
Toll-Like Receptor Signalling Pathways Regulate Hypoxic Stress Induced Fibroblast Growth Factor but Not Vascular Endothelial Growth Factor-A in Human Microvascular Endothelial Cells

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Normoxia and Hypoxia Treatment
2.3. Inhibition of Key Signal Transduction Pathways
2.4. Cell Viability Assay
2.5. Total RNA Isolation
2.6. Reverse Transcription into Complementary DNA (cDNA)
2.7. Quantitative Polymerase Chain Reaction (qPCR)
2.8. Flow Cytometry
2.9. ELISA
2.10. Protein Estimation for Immunoblotting
2.11. Western Blot
2.12. Data Analysis
3. Results
3.1. Effect of Hypoxia on HMEC-1 Viability
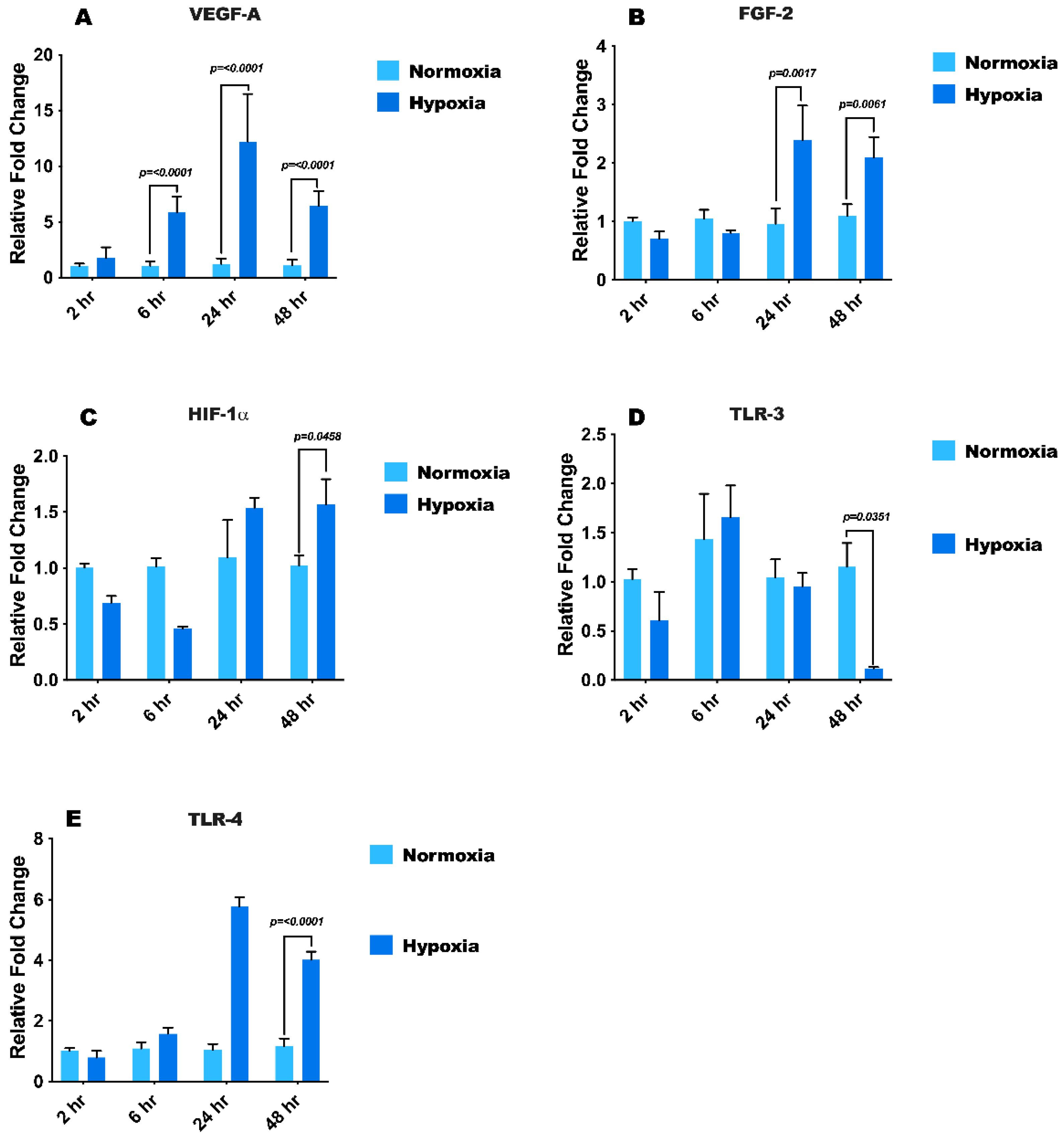
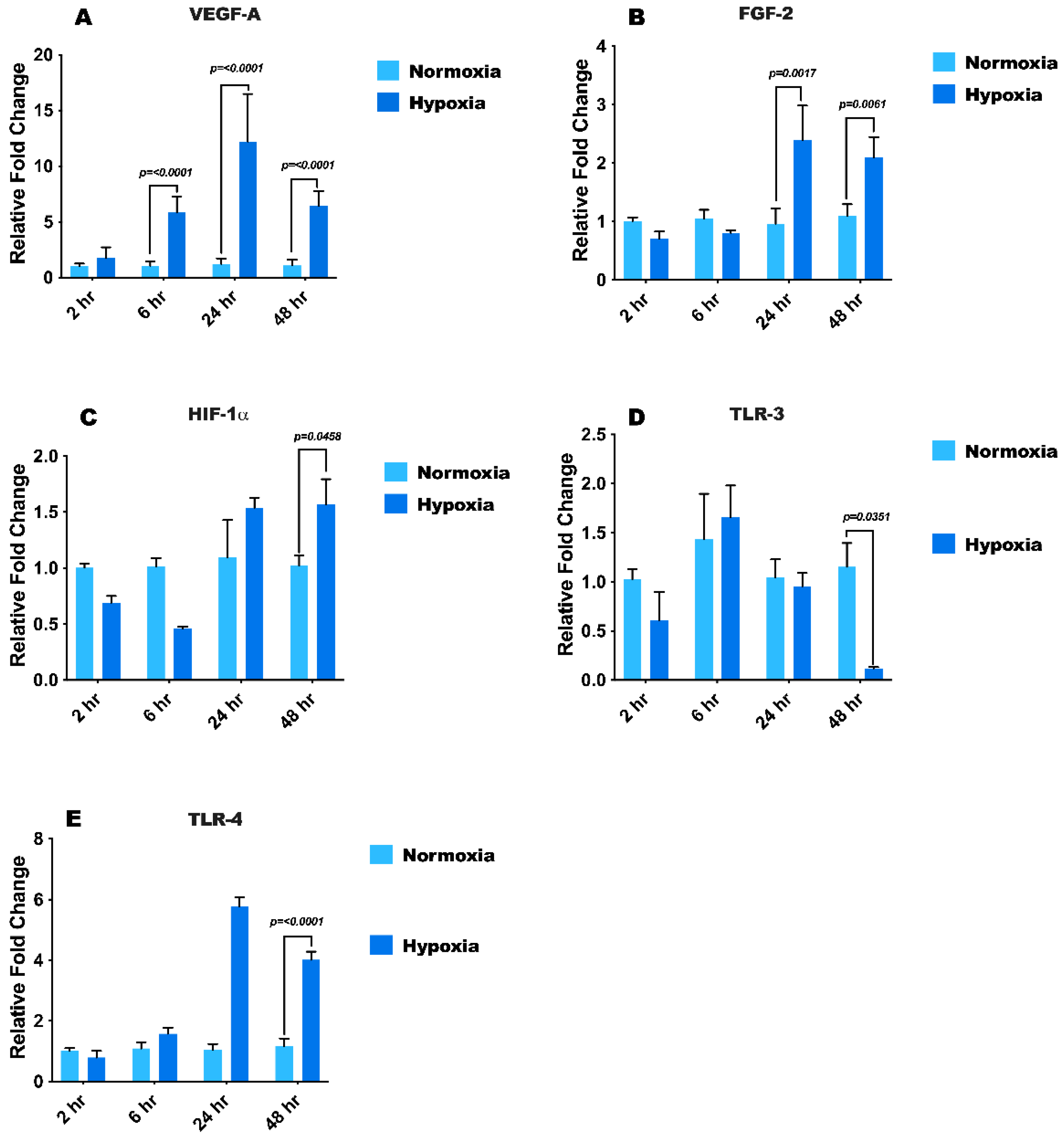
3.2. Hypoxia Treatment Modulates mRNA Expression of Growth Factors, HIF1-α, and Toll-Like Receptors
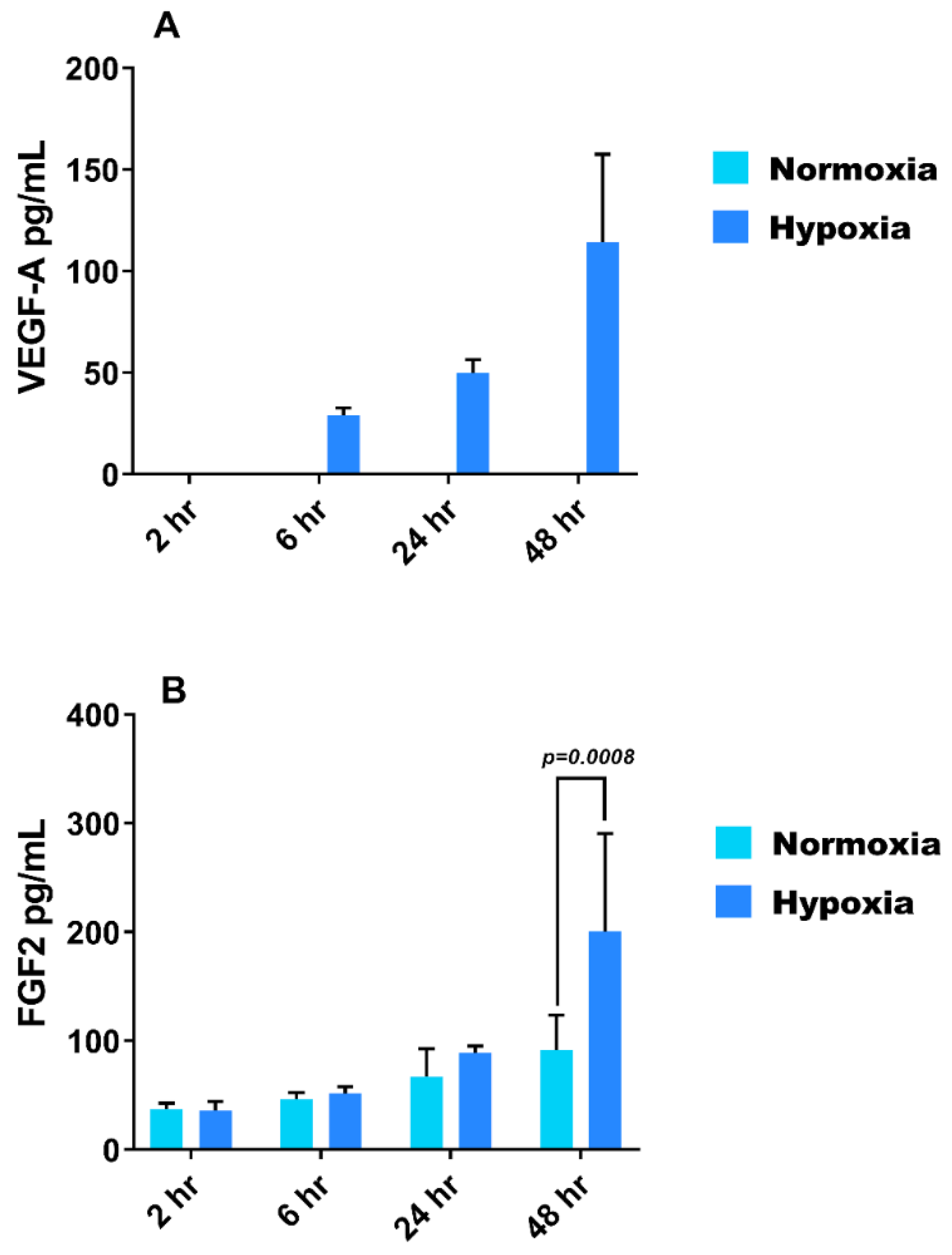
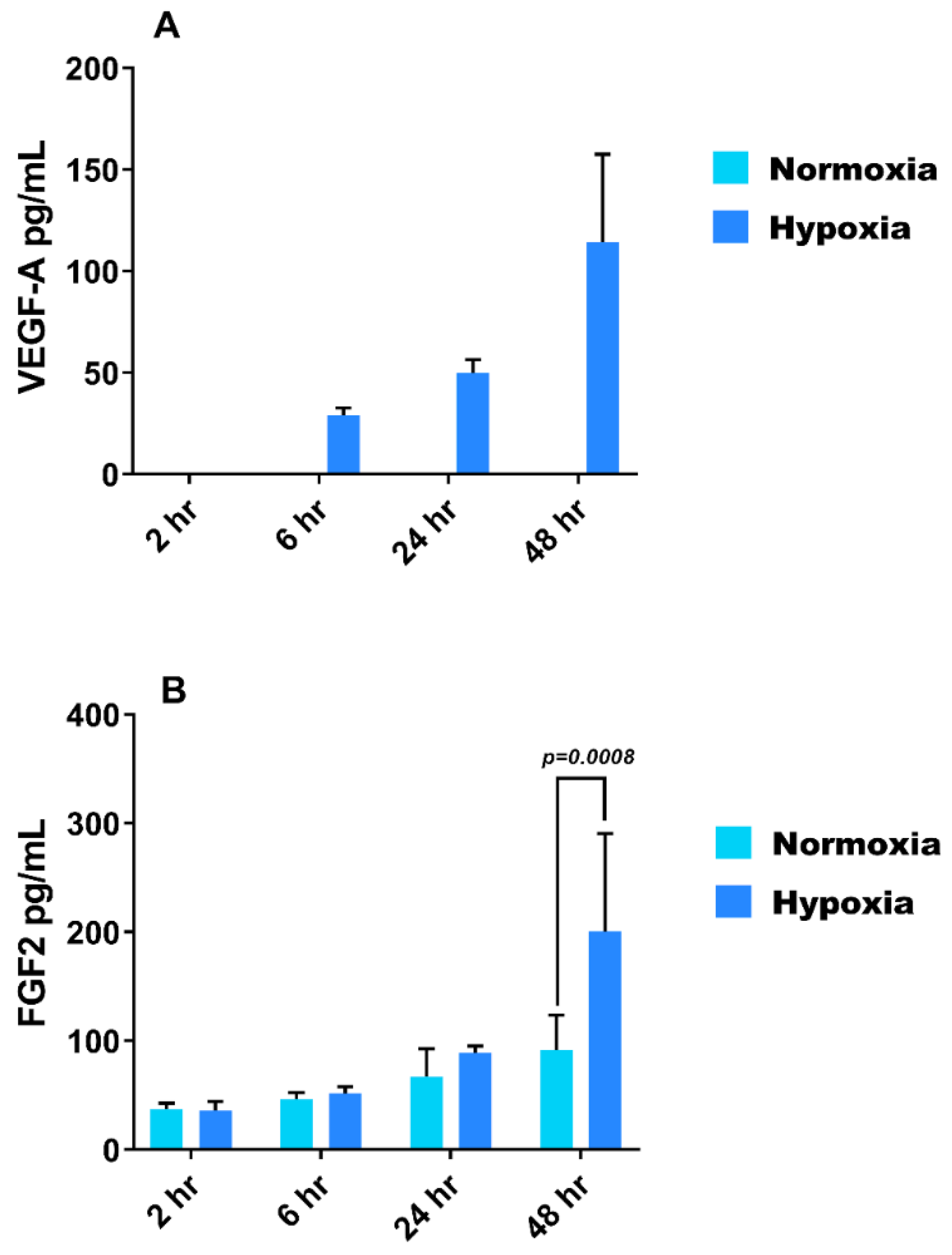
3.3. Effect of Hypoxia on VEGF-A and FGF-2 Secretion from HMEC-1
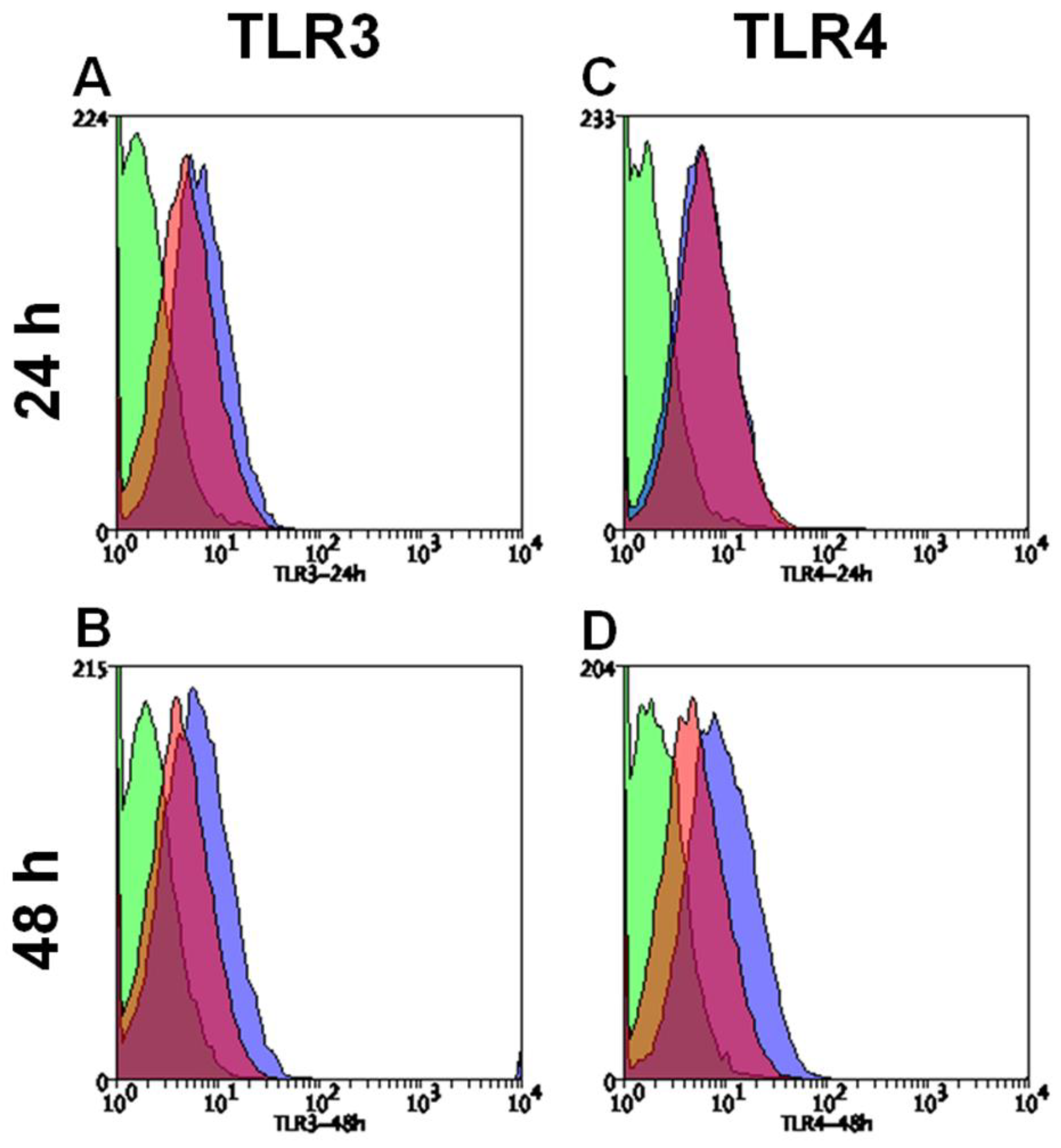
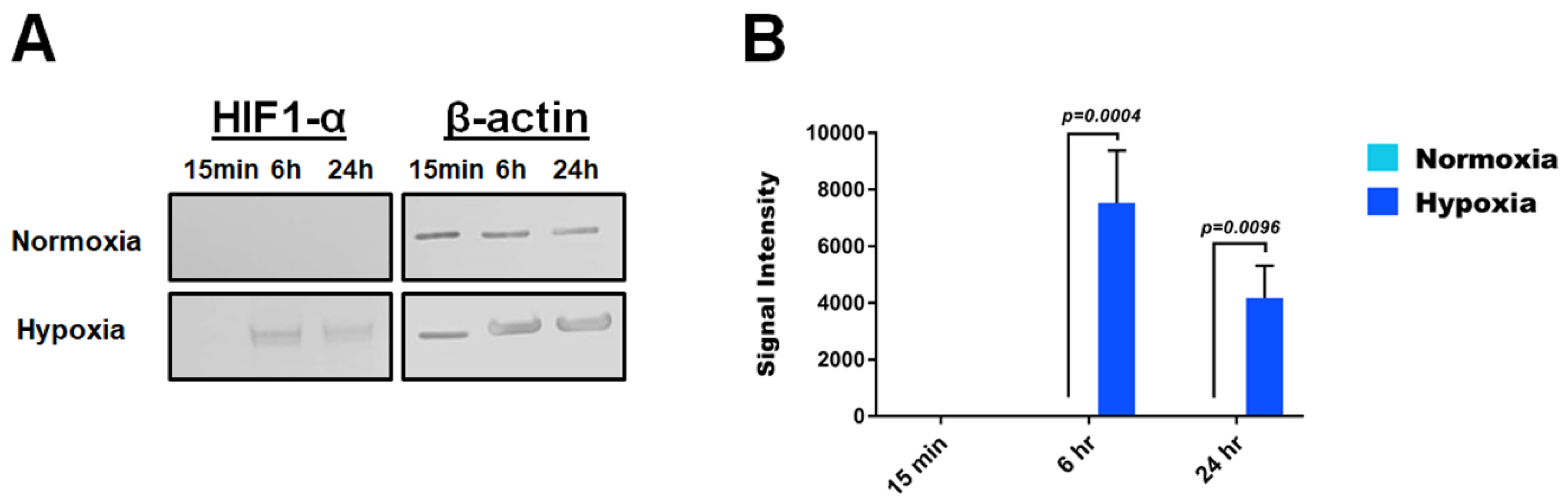
3.4. Hypoxia Modulates Protein Expression of TLR3, TLR4 and HIF1-α in HMEC-1
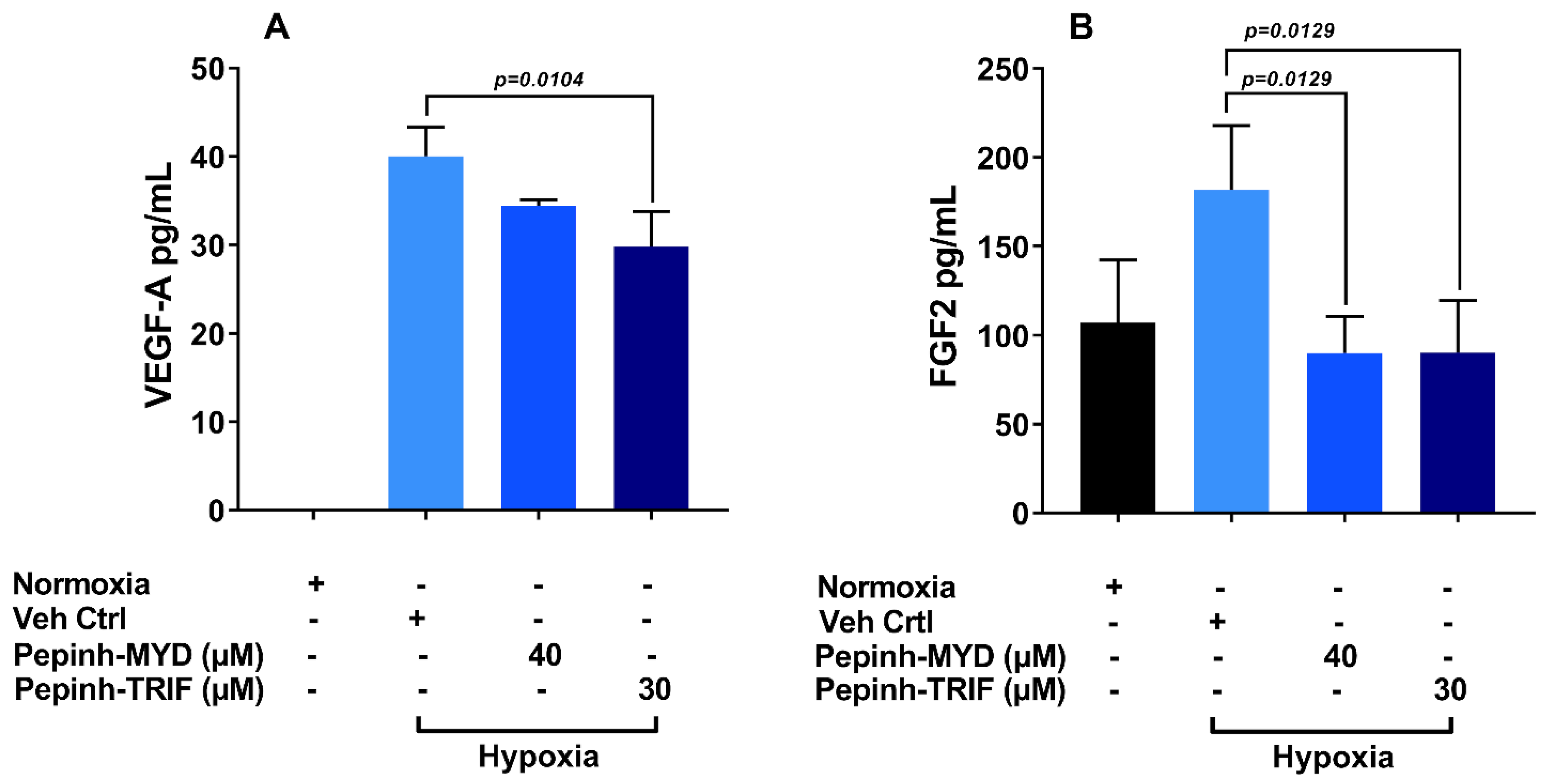
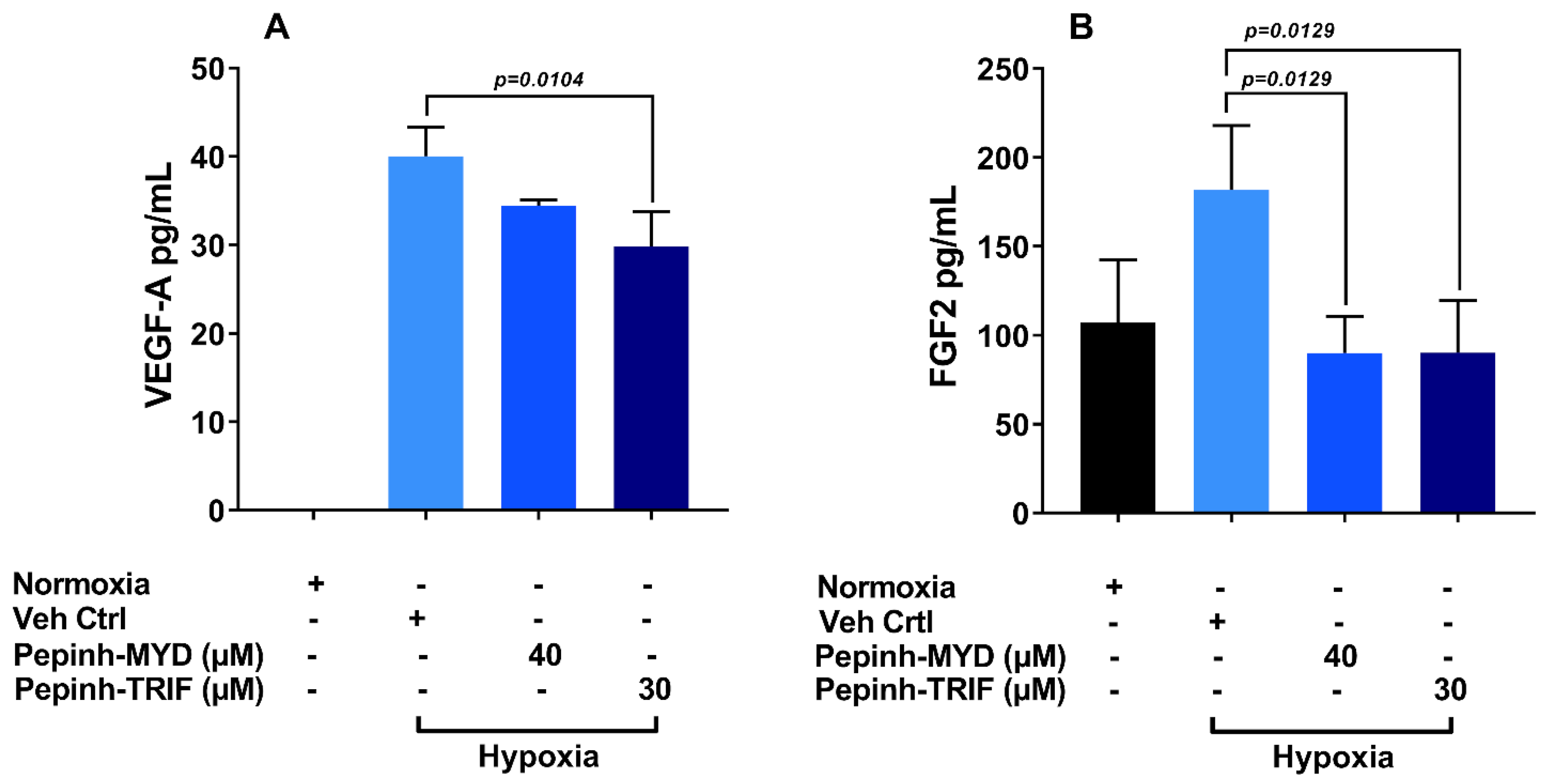
3.5. TRIF Activation Pathway Plays a Key Role in Induction of VEGF-A and FGF-2 under Hypoxia
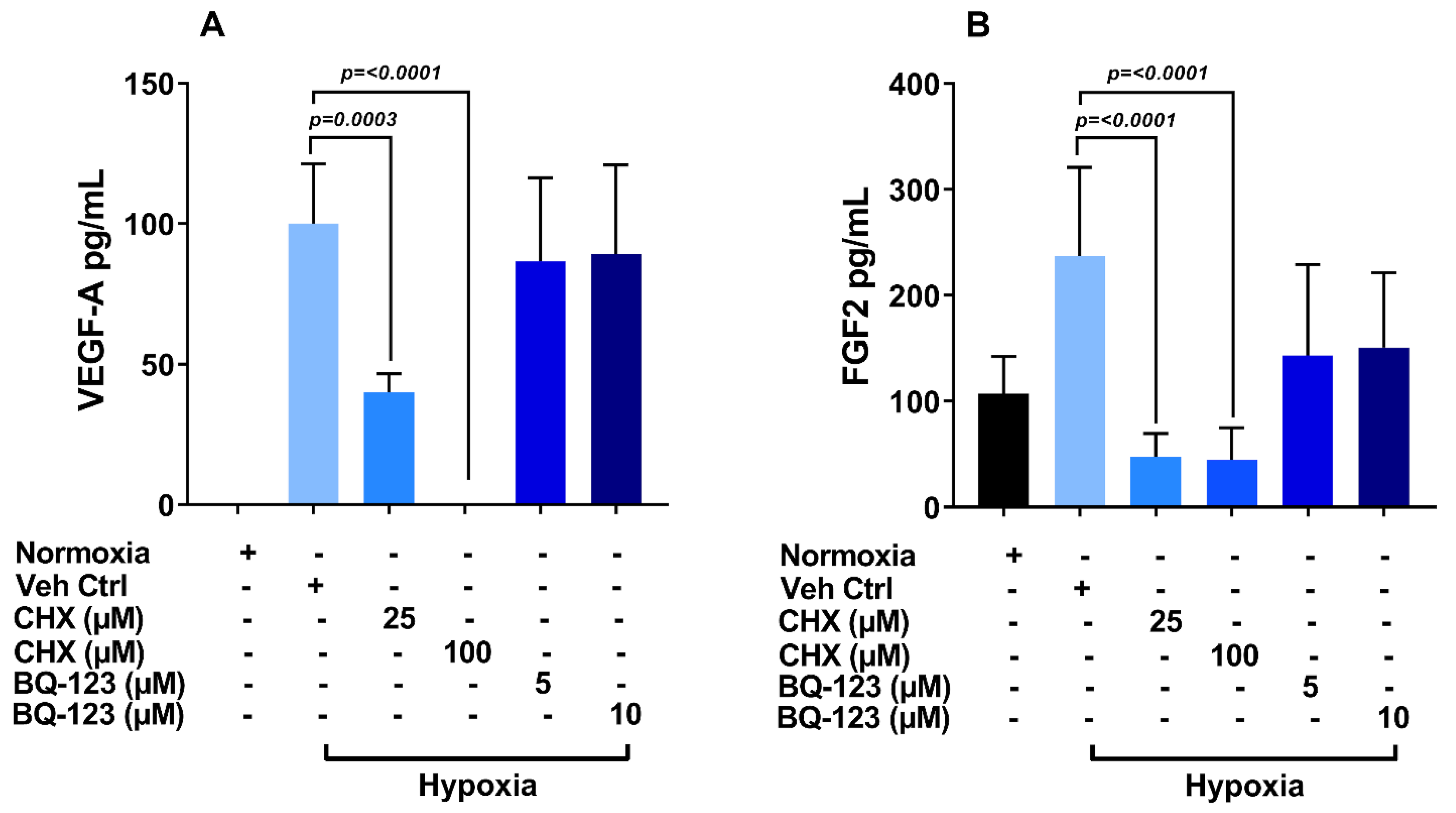
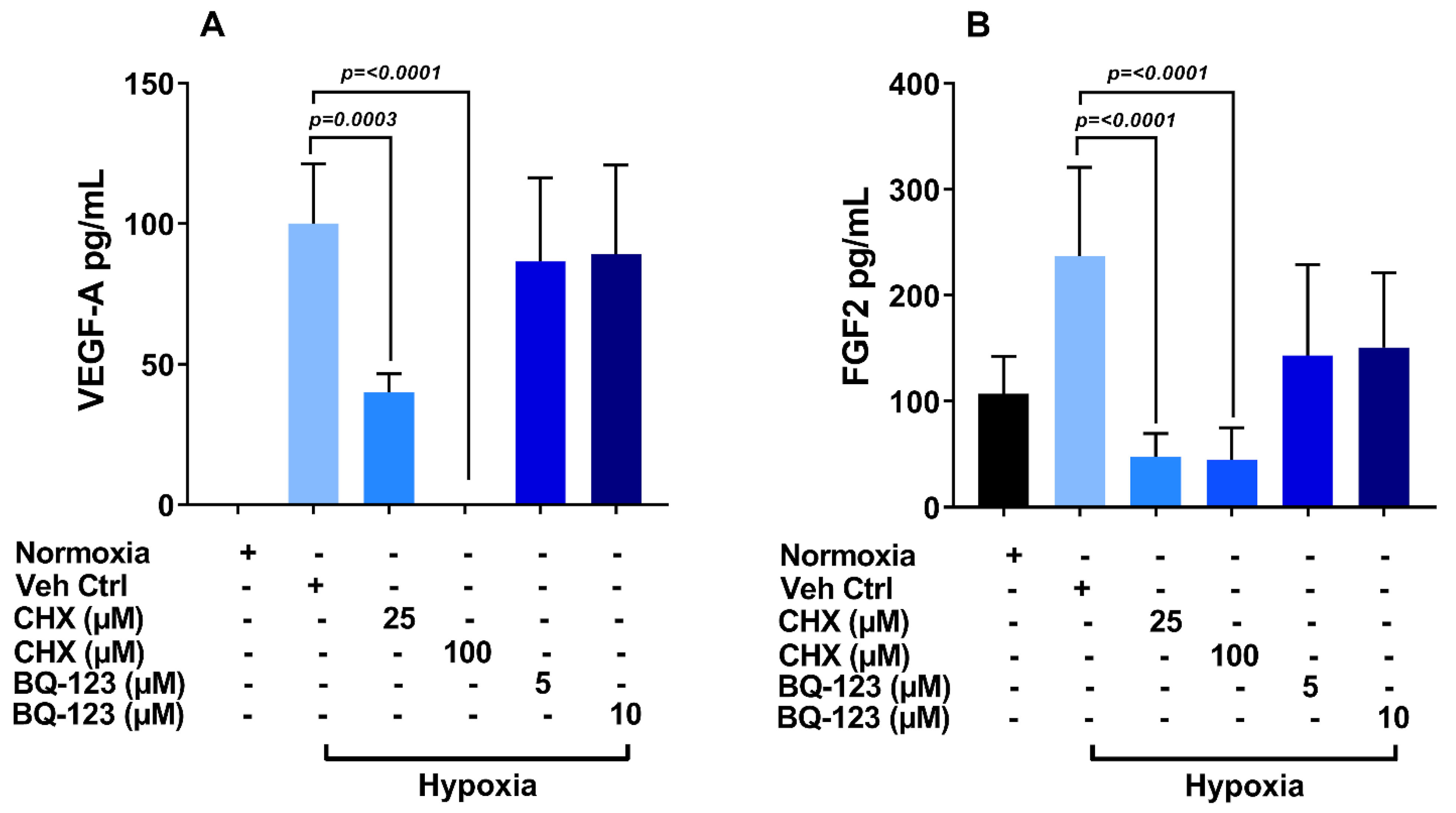
3.6. Role of Endothelin Receptor in Regulation of VEGF-A and FGF-2 Production
