
The Basic Theorem of Temperature-Dependent Processes
by
 , and
, and
Valentin N. Sapunov
1,* ,
,
Eugene A. Saveljev
1,
Mikhail S. Voronov
1,
Markus Valtiner
2,* and
Wolfgang Linert
2,* 1
Department of Chemical Technology of Basic Organic and Petrochemical Synthesis, Mendeleev University of Chemical Technology of Russia, Miusskaya sq. 9, 125047 Moscow, Russia
2
Department of Applied Physics, Vienna University of Technology, Wiedner Hauptstrasse 8-10/134-AIP, 1040 Vienna, Austria
*
Authors to whom correspondence should be addressed.
Thermo 2021, 1(1), 45-60; https://0-doi-org.brum.beds.ac.uk/10.3390/thermo1010004
Submission received: 26 January 2021
/
Revised: 19 February 2021
/
Accepted: 23 February 2021
/
Published: 19 March 2021
{kind=link}
{kind=link}
{kind=link}
Abstract
:The basic theorem of isokinetic relationships is formulated as “if there exists a linear correlation “structure∼properties” at two temperatures, the point of their intersection will be a common point for the same correlation at other temperatures, until the Arrhenius law is violated”. The theorem is valid in various regions of thermally activated processes, in which only one parameter changes. A detailed examination of the consequences of this theorem showed that it is easy to formulate a number of empirical regularities known as the “kinetic compensation effect”, the well-known formula of the Meyer–Neldel rule, or the so-called concept of “multi-excitation entropy”. In a series of similar processes, we examined the effect of different variable parameters of the process on the free energy of activation, and we discuss possible applications.
1. Introduction
Exponential models of the temperature dependence of the properties of certain processes appeared in the scientific literature as early as the end of the 19th century. Such a functional dependence was proposed by Reynolds (1884) to describe the change in viscosity as a function of temperature. Almost at the same time, Arrhenius assumed (1889) similar equations for the temperature dependence of reaction rates. Now, exponential functions are the most important and widespread functions in physics, chemistry and biology.
Similarly, in physics, exponential functions describe the decay of radioactive nuclei [1], the emission of light by atoms [2], and electrical conductivity in various semiconductors [3,4,5] and even in superconductors [6]. In biology, exponential functions describe the growth of bacterial, viral or animal populations [7,8].
However, attempts to find any non-exponential dependence of the rates of chemical reactions on temperature continue to this day. For example, to improve the description of the temperature dependence of the rate constants for functions that differ from Arrhenius’ theory, work based on empirical two-parameter functions with two “non-Arrhenian” temperatures was performed [9,10,11,12,13,14]. However, these models and aspects are not the focus of this review.
Empirical models of processes usually work for a limited range of temperatures, and are expressed in a universal form or the universal Arrhenius equation [15]:
where is, as a rule, the rate of a thermally activated process; is the Boltzmann constant; E is the sensitivity of the process to the temperature or the activation energy (in the case of reaction rates). The physical meaning of all the above-named exponential dependencies becomes more obvious from the point of view of statistical physics. The model presented (Equation (1)) has a striking similarity to the Gibbs canonical distribution formula. In this sense, the function and, precisely, the exponential factor represent the probability of the existence of a state of a thermodynamic system, with the energy E, in thermal equilibrium with a heat bath (the environment) at a fixed temperature T. Furthermore, if this state is a transition state, the above exponential dependency then reflects the probability that a system can transition along a reaction coordinate, or in other words, it reflects the probability of a process proceeding at different temperatures. This is only true if, and only if, the system reaches a point of no return at this state, which is an explicit assumption (no-recrossing assumption) in classical chemical thermodynamics such as the Eyring–Polanyi theory.
The most intensive use of the universal Arrhenius Equation (1) began in the field of physical organic chemistry, namely, in chemical kinetics, making it the basic equation of chemical kinetics. Since the middle of the twentieth century, a quantitative approach to the interpretation of reaction mechanisms began to develop [16,17,18]. Here, we are talking about the correlation analysis of the relationship between the rate constants of some series of reactions and certain parameters of the reaction medium or the physicochemical properties of the reactants involved [19,20]. The use of regression analysis with the help of the so-called QSAR models (models of quantitative structurally active relationships) made it possible to perform a unified quantitative characterization of a series of similar reactions [21], or the influence of solvents according to Edwards [22], Grunwald-Winstein [23,24] and many others. Many linear correlation dependencies appear that connect the properties (in particular, the reactivity) of a single series of chemical compounds with parameters that uniquely correspond to the structural features of this series. The prototype of the majority of correlations is the Brönsted equation, which establishes a quantitative relationship between the strengths of acids and bases and their catalytic activity [25]. There appeared correlation dependencies of Hammett [17] and Taft [26,27,28], based on the same principle—the relationship between the structure and reactivity. Subsequently, many variations of the above-mentioned correlation equations appeared, such as the Swain–Lupton equation [29] and Yukawa–Tsuno equation [30]. Similar correlations are widely used to describe the influence of certain parameters, generalized (e.g., the nature of the substrate or properties of the medium) in a series of thermally activated processes when this parameter is changed. In this case, the variable (T) in Equation (1) is a function of two parameters, the temperature and the variable parameter (Equation (2)):
The correlation analysis of the temperature dependencies of different processes using the two-parameter Equations (1) and (2) made it possible to reveal another regularity for which statistical physics cannot answer the question of the reasons for its existence.
This is the so-called “compensation effect”. This concept covers such regularities as, for example, the establishment of a relationship between the numerical values of the exponent and the pre-exponent, among which are the Barclay–Butler rule [31], the theta rule [32], the Smith–Topley effect [33], the Constable–Cremer relation [34,35], and many others.
Particularly widespread was the establishment by Meyer and Neldel of the fact that the logarithm of the pre-exponential coefficient (or “factor”) vs. exhibits very good correlation in certain series [36]. The mentioned relation is known as the Meyer–Neldel rule (MNR), which opened up new possibilities for a further step in development, especially in theoretical physical chemistry [37,38]. A general form, Equation (3), expresses the MNR as follows:
Here, is a constant pre-exponential factor, and EMN is the so-called Meyer–Neldel energy. In turn, Equation (4) expresses the Meyer–Neldel energy (E).
where is an isokinetic temperature determined by the Meyer–Neldel energy and represents a temperature at which all the processes of a certain series are insensitive to variation of the parameter [39]. All the above-mentioned correlations reflect the fundamental principle of physical organic chemistry—the linearity of the dependencies of free energies, LFER (a linear free-energy relationship) [40,41], or according IUPAC-Gibbs energy relation [42]. Mathematically, the LFER principle generalizes the correlation dependencies considered above as the relationship between enthalpy () and entropy () in the form of Equation (5):
In accordance with the recommendations of IUPAC (IUPAC Recommendations 1994), it was called an “isokinetic relationship”, or “compensation effect”, and parameter () was called the “isokinetic temperature”, i.e., = (see Equation (5)). In heterogeneous catalysis, this correlation is often referred to as “the kinetic compensation effect” [43].
The idea of the “kinetic compensation effect” was developed back in 1938 [44,45] and has been elaborated in a number of articles; especially noteworthy is the review by Koga [46]. Moreover, kinetic compensation effects were obtained analytically by Roura and Farjas [47], and particularly interesting is the work by Holba [48] and very recent work [49,50].
Quite often, relation Equation (5) is replaced (not arising analytically) by another Equation (6), which represents, in essence, the same:
where is the pre-exponential factor of the Arrhenius equation and is the Arrhenius energy.
The idea of a compensation of “entropy∼enthalpy” proved to be very fruitful for numerous situations related to chemistry, biology or, precisely, biochemical reactions, and materials science [51,52,53]. Biochemists perceived the presence of isokinetic correlations with particular enthusiasm, since the linear approximation vs. for complex biological systems offers a simple and successful interpretation of the mechanisms of interaction of reagents [54,55,56,57].
At the same time, the authors of many articles emphasize that it is necessary to distinguish correlations that are purely mathematical in nature and those that have a physical meaning [58,59]. However, a statistical analysis of numerous correlations has shown that experimental errors cannot be the basis of this phenomenon, and therefore, it can serve as a good tool for determining the nature of the processes that occur [60,61,62,63]. For interpreting the kinetic data of chemical reactions, it was suggested that the isokinetic relationship is a result of a certain energy resonance between the reacting molecules and their environments [41]. The isokinetic temperature can be interpreted as an active vibrational frequency of the surroundings of the reaction site. The corresponding frequency was found in the vibrational spectra of the solvent in the case of liquid-phase reactions [64,65,66]. In the case of heterogeneous reactions, the resonance interaction can occur between the quantum of the catalyst vibration and the quantum of the reactant vibration [67,68]. On all these occasions, the activation energy is expressed as a sum of the vibrational quanta of a vibrational mode that will bring the molecule to reaction [67]. It has been suggested that the observation of isokinetic relationships is evidence in favor of an identical reaction mechanism for a series of related reactions [69]. In the case of other temperature-dependent physical processes, the physical origin of the Meyer–Neldel rule is still not well explained. However, everyone agrees that there is an explanation, namely, the exponential distribution of activation barriers [15,70,71,72,73]. In this way, in spite of the fact that both the MNR and concept of the isokinetic relationship are widely used, the microscopic origin of the MNR and, therefore, the physical meaning of the EMN are still a topic of discussion in the literature [74,75,76]. Many authors look forward to further developments in this interesting and important area [77]. One of the steps in this direction is the proof of the existence of a basic theorem of the isokinetic relationship (vide infra).
