
Comprehension of the Relationship between Autophagy and Reactive Oxygen Species for Superior Cancer Therapy with Histone Deacetylase Inhibitors


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
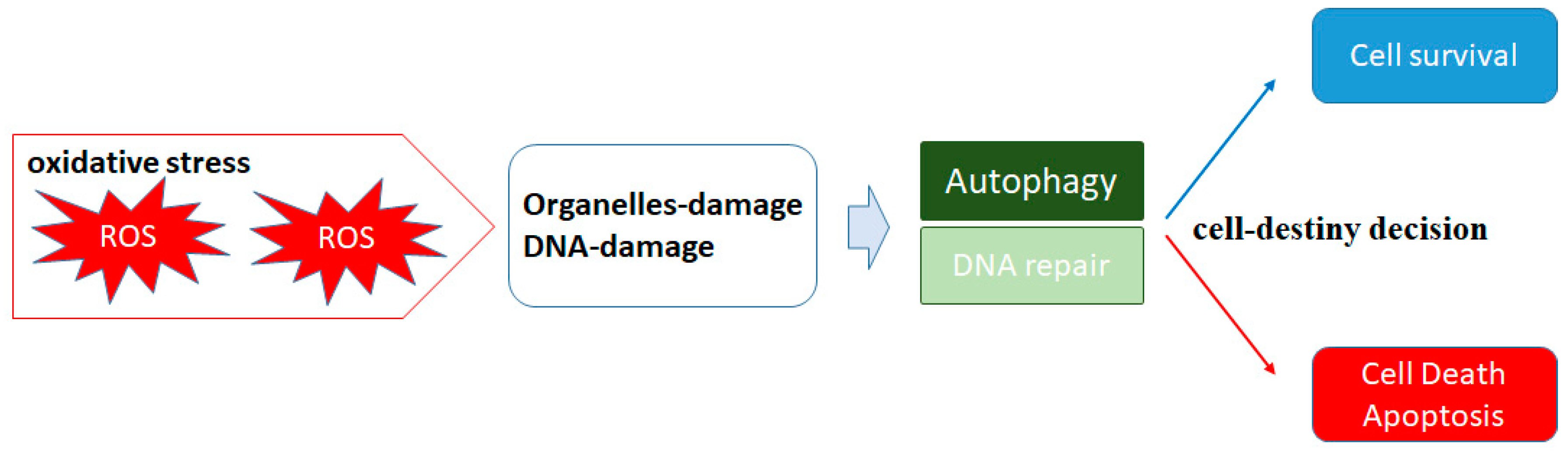
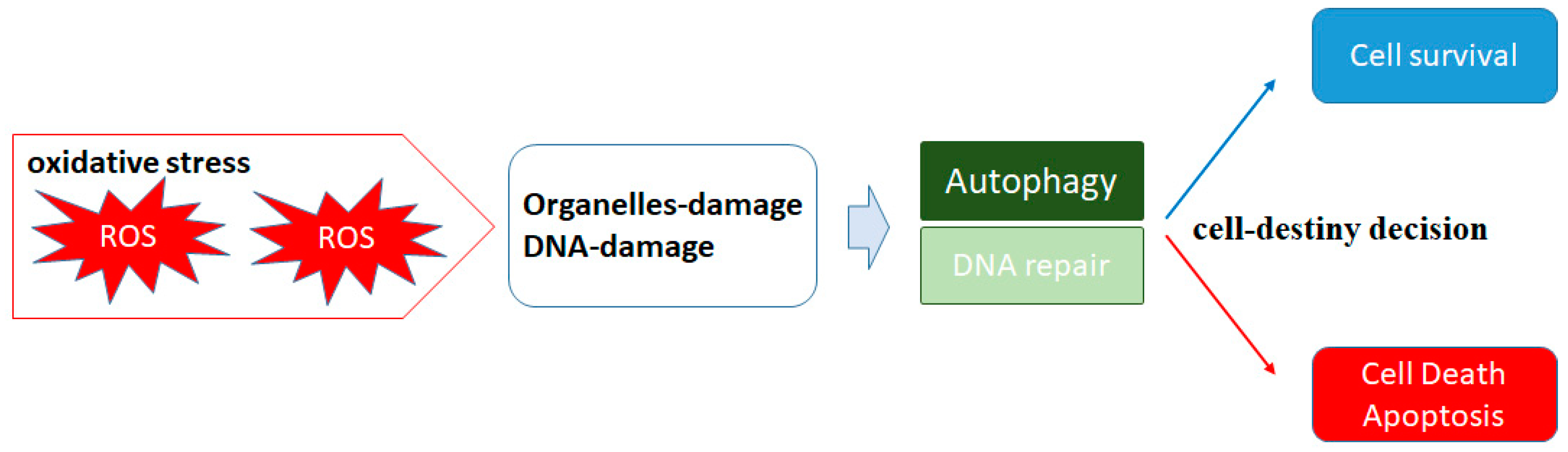
2. ROS and Autophagy
3. Hypoxia and ROS in Tumor Progression
4. HDAC Inhibitors in Cancer Therapy
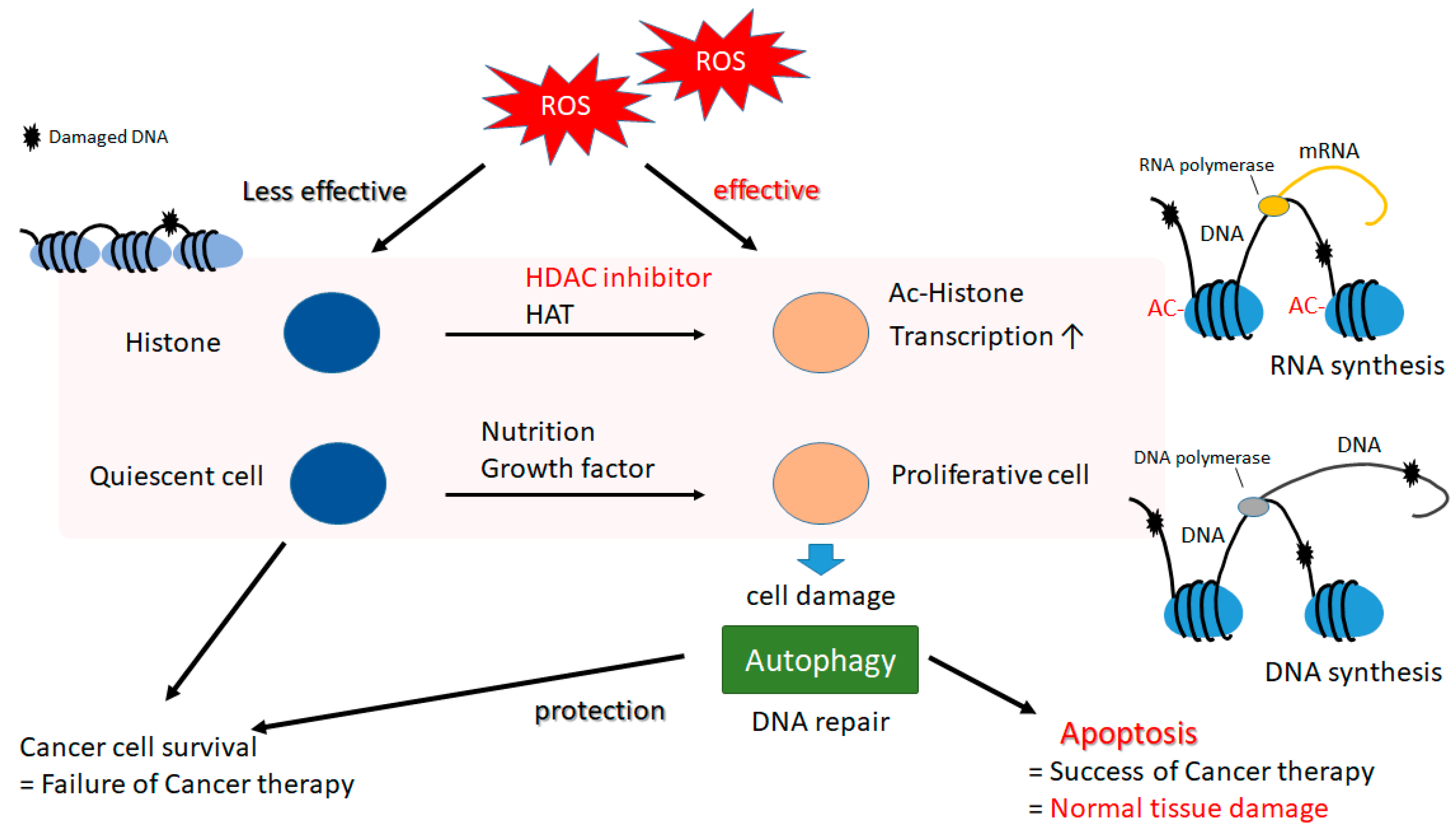
5. A Matter of Cell Life or Cell Death Could Be Determined by Autophagy in Both Cancerous and Normal Types of Cells
6. Histone Modification in Cancer and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef] [Green Version]
- Son, J.M.; Lee, C. Mitochondria: Multifaceted regulators of aging. BMB Rep. 2019, 52, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.S.; Singh, B.; Sharma, D.; Sandur, S.K. Role of protein S-Glutathionylation in cancer progression and development of resistance to anti-cancer drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef] [PubMed]
- Adampourezare, M.; Dehghan, G.; Hasanzadeh, M.; Hosseinpoure Feizi, M.A. Application of lateral flow and microfluidic bio-assay and biosensing towards identification of DNA-methylation and cancer detection: Recent progress and challenges in biomedicine. Biomed. Pharmacother. 2021, 141, 111845. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.; Hashemy, S.I. MicroRNA-mediated redox regulation modulates therapy resistance in cancer cells: Clinical perspectives. Cell. Oncol. 