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheong, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. BioMed Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Ioanna, Z.; Christian, S.; Christian, G.; Daniel, B. Plasma levels of hypoxia-regulated factors in patients with age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Spinella, F.; Caprara, V.; Di Castro, V.; Rosanò, L.; Cianfrocca, R.; Natali, P.G.; Bagnato, A. Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J. Mol. Med. 2013, 91, 395–405. [Google Scholar] [CrossRef]
- Totan, Y.; Koca, C.; Erdurmus, M.; Keskin, U.; Yigitoglu, R. Endothelin-1 and Nitric Oxide Levels in Exudative Age-Related Macular Degeneration. J. Ophthalmic Vis. Res. 2015, 10, 151–154. [Google Scholar]
- Stewart, E.A.; Wei, R.; Branch, M.J.; Sidney, L.E.; Amoaku, W.M. Expression of Toll-like receptors in human retinal and choroidal vascular endothelial cells. Exp. Eye Res. 2015, 138, 114–123. [Google Scholar] [CrossRef]
- Yi, H.; Patel, A.K.; Sodhi, C.P.; Hackam, D.J.; Hackam, A.S. Novel role for the innate immune receptor Toll-like receptor 4 (TLR4) in the regulation of the Wnt signaling pathway and photoreceptor apoptosis. PLoS ONE 2012, 7, e36560. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liang, L.; Qian, D.; Yu, H.; Yang, P.; Lei, B. Increase in peripheral blood mononuclear cell Toll-like receptor 2/3 expression and reactivity to their ligands in a cohort of patients with wet age-related macular degeneration. Mol. Vis. 2013, 19, 1826. [Google Scholar]