2. Discussion
As can be concluded from the above review, many series of similar physical processes, including a series of chemical reactions of one and the same type, are described not only by exponential functions of the type of Equation (1). They also correspond to the concept of the “compensation effect” or, in particular, “isokinetic relations” in accordance with Equation (3). Here, it should be noted that the validity of this concept refers, apparently, to all physically temperature-dependent processes, or at least to all the elementary stages of any complex reaction mechanism. Generally, chemists talk about the isokinetic relation in terms of the importance of the isokinetic (or isoequilibrium) temperature, condensed matter physicists and material scientists use the Meyer–Neldel rule, and biochemists use the compensation effect or rule. For the convenience of understanding the further material, let us return to the more familiar form for chemists’ designation of the process characteristic—the reaction rate constant:
However, we will consider a more generalized case in which the functional dependence of this constant, k(T,), on temperature is expressed by Equation (1), and the pre-exponential factor is insensitive to temperature. This consideration covers all cases falling under the description of the Meyer–Neldel rule but is somewhat divergent from the Eyring–Polanyi transition state theory [78]. The dependence of the exponential factor on the temperatures can be neglected for several reasons. The general form of the Eyring–Polanyi equation resembles the Arrhenius equation:
where is the Gibbs energy of activation, is the transmission coefficient, is Boltzmann’s constant, and h is Planck’s constant. It should be noted that the transmittance (), the value of which is arbitrary, often evens out the influence of another cofactor—the temperature. Both cofactors are included in the pre-exponential coefficient of Equation (8) as one effective coefficient, which does not depend much on temperature. The main reason is that the Eyring–Polanyi theory is very difficult to apply to other (except for chemical kinetics) thermally activated processes.
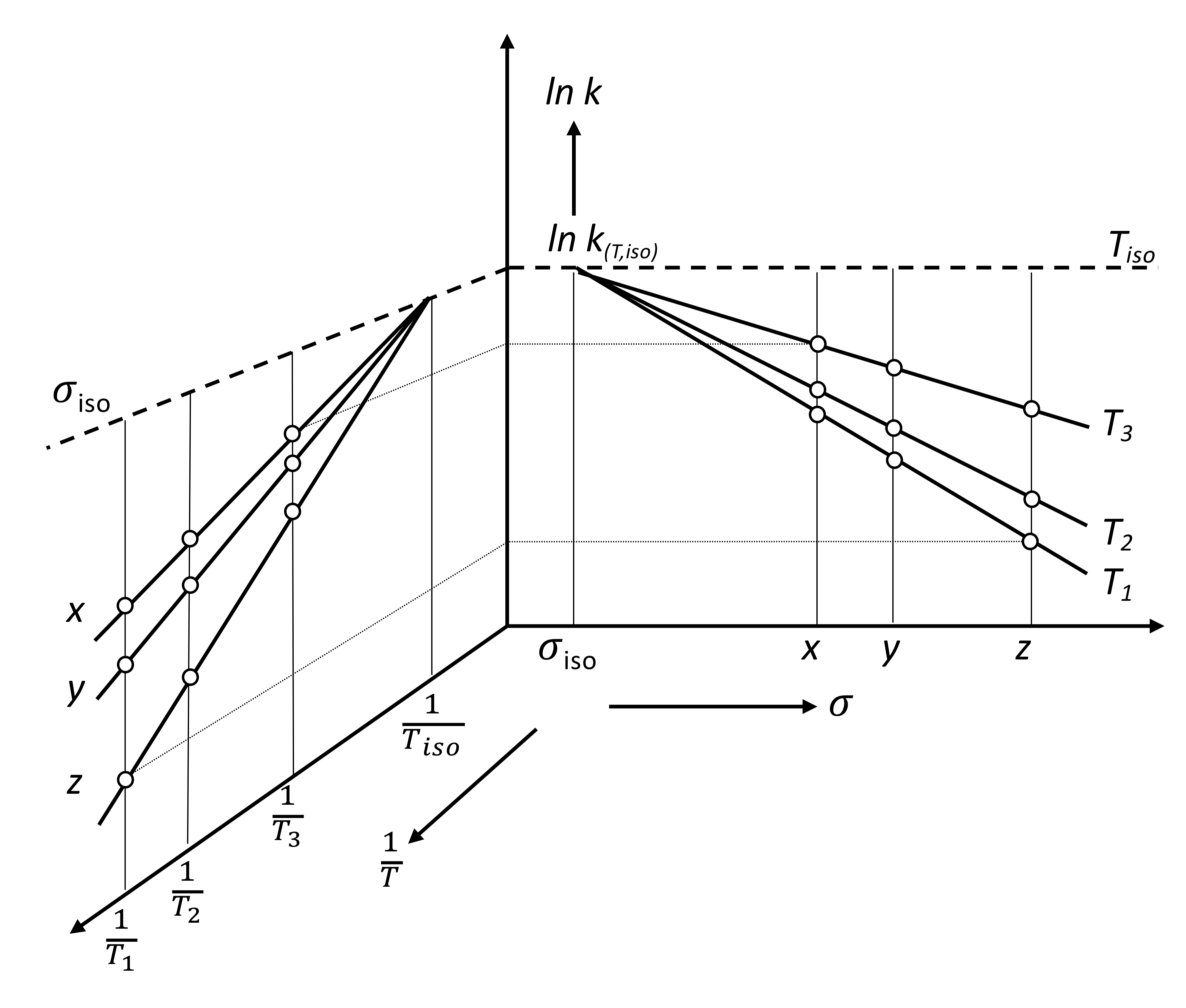
The two-parameter Equation (3) in logarithmic form can be represented graphically either by the temperature dependence of the slope of an “-parameter line” or by the -parameter dependence of the slope of an “Arrhenius line”. In the first case, we are talking about typical LFER correlations (“structure–properties”), such as Hammett diagrams, the influence of solvents and so on. In the second case, we refer to typical Arrhenius diagrams for a series of similar reactions with a variable parameter (). The relationship between these two representations is shown in Figure 1 using the form of an open-book double-diagram [79] (Figure 2 in [66]). Since both diagrams are based on the same data, the rate constant of the process for a certain parameter i and certain temperature , they can be considered as projections of a three-dimensional space figure in the corresponding two-dimensional planes.
It is obvious that the point of intersection of the lines on both plots reflects the same value and corresponds to the isokinetic case. The Arrhenius diagram (left-hand side plot) shows the intersection point, , when the values of the rate constants k(T, ) at are the same and are insensitive to parameter variation, (for example x, y, and z). The LFER-correlation plot (right-hand side) shows the intersection point when the values of the rate constants, , at are insensitive to temperature. In other words, for the process with the parameter, the Arrhenius activation energy is equal to zero. The slopes of the lines in the LFER diagrams are interpreted as a measure of the sensitivity of the process to variable parameters, , although their values characterize the closeness of the actual temperature of the process to the isokinetic temperature .
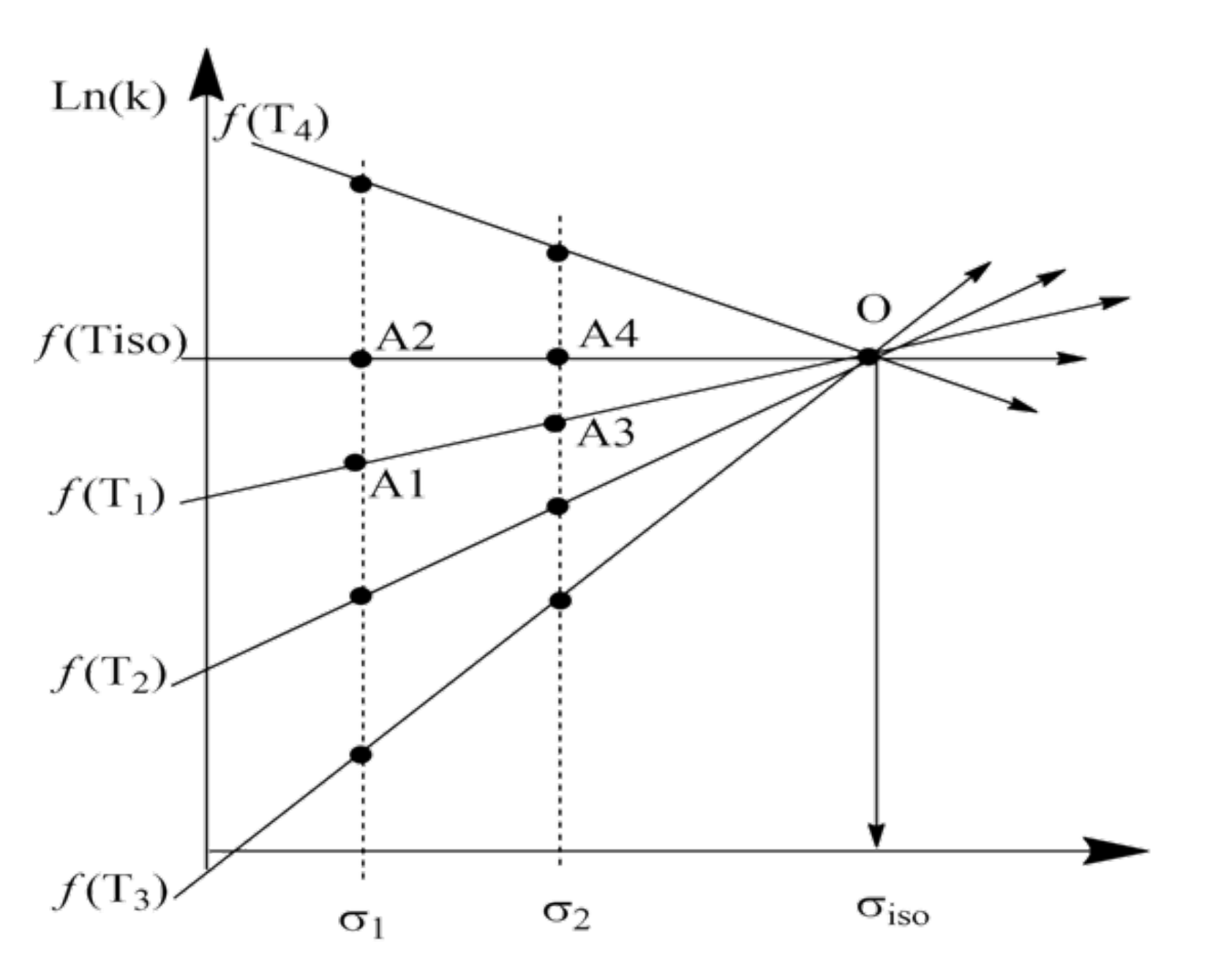
Usually, the experimental realization of the process at (or near) the isokinetic temperature () is not particularly difficult; then, the process with the isokinetic parameter () is most often impossible. In this case, we should use the theorem that says: “if there exists a linear correlation for “structure–properties” at two temperatures, the point of their intersection will be a common point for the same correlation at other temperatures, until the Arrhenius law is violated”. To prove the theorem, we consider the right-hand side plot in Figure 1 separately, which is a typical case of an LFER -correlation plot in the Arrhenius equation approximation presented in Figure 2.