2019, 42, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.O.; Salem, K.; Wagner, B.A.; Bera, S.; Singh, N.; Tiwari, A.; Choudhury, A.; Buettner, G.R.; Goel, A. Interleukin-6 counteracts therapy-induced cellular oxidative stress in multiple myeloma by up-regulating manganese superoxide dismutase. Biochem. J. 2012, 444, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2alpha Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zang, Y.; Qu, J.; Tang, M.; Zhang, T. The Toxicity of Metallic Nanoparticles on Liver: The Subcellular Damages, Mechanisms, And Outcomes. Int. J. Nanomed. 2019, 14, 8787–8804. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, J. Granzyme A activates another way to die. Immunol. Rev. 2010, 235, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; St. Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.C.; Sofiyatun, E.; Chen, W.J.; Wang, L.C. Life as a Vector of Dengue Virus: The Antioxidant Strategy of Mosquito Cells to Survive Viral Infection. Antioxidants 2021, 10, 395. [Google Scholar] [CrossRef]
- Meijles, D.N.; Zoumpoulidou, G.; Markou, T.; Rostron, K.A.; Patel, R.; Lay, K.; Handa, B.S.; Wong, B.; Sugden, P.H.; Clerk, A. The cardiomyocyte “redox rheostat”: Redox signalling via the AMPK-mTOR axis and regulation of gene and protein expression balancing survival and death. J. Mol. Cell. Cardiol. 2019, 129, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Shimron, F.; Peleg, Y.; Elazar, Z. Fatty acid synthase is preferentially degraded by autophagy upon nitrogen starvation in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 1434–1439. [Google Scholar] [CrossRef] [Green Version]
- Che, J.; Wang, W.; Huang, Y.; Zhang, L.; Zhao, J.; Zhang, P.; Yuan, X. miR-20a inhibits hypoxia-induced autophagy by targeting ATG5/FIP200 in colorectal cancer. Mol. Carcinog. 2019, 58, 1234–1247. [Google Scholar] [CrossRef]