- Kumar, M.V.; Nagineni, C.N.; Chin, M.S.; Hooks, J.J.; Detrick, B. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J. Neuroimmunol. 2004, 153, 7–15. [Google Scholar] [CrossRef]
- Klettner, A.; Koinzer, S.; Meyer, T.; Roider, J. Toll-like receptor 3 activation in retinal pigment epithelium cells—Mitogen-activated protein kinase pathways of cell death and vascular endothelial growth factor secretion. Acta Ophthalmol. 2013, 91, e211–e218. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.G.; Albuquerque, R.J.; Kleinman, M.E.; Tarallo, V.; Greco, A.; Nozaki, M.; Green, M.G.; Baffi, J.Z.; Ambati, B.K.; De Falco, M.; et al. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc. Natl. Acad. Sci. USA 2009, 106, 7137–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.K.; Hackam, A.S. Toll-like receptor 3 (TLR3) protects retinal pigmented epithelium (RPE) cells from oxidative stress through a STAT3-dependent mechanism. Mol. Immunol. 2013, 54, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Stratton, C.; Francis, P.J.; Kleinman, M.E.; Tan, P.L.; Gibbs, D.; Tong, Z.; Chen, H.; Constantine, R.; Yang, X.; et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N. Engl. J. Med. 2008, 359, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Taylor, B.; Ourthiague, D.R.; Hoffmann, A. Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci. Signal 2015, 8, ra69. [Google Scholar] [CrossRef] [Green Version]
- Dvoriantchikova, G.; Barakat, D.J.; Hernandez, E.; Shestopalov, V.I.; Ivanov, D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol. Vis. 2010, 16, 1907–1912. [Google Scholar]