This shows a plot of two conditional experimental dependencies, ) and ), of one series of experiments, obtained for the temperatures T and T, respectively. The abscissa reflects the characteristic values of the variable parameter in the process. The lines intersect at point [0], which corresponds to the parameter and the logarithm of the rate constants ) at any temperature T. According to the theorem, the experimental points obtained at other temperatures must be on straight lines (for example, straight lines or that also pass through point [0]).
To prove the theorem, we draw a horizontal line through the point [0] and consider two triangles, and . The tangent of the acute angle at the vertex (0) in this triangle is expressed by Equation (9):
If we now replace the letter designations with the corresponding parameters of the Arrhenius equation, we obtain Equation (10):
where and are the Arrhenius activation energies for the reactions at parameters and , respectively.
It is not difficult to verify that we obtain such equations when considering LFER -correlation plots for other temperatures (lines and so on). On the one hand, for any temperature, , we get the tangent of the angle of a straight line between two points located on verticals reconstructed from points and on the abscissa, respectively, which passes through point [0]. The horizontal line taken from the point [0] turns out to be , which corresponds to the process at and whose logarithm of rate constants does not depend on the parameter .
On the other hand, the point [0] corresponds to the logarithm of the rate constant ) for and T, and to the logarithms of the rate constants ) for and any temperature T.
The above discussion shows that all linear correlations of the same type of processes obtained at different temperatures will intersect at one point until the Arrhenius law is violated. The establishment of this fact can be regarded as a proof of the above theorem as a “basic theorem of isokinetic relationships”.
3. Some Consequences of the Theorem
3.1. Interpretation of Known Empirical Regularity
The main conclusion of the theorem can be considered the obtaining of equalities reflected by Equation (11). We see that there is an equality:
As the Arrhenius activation energy for the process with the parameter is equal to zero, the rate constant of the process is expressed by the pre-exponential factor only (Equation (12)):
where is the pre-exponential factor in the Arrhenius equation at parameter . Moreover, from Equation (11), we can derive a number of known empirical rules and laws. For example, from the combination of Equations (11) and (12), we can obtain:
This equation is easily transformed into a more familiar form of the relation (Equation (6)), often called the “kinetic compensation effect” [43]:
This equation can serve as a basis for the derivation of some solid empirical rules. If we add to two parts of Equation (14) the energy term , we obtain the following expression:
which can be easily transformed into the well-known Meyer–Neldel rule (MNR)
or
Alternatively, if Equation (14) is expressed in terms of Eyring–Polanyi, we obtain an expression often used to explain the concept of “multi-excitation entropy” [65,80,81]. Specifically, A. Yelon et al. argue that a large number of excitations may be collected in a large number of ways and that this multiplicity gives rise to entropy:
The authors have called the first term in Equation (18) a barrier entropy, and the second term, the multi-excitation entropy. Equation (14), expressed in terms of Eyring–Polanyi, practically coincides with the equation that A. Yelon proposed as follows:
We can shed light on the physical meaning of the terms in the Yelon Equation (18). The barrier entropy is the entropy of the process with the parameter when the Arrhenius activation energy is zero. In other words, any process passes without an energy supply from the outside if the value of its entropy has reached the barrier entropy. Otherwise, a process passes with energy feeding from the outside, and this energy must compensate for the lack of entropy to its baseline value (barrier entropy). Such feeding of energy is provided in the form of multi-excitation entropy, which is part of the activation entropy. As approaches , for any parameter , or as the parameter approaches , independently of the temperature, the multi-excitation entropy decreases to zero. Apparently, this is precisely the effect of the compensation “enthalpy-entropy”.
Thus, in the course of the proof of the theorem, we obtained a direct relationship with linear correlation dependencies reflecting the LFER (a linear free-energy relationship) principle and their energy parameters, the isokinetic relationship or compensation effect. In addition, the consequences of the theorem enable us to understand the physical meaning of certain empirical regularities. Equally interesting and useful in a practical sense is considering the regions diagram shown in Figure 2. It is about the patterns of change in the same type of processes in which the operating temperature (T) changes in regions before and after , or in which the parameter changes in regions before and after .
3.2. The Effect of the Temperature on the Impact of Variable Parameter
The first correlations that appeared in the scientific literature, as a rule, did not cover the temperature range or a range of process parameters (), close to either the T or to . With increasing interest, especially in the field of heterogeneous catalysis, the operating temperature ranges or parameters expanded and experiments appeared near or even on the other side of the “isokinetic” temperature, T [82,83]. Upon reaching the reaction temperature, T, the effect of the varied parameter level due to the mutual compensation of entropy (T) and enthalpy () in the free energy of activation () correspond to the process at T (Figure 2).
A further change in the reaction temperature leads to the fact that the contributions of these constituents of the free activation energy again become different. In this way, during the transition of operating temperatures through T occurs a complete inversion of the influence of the parameters (compare the lines of and in Figure 2). This results, for example, in the order of the influence of the substituents in pyridines, which catalyze the reaction of phenyloxirane with nitrobenzoic acid [84,85]. A similar fact was established when studying the activity of a number of heterogeneous catalysts [86] or enzymes [87]. Many authors consider such an inversion of the influence of the variable parameter as a change in the mechanism of the process, whereas in reality, this is only a “game” of thermodynamic activation parameters [88].
Here, we can formulate the following conclusion of the theorem: the order of the effect of the variable in a series of reactions of the same type reverses during the transition of the operating temperatures through T. We want to emphasize that this conclusion arises from a mathematical analysis of the obtained dependencies, assuming that they are preserved when passing through a point T. To date, there is almost no experimental evidence for this [84,85,86,87], and more experimental evidence is clearly needed.
3.3. The Effect of the Variable Parameter on the Free Energy of Activation
The revealed regularity of the changing of the order of the effect of variable is preserved for a given series of experiments up to . It should be noted that with this change in the effect of on the process, the activation energy decreases. These results indicate that the entropy difference between the transition and the ground states contributes significantly to the free energy of activation. In such cases, one can establish the decisive role of entropy in the free Gibbs activation energy G [89,90]. In most cases, this is established for biochemical processes, [91,92] or for heterogeneous catalytic processes [93].
3.4. The Negative Activation Energies
Another poorly understood area of the isokinetic relationship relates to experimental data relating to the study of the temperature effect on a series of reactions near the isokinetic parameter and on the other side of it (Figure 2). If the linearity of the correlation is maintained, the course of the correlation curves must correspond to a negative activation energy.
Reactions with negative activation enthalpy are generally considered to have a pre-equilibrium. In this case, the negative effect of temperature affects the position of the preliminary equilibrium with the formation of an intermediate. If the process of equilibrium formation of the intermediate is exothermic, then as the temperature rises, the concentration of the intermediate product decreases more than the rate of the main reaction, thus imitating the effect of a negative activation energy [94].
This case, apparently, cannot be considered, since the process speed is not determined by one exponential dependence, such as Equation (1), since the total activation energy is a function of three processes [95].
However, simple reactions with a negative activation energy (so-called anti-Arrhenius kinetics or anti-Arrhenius behavior [96]) are still possible and occur [97]. Such reactions are a consequence in the fields of radical chemistry and atmospheric science [98,99,100]. Often, they are considered barrier-free reactions [101,102]. The interpretation of the unusual temperature dependence requires considerations that extend beyond the purely enthalpic ones that are commonly presented in undergraduate text books; it is also necessary to take into account entropic factors that influence the fate of the reaction intermediate(s) [94,103].
