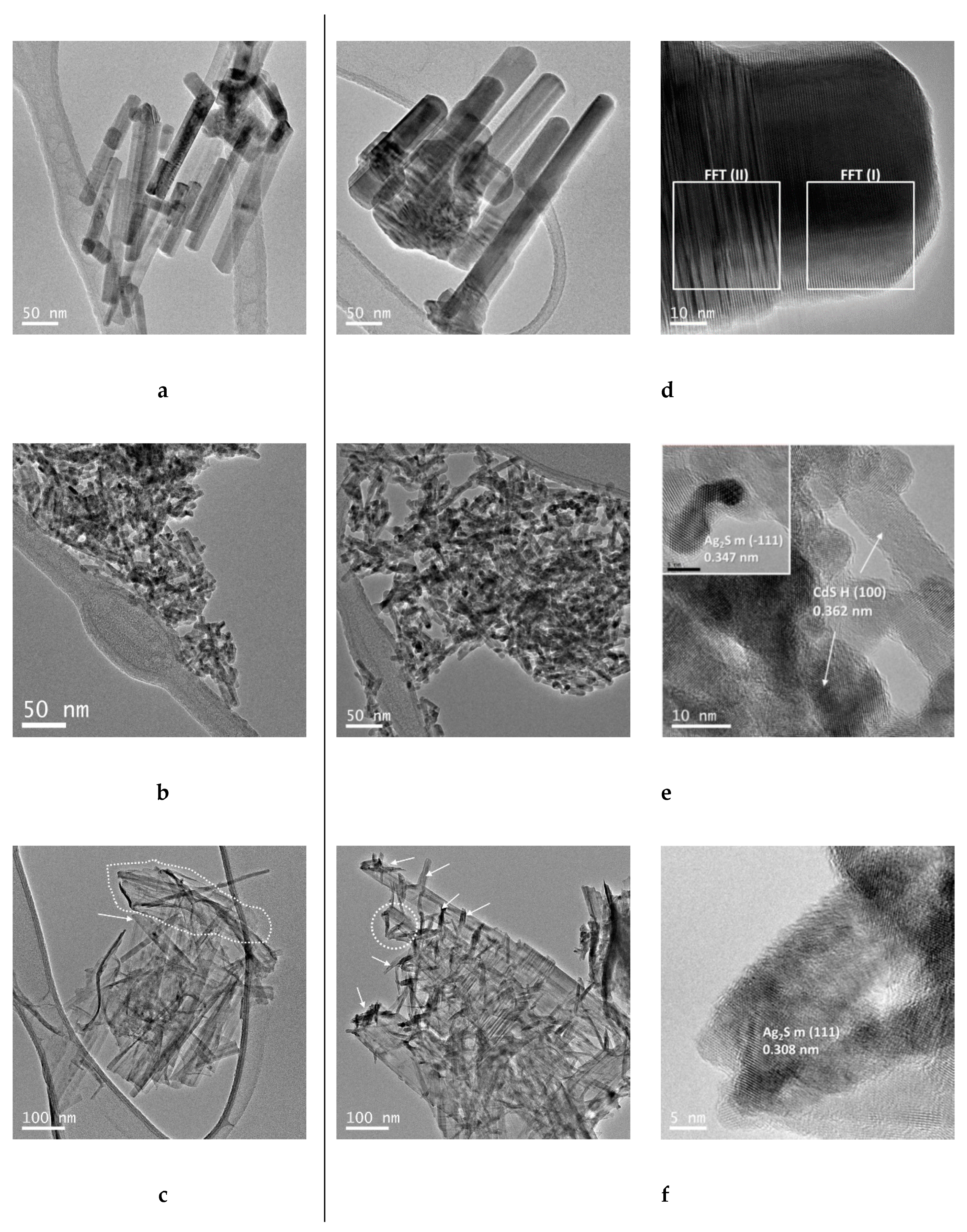
In the case of the sample, prepared with elemental sulphur (AgCdS-S,
Figure 13a), highly crystalline CdS nanobars, in poor contact with AgCdS particles of high size, were obtained. The improvement in the photoactivity of the AgCdS-S sample respect its CdS-S counterpart lies in the formation of AgCdS particles whose band gap allows the absorption of a wider range of the solar spectrum. The improvement in the photoactivity of the AgCdS-S sample is limited by the formation of AgCdS particles in scarce number and with high size. For the sample prepared with L-Cysteine (AgCdS-L,
Figure 13b), the insertion of silver was extended to the whole CdS structures and the size of the AgCdS particles was lower than in the previous case prepared with elemental sulphur. Therefore, the better development of AgCdS particles with lower size obtained in this AgCdS-L sample justifies its improvement in photoactivity respect to that obtained for the sample prepared with elemental sulphur. The sample prepared with thiourea (AgCdS-T,
Figure 13c), shows the combination of CdS, Ag
2S and small crystalline structures of CdS (quantum dots). These three components contribute to the improvement of the photoactivity observed in the AgCdS-T sample. Part of the improvement in photoactivity is related to the existence of small crystalline domains (QD) with strong confinement effect because these nanostructures are able to absorb solar light and quickly generate surface electrons and holes, and also because these nanostructures significantly enhance the surface amplitude of electrons and holes facilitating their interfacial reactions. In addition to this, the segregation of Ag