- Xu, W.Q.; Wang, Y.S. The role of Toll-like receptors in retinal ischemic diseases. Int. J. Ophthalmol. 2016, 9, 1343–1351. [Google Scholar]
- Kindzelskii, A.L.; Elner, V.M.; Elner, S.G.; Yang, D.; Hughes, B.A.; Petty, H.R. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J. Gen. Physiol. 2004, 124, 139–149. [Google Scholar] [CrossRef]
- Zareparsi, S.; Buraczynska, M.; Branham, K.E.; Shah, S.; Eng, D.; Li, M.; Pawar, H.; Yashar, B.M.; Moroi, S.E.; Lichter, P.R.; et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2005, 14, 1449–1455. [Google Scholar]
- Dua, H.S.; Otri, A.M.; Hopkinson, A.; Mohammed, I. In vitro studies on the antimicrobial peptide human beta-defensin 9 (HBD9): Signalling pathways and pathogen-related response (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2014, 112, 50–73. [Google Scholar] [PubMed]
- Mohammed, I.; Kulkarni, B.; Faraj, L.A.; Abbas, A.; Dua, H.S.; King, A.J. Profiling ocular surface responses to preserved and non-preserved topical glaucoma medications: A 2-year randomized evaluation study. Clin. Exp. Ophthalmol. 2020, 48, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Yeung, A.; Abedin, A.; Hopkinson, A.; Dua, H.S. Signalling pathways involved in ribonuclease-7 expression. Cell. Mol. Life Sci. 2011, 68, 1941–1952. [Google Scholar] [CrossRef]