It is believed that an elementary reaction may exhibit a negative enthalpy of activation if the reaction entropy is negative enough [104]. This situation is more common for enzymatic reactions, i.e., enzyme catalysis [105,106]. Anti-Arrhenian behavior is connected with the conformational structure of the protein, which promotes the contact during its fragments’ interaction [107]. In all cases, against the background of reducing the enthalpy of activation in the form of the Arrhenius energy and even the appearance of its negative value, the entropy component of the free energy of activation is a dominant factor in the implementation process.
To interpret the process, it is obviously not enough to consider it only in terms of thermal energy. It is necessary to consider the potential component—the change in the entropy of the process, i.e., creating the conditions that are most suitable for the necessary change in the geometry when the system goes from one state to another. An increase in temperature leads to a decrease in the probability of colliding molecules falling into each other, or to difficulty in forming new connections, expressed as the cross section of the process, which decreases with increasing temperature. This, apparently, is the basis for the emergence of such concepts as exergonic and endergonic processes. The names describe what happens with any form of free energy during the process, while endothermic and exothermic relate only to heat or thermal energy.
Summing up, it can be stated that the negative value of the activation entropy is inherent in processes when the temperature increases all the degrees of freedom while it does not allow accepting the necessary configuration for a barrier-free transition to a new state. To complete the process, the system must transmit some energy to the external environment, i.e., a typical exergonic process.
4. Outlook and Possible Applications
Although the presence of an isokinetic ratio or enthalpy∼entropy compensation along with the observation of the Meyer–Neldel rule has been observed for a long time in various regions of thermally activated processes, there is still no single model for this phenomenon. In each case, various plausible models have been proposed. The explanation of phenomena in terms of statistical thermodynamics did not lead to success [77,108].
The approach based on the exponential density distribution of states that induces a shift at the Fermi level is very general, but it cannot be a universal explanation for the MNR [109]. Among the explanations, the more acceptable idea is the notion of a certain energy resonance between the reacting molecules and their environment [41]. The idea brilliantly interprets the essence of the compensation effect in chemical reactions, but it is not suitable for explaining the temperature dependence of other processes, such as conductivity, glass crystallization, etc.
In the latter cases, many authors believe that the Meyer–Neldel compensation in these systems is a joint consequence of (1) the existence of localized states and (2) the energy dependence of the electron–phonon coupling [110] and is associated with a breach of any form in amorphous materials (glasses) because of their short-range structural orders [74,111].
In our opinion, the similarity of all the mentioned temperature-dependent processes and a universal explanation for the MNR must be based on the decisive role of the entropic factors of the process. All the cases of temperature-dependent processes and the observance of the MNR rule in them are ultimately connected with the geometric reorganization of the reacting system and the cost of this rearrangement of the corresponding energy from the outside. Summarizing what was said above, we established that in all cases, it is a question of mutual compensation for the change in entropy by an external source of energy.
For clarity and simplicity, let us consider chemical processes. Above, we obtained a generalized model of similar processes with a single parameter change. It has the form of Equation (17). The transformation of this model from the point of view of the Eyring–Polanyi theory can be represented in the form of the following equation:
where is the Gibbs free activation energy for a reaction at the temperature T and parameter ; is the Gibbs free activation energy for the reaction at parameter ; is the activation enthalpy for the reaction at parameter .
The last term on the right-hand side of Equation (21) is part of the total activation entropy of the process in terms of a Gibbs free activation energy , which depends on the parameter . We denote it as
Combining Equations (21) and (22) provides a very important result, which is that the Gibbs free activation energy of the process consists of two parts.
The first part in Equation (23) is defined as the free Gibbs activation energy of a barrier-free process at parameter , which remains constant for the entire series of processes with different parameters , and consists of only one activation entropy term as follows:
On the contrary, the second part is the variable unit of the Gibbs free activation energy of the process at the temperature T and parameter :
Here, both components (enthalpy and entropy) depend on the parameter , the effect of which reduces to a change in the pumping of energy from the outside.
On the other hand, it is easy to understand that the total activation entropy also consists of two parts, the first of which corresponds to the activation entropy and remains constant for the whole series of processes with parameters :
The compensation of entropy∼enthalpy (22) implies that the variable part of the entropy, i.e., , must be greater than zero. This corresponds to an increase in the degrees of freedom of the reaction system (an increase in the “disorder” of the system) during the formation of the activated complex. Apparently, this excess part of the activation entropy does not allow the reacting system to reach the reaction conditions. To increase the degree of order in the system, it is necessary to expend energy by introducing it from outside as an enthalpy of activation, and as much as necessary to compensate for the increased entropy.
The effectiveness of the parameter is reduced to a change in a variable part of entropy, that is, a change in the degrees of freedom of the system. When the process is performed with an isokinetic parameter (), the system acquires the required structure of the degrees of freedom and does not require additional energy (in the form of enthalpy), and Equation (25) becomes zero.
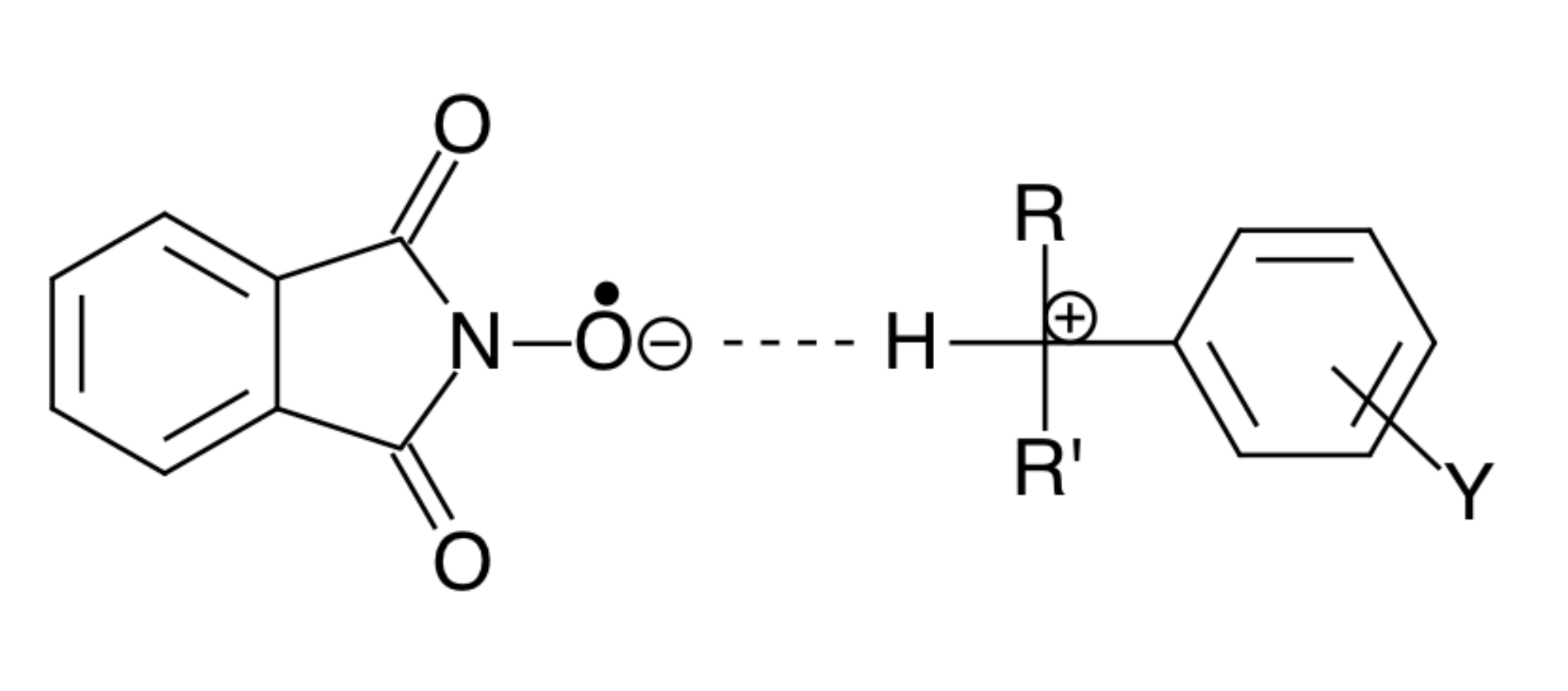
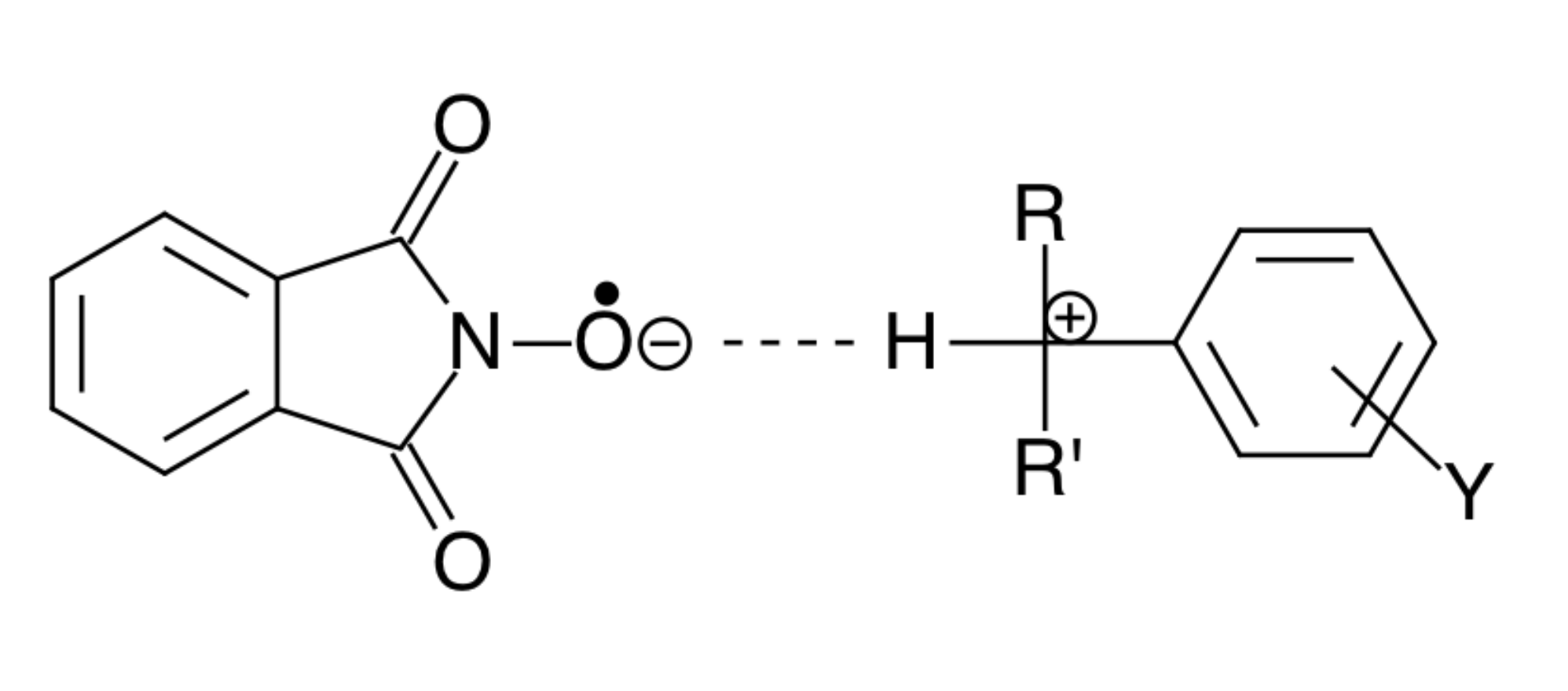
In other words, the normal vibrations of the reacting system coincide with the vibrations of the activated complex. In Figure 3, this is indicated for the example of a transition-state structure of the reaction between the radical PINO• and cumene.