2S and its junction with CdS allows the absorption of a wider range of the solar spectrum. Nevertheless, the effective management of the charge carriers to produce the redox reactions strongly depends on the relative position of the valence band and conduction band. The relative position of the valence and conduction band of CdS and Ag
2S in the sample AgCdS-T has been calculated theoretically (see Materials and Methods section in
Supplementary Info) and the results are collected in
Table 7 [
75,
76,
77,
78].
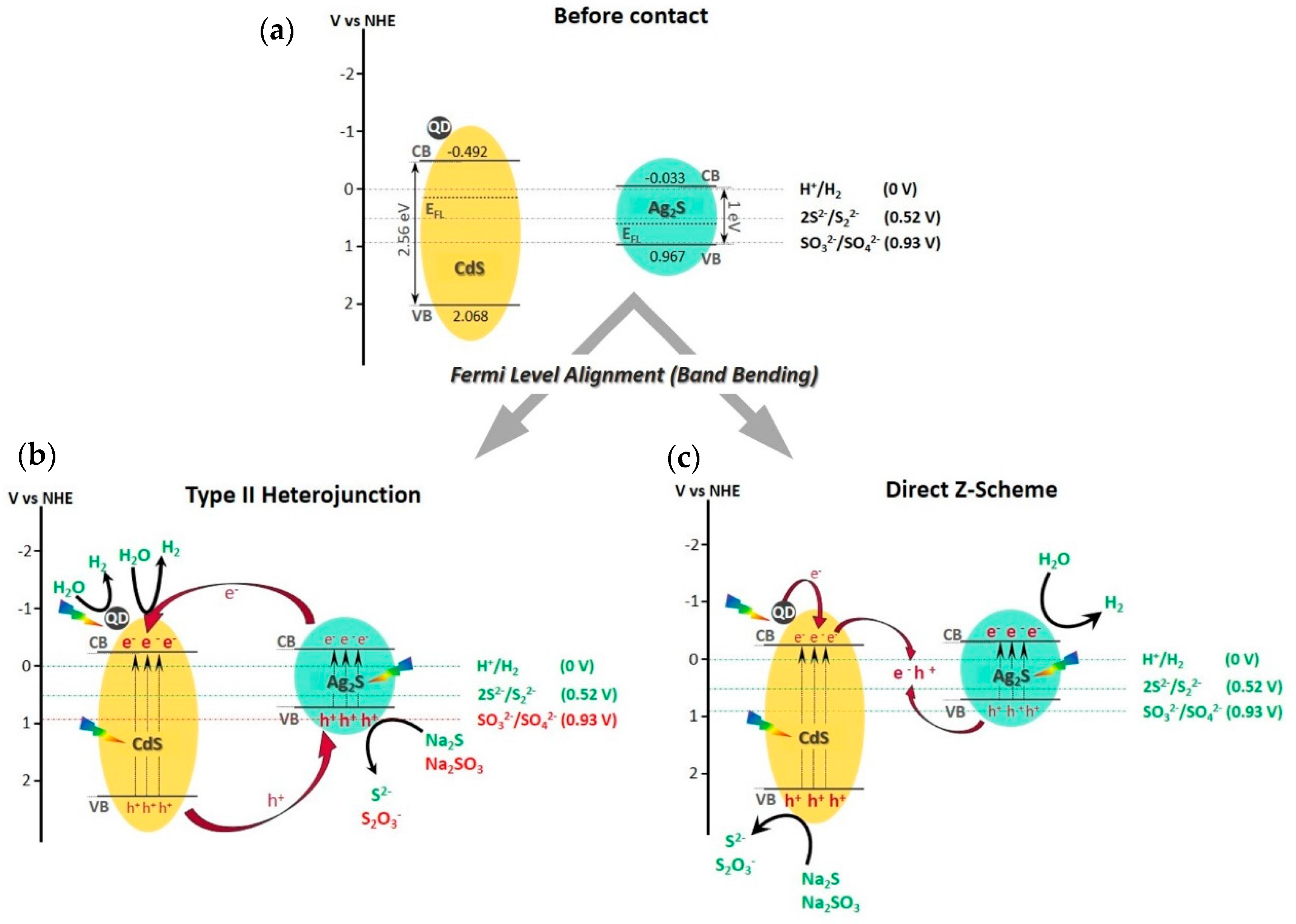
As it is depicted in
Figure 14a, the CdS acts as a n-type semiconductor while the Ag
2S is a p-type material, therefore the Fermi level (E
FL) for CdS is near the conduction band and for Ag
2S is near the valence band [
79]. The close contact between CdS and Ag
2S in the AgCdS-T sample promotes the Fermi level alignment through the flow of electrons from the CdS conduction band to that of Ag
2S, leading to the band bending of both semiconductors, as it is shown in
Figure 14b,c.
After the band bending, the electronic transfer mechanism between CdS and Ag
2S can be produced in two different ways, forming a p-n heterojunction or a direct Z-scheme (
Figure 14b,c). In the p-n heterojunction, the conduction band of Ag
2S is located at higher potential respect to that of CdS, so it favors the flow of electrons from Ag
2S to CdS, and on the contrary, the holes moves from the valence band of CdS to the valence band of Ag
2S [
80]. In this case, the quantum dots contributed to both improve the light absorption and to enhance the surface amplitude of electrons and holes facilitating their interfacial reactions. However, this p-n scheme showed two main disadvantages that should be take into account. On one hand, and according to the work of Di et al. [