- Spinella, F.; Caprara, V.; Cianfrocca, R.; Rosanò, L.; Di Castro, V.; Garrafa, E.; Natali, P.G.; Bagnato, A. The interplay between hypoxia, endothelial and melanoma cells regulates vascularization and cell motility through endothelin-1 and vascular endothelial growth factor. Carcinogenesis 2014, 35, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Choi, Y.J.; Joung, S.M.; Lee, B.H.; Jung, Y.S.; Lee, J.Y. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 2010, 129, 516–524. [Google Scholar] [CrossRef]
- Crifo, B.; Taylor, C.T. Crosstalk between toll-like receptors and hypoxia-dependent pathways in health and disease. J. Investig. Med. 2016, 64, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spirig, R.; Djafarzadeh, S.; Regueira, T.; Shaw, S.G.; von Garnier, C.; Takala, J.; Jakob, S.M.; Rieben, R.; Lepper, P.M. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS ONE 2010, 5, e0010983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, A.; Galli, R.; Gabellini, C.; Lukashev, D.; Starace, D.; Gorlach, A.; De Cesaris, P.; Ziparo, E.; Del Bufalo, D.; Sitkovsky, M.V.; et al. Toll-like receptor 3 regulates angiogenesis and apoptosis in prostate cancer cell lines through hypoxia-inducible factor 1 alpha. Neoplasia 2010, 12, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid. Redox Signal 2008, 10, 983–995. [Google Scholar] [CrossRef]
- Browning, A.C.; Halligan, E.P.; Stewart, E.A.; Swan, D.C.; Dove, R.; Samaranayake, G.J.; Amoaku, W.M. Comparative gene expression profiling of human umbilical vein endothelial cells and ocular vascular endothelial cells. Br. J. Ophthalmol. 