As a result of this resonance, the amplitude of these oscillations increases sharply, and, since chemical bonds are weakened, conditions are created for the appearance of “imaginary” oscillations of the activated complex. This corresponds to conditions for the passage of the reaction system through the activation barrier without the activation energy.
The total free activation energy (Equation (17)) becomes equivalent to , which allows the process with zero activation energy to proceed. If the process is performed with the parameter , the system needs energy from outside. When the reaction temperature approaches the isokinetic temperature, the need for this disappears, and with T T, the effects of entropy and enthalpy are compensated for, and the free energy of activity becomes . At the isokinetic temperature, the complete compensation of the entropy and enthalpy takes place; that is, the second term in Equation (23) reduces to zero.
We have only considered the regularities that accompany a series of one-parametric chemical processes. However, the foregoing reasoning will remain the same when considering a series of other physicochemical or physical-temperature-dependent and one-parameter processes () if we consider that they are also determined by the change in the Gibbs free activation energy defined as follows:
is the pattern of a thermally activated process, k is the Boltzmann constant, and is a coefficient of proportionality.
is the Gibbs free activation energy for a process at the temperature T and parameter , in accordance with Equation (23).
For any such series of experiments, the effect of the parameter is reduced to the effect on the Gibbs free activation energy, changing the necessary pumping of energy from the outside. In this case, a situation (at least theoretical) is possible where the system ceases to need additional energy. In this case, the free activation energy consists only of the entropy term (Equation (24)). In all other cases, the energy is pumped through the variable part of the free energy , represented by Equation (25), for the process to proceed. When the isokinetic temperature is reached, with TT, the effects of entropy and enthalpy are compensated for, and the free energy of the activity becomes .
5. Conclusions
In this review, we discussed the universal applicability of Arrhenius’ law, as the key equation of the theory of the temperature dependence of the flow of various chemical and physical processes. We understand that the linearization of thermodynamic parameters can be carried out in a very restricted range, deviations being well studied and quantitatively accounted for once the temperature range confining the system is known [112]. Still, the regularities of the relationships of thermodynamic functions, even in narrow ranges, provide insight into various phenomena. These include the “tunnel effect”, where near the “isokinetic” parameter of the process, the course loses its dependence on temperature or there is change in the influence of process parameters in an opposite manner when passing a certain temperature ().
We believe that the idea of a correlation analysis of temperature-dependent processes will provide insight into various processes including electrochemical reactions, molecular interactions and specific interactions such as ligand/receptor binding. How the variable parameter changes will allow finding regions with the maximum or minimum effect of temperature on the process under study.
Author Contributions
Conceptualization, writing and draft preparation, review and editing was performed by all authors. All authors have read and agreed to the published version of the manuscript.
Funding
M.V. acknowledges support by the European Research Council (Grant: CSI.interface, ERC-StG 663677). The reported study was also funded by RFBR through the research project No. 18-29-24009.
Data Availability Statement
All data that is basis for this work will be made available upon resaonable request.
Acknowledgments
The authors thank Bianca S. Ranovsky (TU Wien) for supporting the drafting of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Silverman, M.P. Effects of a Periodic Decay Rate on the Statistics of Radioactive Decay: New Methods to Search for Violations of the Law of Radioactive Change. J. Mod. Phys. 2015, 6, 1533–1553. [Google Scholar] [CrossRef] [Green Version]
- Tarvainen, V.; Hakola, H.; Hellen, H.; Back, J.; Hari, P.; Kulmala, M. Temperature and light dependence of the VOC emissions of Scots pine. Atmos. Chem. Phys. 2005, 5, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Lubianiker, Y.; Balberg, I. A comparative study of the Meyer-Neldel rule in porous silicon and hydrogenated amorphous silicon. J. Non-Cryst. Solids 1998, 227, 180–184. [Google Scholar] [CrossRef]
- Yan, H.; Abdelhadi, M.M.; Jung, J.A.; Willemsen, B.A.; Kihlstrom, K.E. Exponential dependence of the vortex pinning potential on current density in high-T-c superconductors. Phys. Rev. B 2005, 72. [Google Scholar] [CrossRef]
- Mehta, N.; Kushwaha, V.S.; Kumar, A. Further Meyer-Neldel Rule for Thermally Activated High Field Conduction in Thin Films of Glassy Se100-xSbx Alloys. Int. J. Appl. Glass Sci. 2010, 1, 172–179. [Google Scholar] [CrossRef]
- Widenhorn, R.; Mundermann, L.; Rest, A.; Bodegom, E. Meyer-Neldel rule for dark current in charge-coupled devices. J. Appl. Phys. 2001, 89, 8179–8182. [Google Scholar] [CrossRef] [Green Version]
- Cooper, V.S.; Bennett, A.F.; Lenski, R.E. Evolution of thermal dependence of growth rate of Escherichia coli populations during 20,000 generations in a constant environment. Evolution 2001, 55, 889–896. [Google Scholar] [CrossRef]
- Englund, G.; Ohlund, G.; Hein, C.L.; Diehl, S. Temperature dependence of the functional response. Ecol. Lett. 2011, 14, 914–921. [Google Scholar] [CrossRef]
- Simon, P. Single-step kinetics approximation employing non-Arrhenius temperature functions. J. Therm. Anal. Calorim. 2005, 79, 703–708. [Google Scholar] [CrossRef]
- Šimon, P.; Thomas, P.; Dubaj, T.; Cibulková, Z.; Peller, A.; Veverka, M. The mathematical incorrectness of the integral isoconversional methods in case of variable activation energy and the consequences. J. Therm. Anal. Calorim. 2014, 115, 853–859. [Google Scholar] [CrossRef]
- Šimon, P.; Dubaj, T.; Cibulková, Z. Equivalence of the Arrhenius and non-Arrhenian temperature functions in the temperature range of measurement. J. Therm. Anal. Calorim. 2015, 120, 231–238. [Google Scholar] [CrossRef]
- Maitra, S.; Bandyopadhyay, N.; Pal, J. Application of Non-Arrhenius Method for Analyzing the Decomposition Kinetics of SrCO3 and BaCO3. J. Am. Ceram. Soc. 2008, 91, 337–341. [Google Scholar] [CrossRef]
- Chen, H.; Liu, N. Application of Non-Arrhenius Equations in Interpreting Calcium Carbonate Decomposition Kinetics: Revisited. J. Am. Ceram. Soc. 2009, 93, 548–553. [Google Scholar] [CrossRef]
- Dubaj, T.; Cibulková, Z.; Šimon, P. An incremental isoconversional method for kinetic analysis based on the orthogonal distance regression. J. Comput. Chem. 2015, 36, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Li, J.V.; Johnston, S.W.; Yan, Y.; Levi, D.H. Measuring temperature-dependent activation energy in thermally activated processes: A 2D Arrhenius plot method. Rev. Sci. Instrum. 2010, 81. [Google Scholar] [CrossRef] [Green Version]