81], from the kinetic point of view the electrostatic repulsions in the conduction band could avoid the electron transfer to produce hydrogen, and thus, the hole transfer between the valence bands was also limited. While, on the other hand, the effectiveness of the p-n heterojunction also resides in the redox ability to carry out the desired oxidation/reduction reactions. In this sense, the
Figure 14a shows that the oxidation of SO
32− in the valence band of Ag
2S is not possible due to the redox potential. According to the equilibrium reactions of the sacrificial reagents (see
supplementary information, Figure S9) this fact can negatively affect the photoactivity due to the mismatch of the equilibrium balances of S
2− and SO
32− species, which could lead to an increase in disulphide species (S
22−) that acts as an optical filter and therefore worsen the capacity to absorb light [
82].
In the charge transfer route of a direct Z-scheme (
Figure 14c), the electrons of the n-type semiconductor (CdS) together with the electrons injected by the quantum dots are directly recombined with the holes of the p-type semiconductor (Ag
2S). Therefore, the most reductant electrons remain in Ag
2S to produce hydrogen, and the most oxidant holes in the CdS oxidize the S
2−/SO
32− species. In this structure it is allowed a better spatial separation of the charge carriers together with the adequate location of the valence band and conduction band to carry out the oxidation/reduction processes. Based on this, it seems that in this scheme, the junction between CdS and Ag
2S allows a better management of the energy carriers, and therefore it can be justified the improvement of the hydrogen production in the AgCdS-T sample.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}