2012, 96, 128–132. [Google Scholar] [CrossRef]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Paichha, M.; Lenka, S.S.; Chakrabarty, R.; Samanta, M. Hypoxic stress: Impact on the modulation of TLR2, TLR4, NOD1 and NOD2 receptor and their down-stream signalling genes expression in catla (Catla catla). Mol. Biol. Rep. 2016, 43, 1–9. [Google Scholar] [CrossRef]
- Cole, J.E.; Navin, T.J.; Cross, A.J.; Goddard, M.E.; Alexopoulou, L.; Mitra, A.T.; Davies, A.H.; Flavell, R.A.; Feldmann, M.; Monaco, C. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc. Natl. Acad. Sci. USA 2011, 108, 2372–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.S.; Suh, Y.G.; Yi, H.S.; Lee, Y.S.; Jeong, W.I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J. Hepatol. 2013, 58, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.; Thompson, A.A.R.; Bhagwani, A.R.; Hultman, S.; Ji, H.; Kotha, N.; Farr, G.; Arnold, N.D.; Braithwaite, A.; Casbolt, H. Toll-like Receptor 3 Is a Therapeutic Target. for Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, H.; Zhang, J.; Wang, G.; Zhang, J.; Jiang, F.; Xiao, Q. Toll-like receptor 4 in bone marrow-derived cells contributes to the progression of diabetic retinopathy. Mediat. Inflamm. 2014, 2014, 858763. [Google Scholar] [CrossRef]
- Cai, M.; Zhang, X.; Li, Y.; Xu, H. Toll-like receptor 3 activation drives the inflammatory response in oxygen-induced retinopathy in rats. Br. J. Ophthalmol. 2015, 99, 125–132. [Google Scholar] [CrossRef]