- Bronsted, J.N.; Pedersen, K. The catalytic disintegration of nitramide and its physical-chemical relevance. Z. Phys. Chem. Stochiom. Verwandtschaftslehre 1924, 108, 185–235. [Google Scholar]
- Hammett, L.P. Some relations between reaction rates and equilibrium constants. Chem. Rev. 1935, 17, 125–136. [Google Scholar] [CrossRef]
- Chapman, N.B.; Shorter, J. (Eds.) Advances in Linear Free Energy Relationships; Springer: New York, NY, USA, 1972. [Google Scholar] [CrossRef]
- Palm, V.A. Structure and reactivity of organic compounds (quantitative laws). Russ. Chem. Rev. 1961, 30, 471–498. [Google Scholar] [CrossRef]
- Koppel, A.; Palm, V. The Influence of the Solvent on Organic Reactivity. In Advances in Linear Free Energy Relationships; Chapman, N.B., Shorter, J., Eds.; Springer: New York, NY, USA, 1972. [Google Scholar]
- Swain, C.G.; Swain, M.S.; Powell, A.L.; Alunni, S. Solvent effects on chemical-reactivity-evaluation of anion and cation solvation components. J. Am. Chem. Soc. 1983, 105, 502–513. [Google Scholar] [CrossRef]
- Edwards, J.O. Correlation of relative rates and equilibria with a double basicity scale. J. Am. Chem. Soc. 1954, 76, 1540–1547. [Google Scholar] [CrossRef]
- Grunwald, E.; Winstein, S. The correlation of solvolysis rates. J. Am. Chem. Soc. 1948, 70, 846–859. [Google Scholar] [CrossRef]
- Winstein, S.; Grunwald, E.; Jones, H.W. The correlation of solvolysis rates and the classification of solvolysis reactions into mechanistic categories. J. Am. Chem. Soc. 1951, 73, 2700–2707. [Google Scholar] [CrossRef]
- Bronsted, J.N. Einige Bemerkungen uber den Begriff der Sauren und Basen. Recl. Trav. Chim. Pays-Bas 2010, 42, 718–728. [Google Scholar] [CrossRef]
- Taft, R.W. Linear steric energy relationships. J. Am. Chem. Soc. 1953, 75, 4538–4539. [Google Scholar] [CrossRef]
- Taft, R.W. Linear free energy relationships from rates of esterification and hydrolysis of aliphatic and ortho-substituted benzoate esters. J. Am. Chem. Soc. 1952, 74, 2729–2732. [Google Scholar] [CrossRef]
- Taft, R.W. Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters. J. Am. Chem. Soc. 1952, 74, 3120–3128. [Google Scholar] [CrossRef]
- Swain, C.G.; Lupton, E.C. Field and resonance components of substituent effects. J. Am. Chem. Soc. 1968, 90, 4328. [Google Scholar] [CrossRef]
- Yukawa, Y.; Tsuno, Y. Resonance effect in hammett relationship. 2. sigma constants in electrophilic reactions and their intercorrelation. Bull. Chem. Soc. Jpn. 1959, 32, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Barclay, I.M.; Butler, J.A.V. The entropy of solution. Trans. Faraday Soc. 1938, 34, 1445–1454. [Google Scholar] [CrossRef]
- Schwab, G.M. About the mechanism of contact catalysis. Adv. Catal. 1950, 2, 251–267. [Google Scholar] [CrossRef]
- Manche, E.P.; Carroll, B. Compensation and the smith-topley effects. Thermochim. Acta 1979, 31, 387–390. [Google Scholar] [CrossRef]
- Bond, G.C.; Keane, M.A.; Kral, H.; Lercher, J.A. Compensation phenomena in heterogeneous catalysis: General principles and a possible explanation. Catal. Rev. Sci. Eng. 2000, 42, 323–383. [Google Scholar] [CrossRef]
- Bligaard, T.; Honkala, K.; Logadottir, A.; Norskov, J.K.; Dahl, S.; Jacobsen, C.J.H. On the compensation effect in heterogeneous catalysis. J. Phys. Chem. B 2003, 107, 9325–9331. [Google Scholar] [CrossRef]
- Meyer, W.; Neldel, H. Concerning the relationship between the energy constant epsilon and the quantum constant alpha in the conduction-temperature formula in oxydising semi-conductors. Phys. Z. 1937, 38, 1014–1019. [Google Scholar]
- Meijer, E.J.; Matters, M.; Herwig, P.T.; de Leeuw, D.M.; Klapwijk, T.M. The Meyer-Neldel rule in organic thin-film transistors. Appl. Phys. Lett. 2000, 76, 3433–3435. [Google Scholar] [CrossRef]
- Yelon, A.; Movaghar, B.; Branz, H.M. Origin and consequences of the compensation (meyer-neldel) law. Phys. Rev. B 1992, 46, 12244–12250. [Google Scholar] [CrossRef] [PubMed]
- Exner, O. Determination of isokinetic temperature. Nature 1970, 227, 366. [Google Scholar] [CrossRef]
- Wold, S.; Sjostrom, M. Linear free-energy relationships-local empirical rules-or fundamental laws of chemistry-a reply to kamlet and taft. Acta Chem. Scand. Ser. B-Org. Chem. Biochem. 1986, 40, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Guo, Q.X. Isokinetic relationship, isoequilibrium relationship, and enthalpy-entropy compensation. Chem. Rev. 2001, 101, 673–695. [Google Scholar] [CrossRef]
- Pure, I.U. Chemical Terminology. In Gold Book, 2.3.1 ed.; IUPAC: Guaruja, Brazil, 2012; Available online: https://goldbook.iupac.org/ (accessed on 15 March 2021).
- Zsako, J. Kinetic compensation effect. J. Therm. Anal. 1976, 9, 101–108. [Google Scholar] [CrossRef]
- Zawadzki, J.; Bretsznajder, S. Some remarks on the mechanism of reactions of the type: Solid=solid + gas. Trans. Faraday Soc. 1938, 34, 951–959. [Google Scholar] [CrossRef]
- Šesták, J. Errors of kinetic data obtained from thermogravimetric curves at increasing temperature. Talanta 1966, 13, 567–579. [Google Scholar] [CrossRef]
- Koga, N. A review of the mutual dependence of Arrhenius parameters evaluated by the thermoanalytical study of solid-state reactions: The kinetic compensation effect. Thermochim. Acta 1994, 244, 1–20. [Google Scholar] [CrossRef]
- Farjas, J.; Roura, P. Exact analytical solution for the Kissinger equation: Determination of the peak temperature and general properties of thermally activated transformations. Thermochim. Acta 2014, 598, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Holba, P. Temperature dependence of activation energy of endothermic processes and related imperfections of non-isothermal kinetic evaluations. J. Therm. Anal. Calorim. 2017, 129, 609–614. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, N. Kinetic analysis based on the kinetic compensation effect and optimization calculation. Thermochim. Acta 2020, 690, 178686. [Google Scholar] [CrossRef]
- Mianowski, A.; Radko, T.; Siudyga, T. Kinetic compensation effect of isoconversional methods. React. Kinet. Mech. Catal. 2021, 132, 37–58. [Google Scholar] [CrossRef]
- Bel’skii, V.E. Isokinetic relationships for nucleophilic substitution reactions at the saturated carbon atom. Reactions in aqueous solutions. Russ. Chem. Bull. 2000, 49, 806–811. [Google Scholar] [CrossRef]
- Freed, K.F. Entropy-Enthalpy Compensation in Chemical Reactions and Adsorption: An Exactly Solvable Model. J. Phys. Chem. B 2011, 115, 1689–1692. [Google Scholar] [CrossRef]
- Naffakh, M.; Marco, C.; Gomez, M.A.; Jimenez, I. Unique Nucleation Activity of Inorganic Fullerene-like WS2 Nanoparticles in Polyphenylene Sulfide Nanocomposites: Isokinetic and Isoconversional Study of Dynamic Crystallization Kinetics. J. Phys. Chem. B 2009, 113, 7107–7115. [Google Scholar] [CrossRef]