- Singh, K.; Kant, S.; Singh, V.K.; Agrawal, N.K.; Gupta, S.K.; Singh, K. Toll-like receptor 4 polymorphisms and their haplotypes modulate the risk of developing diabetic retinopathy in type 2 diabetes patients. Mol. Vis. 2014, 20, 704–713. [Google Scholar]
- Ishida, I.; Kubo, H.; Suzuki, S.; Suzuki, T.; Akashi, S.; Inoue, K.; Maeda, S.; Kikuchi, H.; Sasaki, H.; Kondo, T. Hypoxia diminishes toll-like receptor 4 expression through reactive oxygen species generated by mitochondria in endothelial cells. J. Immunol. 2002, 169, 2069–2075. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Q.; Lv, H.W.; Tan, W.F.; Fang, B.; Wang, H.; Ma, H. Role of the TLR4 pathway in blood-spinal cord barrier dysfunction during the bimodal stage after ischemia/reperfusion injury in rats. J. Neuroinflamm. 2014, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Egger, M.; Schgoer, W.; Beer, A.G.; Jeschke, J.; Leierer, J.; Theurl, M.; Frauscher, S.; Tepper, O.M.; Niederwanger, A.; Ritsch, A.; et al. Hypoxia up-regulates the angiogenic cytokine secretoneurin via an HIF-1alpha- and basic FGF-dependent pathway in muscle cells. FASEB J. 2007, 21, 2906–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Schlueter, C.; Weber, H.; Meyer, B.; Rogalla, P.; Röser, K.; Hauke, S.; Bullerdiek, J. Angiogenetic signaling through hypoxia: HMGB1: An angiogenetic switch molecule. Am. J. Pathol. 2005, 166, 1259–1263. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Assay ID | Gene Symbol |
|---|---|---|
| Vascular Endothelial Growth Factor A | Hs00900055_m1 | VEGFA |
| Fibroblast Growth Factor 2 | Hs00266645_m1 | FGF2 |
| Hypoxia Inducible Factor 1 alpha | Hs00153153_m1 | HIF1A |
| Toll Like Receptor 3 | Hs01551079_g1 | TLR3 |
| Toll Like Receptor 4 | Hs00152939_m1 | TLR4 |
| Hypoxanthine phosphoribosyl transferase 1 | Hs99999909_m1 | HPRT1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhtar, R.; Tahir, H.; Stewart, E.; Wei, R.; Mohammed, I.; Amoaku, W.M. Toll-Like Receptor Signalling Pathways Regulate Hypoxic Stress Induced Fibroblast Growth Factor but Not Vascular Endothelial Growth Factor-A in Human Microvascular Endothelial Cells. Int. J. Transl. Med. 2021, 1, 25-38. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010003
Akhtar R, Tahir H, Stewart E, Wei R, Mohammed I, Amoaku WM. Toll-Like Receptor Signalling Pathways Regulate Hypoxic Stress Induced Fibroblast Growth Factor but Not Vascular Endothelial Growth Factor-A in Human Microvascular Endothelial Cells. International Journal of Translational Medicine. 2021; 1(1):25-38. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010003
Chicago/Turabian StyleAkhtar, Rukhsar, Husain Tahir, Elizabeth Stewart, Ruoxin Wei, Imran Mohammed, and Winfried M. Amoaku. 2021. "Toll-Like Receptor Signalling Pathways Regulate Hypoxic Stress Induced Fibroblast Growth Factor but Not Vascular Endothelial Growth Factor-A in Human Microvascular Endothelial Cells" International Journal of Translational Medicine 1, no. 1: 25-38. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010003