- Starikov, E.B.; Norden, B. Entropy-enthalpy compensation may be a useful interpretation tool for complex systems like protein-DNA complexes: An appeal to experimentalists. Appl. Phys. Lett. 2012, 100. [Google Scholar] [CrossRef] [Green Version]
- Perez-Galvez, A.; Jaren-Galan, M.; Minguez-Mosquera, M.I. Effect of high-temperature degradative processes on ketocarotenoids present in paprika oleoresins. J. Agric. Food Chem. 2000, 48, 2966–2971. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Ueoka, R. Studies on thermodynamics for hydrolysis. 3. isokinetic temperature related to molecular location of reactants in coaggregates. J. Org. Chem. 1990, 55, 5797–5799. [Google Scholar] [CrossRef]
- Loaiza, A.; Armstrong, K.M.; Baker, B.M.; Abu-Omar, M.M. Kinetics of thermal unfolding of phenylalanine hydroxylase variants containing different metal cofactors (Fe-II, Co-II, and Zn-II) and their lsokinetic relationship. Inorg. Chem. 2008, 47, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- McBane, G.C. Chemistry from telephone numbers: The false isokinetic relationship. J. Chem. Educ. 1998, 75, 919–922. [Google Scholar] [CrossRef]
- Barrie, P.J. The mathematical origins of the kinetic compensation effect: 1. the effect of random experimental errors. Phys. Chem. Chem. Phys. 2012, 14, 318–326. [Google Scholar] [CrossRef]
- Koudriavtsev, A.B.; Linert, W. Do Experimental Errors Really Cause Isoequilibrium and Isokinetic Relationships? Match-Commun. Math. Comput. Chem. 2013, 70, 7–28. [Google Scholar]
- Linert, W.; Yelon, A. Isokinetic relationships preface. Monatshefte Chem. 2013, 144, 1–2. [Google Scholar] [CrossRef]
- Ouvrard, C.; Berthelot, M.; Lamer, T.; Exner, O. A program for linear regression with a common point of intersection: The isokinetic relationship. J. Chem. Inf. Comput. Sci. 2001, 41, 1141–1144. [Google Scholar] [CrossRef]
- Perez-Benito, J.F.; Mulero-Raichs, M. Enthalpy-Entropy Compensation Effect in Chemical Kinetics and Experimental Errors: A Numerical Simulation Approach. J. Phys. Chem. A 2016, 120, 7598–7609. [Google Scholar] [CrossRef]
- Linert, W.; Sapunov, V.N. The isokinetic relationship. 9. connections to linear free-energy relationships. Chem. Phys. 1988, 119, 265–274. [Google Scholar] [CrossRef]
- Yelon, A.; Movaghar, B.; Crandall, R.S. Multi-excitation entropy: Its role in thermodynamics and kinetics. Rep. Prog. Phys. 2006, 69, 1145–1194. [Google Scholar] [CrossRef]
- Pinheiro, L.M.V.; Calado, A.R.T.; Reis, J.C.R. The quaternisation reaction of phosphines and amines in aliphatic alcohols. A similarity analysis using the isokinetic, isosolvent and isoselective relationships. Org. Biomol. Chem. 2004, 2, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Larsson, R. Concluding remarks on the theory of selective energy transfer and exemplification on a zeolite kinetics study. Monatshefte Chem. 2013, 144, 21–28. [Google Scholar] [CrossRef]
- Bratlie, K.M.; Flores, L.D.; Somorjai, G.A. In situ sum frequency generation vibrational spectroscopy observation of a reactive surface intermediate during high-pressure benzene hydrogenation. J. Phys. Chem. B 2006, 110, 10051–10057. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.E. The enthalpy-entropy relationship and its implications for organic chemistry. J. Org. Chem. 1955, 20, 1202–1231. [Google Scholar] [CrossRef]
- Crandall, R.S. Defect relaxation in amorphous-silicon-stretched exponentials, the meyer-neldel rule, and the staebler-wronski effect. Phys. Rev. B 1991, 43, 4057–4070. [Google Scholar] [CrossRef]
- Wang, J.C.; Chen, Y.F. The Meyer-Neldel rule in fullerenes. Appl. Phys. Lett. 1998, 73, 948–950. [Google Scholar] [CrossRef]
- Yelon, A.; Movaghar, B. Microscopic explanation of the compensation (meyer-neldel) rule. Phys. Rev. Lett. 1990, 65, 618–620. [Google Scholar] [CrossRef]
- Dyre, J.C. A phenomenological model for the meyer-neldel rule. J. Phys. C-Solid State Phys. 1986, 19, 5655–5664. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, K. Observation of Meyer Neldel rule and crystallization rate constant stability for Se93-xZn2Te5Inx chalcogenide glasses. Eur. Phys. J. Appl. Phys. 2010, 51. [Google Scholar] [CrossRef]
- Rong, X.; Parolin, J.; Kolpak, A.M. A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution. ACS Catal. 2016, 6, 1153–1158. [Google Scholar] [CrossRef]
- Singh, K.; Mehta, N.; Sharma, S.K.; Kumar, A. Crystallization kinetics of glassy Se90In10-xAgx alloys: Observation of Mayer-Neldel rule. Process. Appl. Ceram. 2016, 10, 137–142. [Google Scholar] [CrossRef]
- Pan, A.; Biswas, T.; Rakshit, A.K.; Moulik, S.P. Enthalpy-Entropy Compensation (EEC) Effect: A Revisit. J. Phys. Chem. B 2015, 119, 15876–15884. [Google Scholar] [CrossRef]
- Laidler, K.J.; King, M.C. The development of transition-state theory. J. Phys. Chem. 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Schmid, R.; Sapunov, V. Non-Formal Kinetics Search of Chemical Reaction Pathways. In Monographs in Modern Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1982; Volume 14, p. 172. [Google Scholar]
- Yelon, A.; Sacher, E.; Linert, W. Multi-Excitation Entropy, Entropy-Enthalpy Relations, and their Impact on Catalysis. Catal. Lett. 2011, 141, 954–957. [Google Scholar] [CrossRef]
- Movileanu, L.; Schiff, E.A. Entropy-enthalpy compensation of biomolecular systems in aqueous phase: A dry perspective. Monatshefte Chem. 2013, 144, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhang, Y.; Xu, W. Single-Molecule Nanocatalysis Reveals Catalytic Activation Energy of Single Nanocatalysts. J. Am. Chem. Soc. 2016, 138, 12414–12421. [Google Scholar] [CrossRef]
- Karunakaran, C.; Chidambaranathan, V. Linear free energy relationships near isokinetic temperature. Oxidation of organic sulfides with nicotinium dichromate. Croat. Chem. Acta 2001, 74, 51–59. [Google Scholar]
- Shpan’ko, I.V.; Sadovaya, I.V. Isoparametricity paradox in the pyridine-catalyzed reaction of phenyloxirane with 3-nitrobenzoic acid. Russ. J. Phys. Chem. A 2013, 87, 1955–1959. [Google Scholar] [CrossRef]
- Shpan’ko, I.V.; Sadovaya, I.V. Phenomenon of isoparametricity in reactions of trans-2,3-diaryloxiranes with arenesulfonic acids. Mechanistic interpretation. Russ. J. Gen. Chem. 2016, 86, 2418–2422. [Google Scholar] [CrossRef]
- Ma, H.; Na, C. Isokinetic Temperature and Size-Controlled Activation of Ruthenium-Catalyzed Ammonia Borane Hydrolysis. Acs Catal. 2015, 5, 1726–1735. [Google Scholar] [CrossRef]
- Sun, D.; Wicker, L. Kinetic compensation and the role of cations in pectinesterase catalysis. J. Agric. Food Chem. 1999, 47, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Vlasov, V.M. Energetics of bimolecular nucleophilic reactions in solution. Uspekhi Khimii 2006, 75, 851–883. [Google Scholar] [CrossRef]
- Armstrong, A.A.; Amzel, L.M. Role of entropy in increased rates of intramolecular reactions. J. Am. Chem. Soc. 2003, 125, 14596–14602. [Google Scholar] [CrossRef]
- Sohtome, Y.; Shin, B.; Horitsugi, N.; Takagi, R.; Noguchi, K.; Nagasawa, K. Entropy-Controlled Catalytic Asymmetric 1,4-Type Friedel-Crafts Reaction of Phenols Using Conformationally Flexible Guanidine/Bisthiourea Organocatalyst. Angew. Chem. Int. Ed. 2010, 49, 7299–7303. [Google Scholar] [CrossRef] [PubMed]
- Laskin, J.; Futrell, J.H. Entropy is the major driving force for fragmentation of proteins and protein-ligand complexes in the gas phase. J. Phys. Chem. A 2003, 107, 5836–5839. [Google Scholar] [CrossRef]
- Strajbl, M.; Sham, Y.Y.; Villa, J.; Chu, Z.T.; Warshel, A. Calculations of activation entropies of chemical reactions in solution. J. Phys. Chem. B 2000, 104, 4578–4584. [Google Scholar] [CrossRef]
- Van der Mynsbrugge, J.; Janda, A.; Sharada, S.M.; Lin, L.C.; Van Speybroeck, V.; Head-Gordon, M.; Bell, A.T. Theoretical Analysis of the Influence of Pore Geometry on Monomolecular Cracking and Dehydrogenation of n-Butane in Bronsted Acidic Zeolites. ACS Catal. 2017, 7, 2685–2697. [Google Scholar] [CrossRef] [Green Version]
- Revell, L.E.; Williamson, B.E. Why Are Some Reactions Slower at Higher Temperatures? J. Chem. Educ. 2013, 90, 1024–1027. [Google Scholar] [CrossRef]
- Hadel, L.M.; Maloney, V.M.; Platz, M.S.; McGimpsey, W.G.; Scaiano, J.C. The absolute kinetics of several reactions of substituted diphenylcarbenes. J. Phys. Chem. 1986, 90, 2488–2491. [Google Scholar] [CrossRef]
- Aquilanti, V.; Coutinho, N.D.; Carvalho-Silva, V.H. Kinetics of low-temperature transitions and a reaction rate theory from non-equilibrium distributions. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2017, 375. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, N.D.; Silva, V.H.C.; de Oliveira, H.C.B.; Camargo, A.J.; Mundim, K.C.; Aquilanti, V. Stereodynamical Origin of Anti-Arrhenius Kinetics: Negative Activation Energy and Roaming for a Four-Atom Reaction. J. Phys. Chem. Lett. 2015, 6, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Varlamov, V.T.; Denisov, N.N.; Nadtochenko, V.A. Negative activation energies and compensation effects for the reactions of diarylaminyl radicals with phenols. Russ. Chem. Bull. 1995, 44, 2282–2286. [Google Scholar] [CrossRef]
- Bowman, D.F.; Gillan, T.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. 3. self-reactions of dialkyl nitroxide radicals. J. Am. Chem. Soc. 1971, 93, 6555. [Google Scholar] [CrossRef]
- Adamic, K.; Bowman, D.F.; Gillan, T.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. 1. self-reactions of diethyl nitroxide radicals. J. Am. Chem. Soc. 1971, 93, 902. [Google Scholar]
- Zavitsas, A.A. Energy barriers to chemical reactions. Why, how, and how much? non-arrhenius behavior in hydrogen abstractions by radicals. J. Am. Chem. Soc. 1998, 120, 6578–6586. [Google Scholar] [CrossRef]
- Li, J.L.; Geng, C.Y.; Huang, X.R.; Sun, C.C. A barrier-free atomic radical-molecule reaction: F + propene. J. Chem. Theory Comput. 2006, 2, 1551–1564. [Google Scholar] [CrossRef]
- Ge, Y. Agreement, Complement, and Disagreement to “Why Are Some Reactions Slower at Higher Temperatures?”. J. Chem. Educ. 2017, 94, 821–823. [Google Scholar] [CrossRef]
- Kapinus, E.I.; Rau, H. Negative enthalpies of activation and isokinetic relationships in the electron transfer quenching reaction of PD-tetraphenylporphyrin by aromatic nitro compounds and quinones. J. Phys. Chem. A 1998, 102, 5569–5576. [Google Scholar] [CrossRef]
- Carlos Aledo, J.; Jimenez-Riveres, S.; Tena, M. The Effect of Temperature on the Enzyme-Catalyzed Reaction: Insights from Thermodynamics. J. Chem. Educ. 2010, 87, 296–298. [Google Scholar] [CrossRef]
- Silverstein, T. The Nuts and Bolts of Michaelis-Menten Enzyme Kinetics: Suggestions and Clarifications. J. Chem. Educ. 2011, 88, 167–168. [Google Scholar] [CrossRef]
- Zheng, W.; Andrec, M.; Gallicchio, E.; Levy, R.M. Simulating replica exchange simulations of protein folding with a kinetic network model. Proc. Natl. Acad. Sci. USA 2007, 104, 15340–15345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starikov, E.B. Valid Entropy-Enthalpy Compensation: Its True Physical-Chemical Meaning. J. Appl. Solut. Chem. Model. 2013, 2, 240–245. [Google Scholar] [CrossRef]
- Widenhorn, R.; Rest, A.; Bodegom, E. The Meyer-Neldel rule for a property determined by two transport mechanisms. J. Appl. Phys. 2002, 91, 6524–6528. [Google Scholar] [CrossRef] [Green Version]
- Abtew, T.A.; Zhang, M.; Pan, Y.; Drabold, D.A. Electrical conductivity and Meyer-Neldel rule: The role of localized states in hydrogenated amorphous silicon. J. Non-Cryst. Solids 2008, 354, 2909–2913. [Google Scholar] [CrossRef]
- Kushwaha, N.; Mehta, N.; Shukla, R.K.; Kumar, D.; Kumar, A. Observation of Meyer-Neldel N rule in amorphous Se100-xTex thin films. J. Optoelectron. Adv. Mater. 2005, 7, 2293–2298. [Google Scholar]
- Carvalho-Silva, V.H.; Coutinho, N.D.; Aquilanti, V. From the Kinetic Theory of Gases to the Kinetics of Rate Processes: On the Verge of the Thermodynamic and Kinetic Limits. Molecules 2020, 25, 2098. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Temperature dependence of a well-behaved linear free-energy relationship (LFER). Left-hand side: Arrhenius plot. Introduction of the isokinetic parameter (iso) would make the specific rate, k(iso), insensitive to temperature (upper broken line). Right-hand side: -correlation plot. At the isokinetic temperature, , the specific rate, , is insensitive to parameter variation. Figure is adapted from R. Schmid et al. [79].
Figure 1.
Temperature dependence of a well-behaved linear free-energy relationship (LFER). Left-hand side: Arrhenius plot. Introduction of the isokinetic parameter (iso) would make the specific rate, k(iso), insensitive to temperature (upper broken line). Right-hand side: -correlation plot. At the isokinetic temperature, , the specific rate, , is insensitive to parameter variation. Figure is adapted from R. Schmid et al. [79].
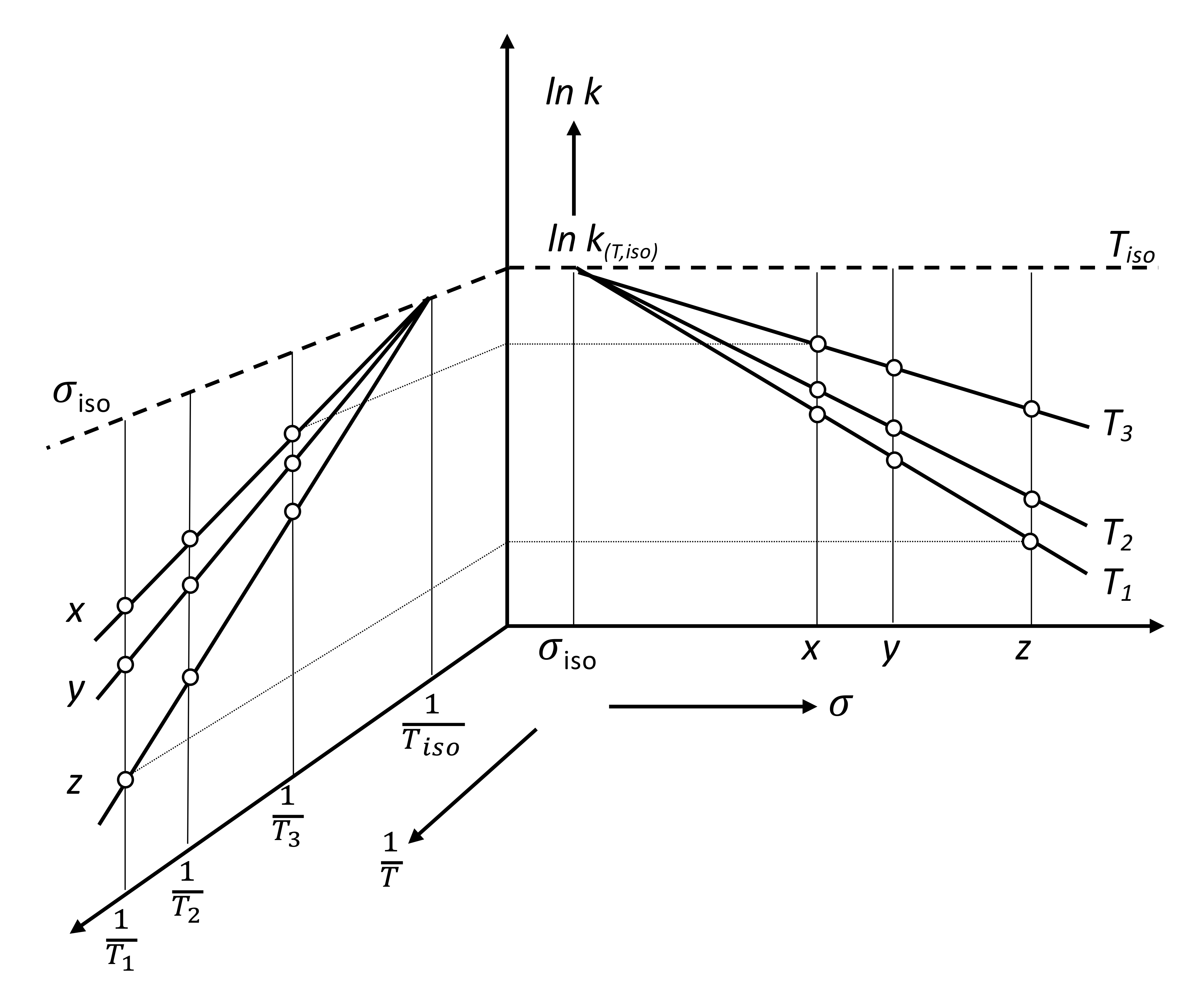
Figure 2.
The LFER-correlation plots of f(T1) to f(T4) and f(T) of one series of conditional experiments with variable parameters in the temperature range T to T, including T, respectively. Groups of points A, A, and A, A correspond to processes with parameters and at temperatures of T and T.
Figure 2.
The LFER-correlation plots of f(T1) to f(T4) and f(T) of one series of conditional experiments with variable parameters in the temperature range T to T, including T, respectively. Groups of points A, A, and A, A correspond to processes with parameters and at temperatures of T and T.

Figure 3.
Resonance structure of the activated complex of a reaction of the radical PINO• and cumene.
Figure 3.
Resonance structure of the activated complex of a reaction of the radical PINO• and cumene.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sapunov, V.N.; Saveljev, E.A.; Voronov, M.S.; Valtiner, M.; Linert, W. The Basic Theorem of Temperature-Dependent Processes. Thermo 2021, 1, 45-60. https://0-doi-org.brum.beds.ac.uk/10.3390/thermo1010004
AMA Style
Sapunov VN, Saveljev EA, Voronov MS, Valtiner M, Linert W. The Basic Theorem of Temperature-Dependent Processes. Thermo. 2021; 1(1):45-60. https://0-doi-org.brum.beds.ac.uk/10.3390/thermo1010004
Chicago/Turabian StyleSapunov, Valentin N., Eugene A. Saveljev, Mikhail S. Voronov, Markus Valtiner, and Wolfgang Linert. 2021. "The Basic Theorem of Temperature-Dependent Processes" Thermo 1, no. 1: 45-60. https://0-doi-org.brum.beds.ac.uk/10.3390/thermo1010004