- Meliton, L.N.; Zhu, X.; Brown, M.; Epshtein, Y.; Kawasaki, T.; Letsiou, E.; Dudek, S.M. Degradation of group V secretory phospholipase A(2) in lung endothelium is mediated by autophagy. Microvasc. Res. 2020, 129, 103954. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Autophagy and oxidative stress in cardiovascular diseases. Biochim. Biophys. Acta 2015, 1852, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, V.; Galluzzi, L.; Bravo-San Pedro, J.M.; Izzo, V.; Maiuri, M.C.; Kroemer, G. Organelle-Specific Initiation of Autophagy. Mol. Cell 2015, 59, 522–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, M.A.; Puglielli, L. Nepsilon-lysine acetylation in the endoplasmic reticulum—A novel cellular mechanism that regulates proteostasis and autophagy. J. Cell Sci. 2018, 131, jcs221747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Song, C.H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Tang, J.; Huang, Y.; Li, G.; Li, Z.; Cai, W.; Yuan, Y.; Liu, J.; Huang, X.; Zhang, H. The Road of Solid Tumor Survival: From Drug-Induced Endoplasmic Reticulum Stress to Drug Resistance. Front. Mol. Biosci. 2021, 8, 620514. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Guan, Q.W.; Chen, F.H.; Xia, Q.X.; Yin, X.X.; Zhou, H.H.; Mao, X.Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxid. Med. Cell. Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar] [CrossRef]
- Song, S.B.; Hwang, E.S. High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules 2020, 10, 761. [Google Scholar] [CrossRef]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol. Med. Rep. 2013, 8, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Lu, M.; Lan, X.B.; Liu, N.; Su, W.K.; Dushkin, A.V.; Yu, J.Q. Neuroprotective Effects of Oxymatrine on PI3K/Akt/mTOR Pathway After Hypoxic-Ischemic Brain Damage in Neonatal Rats. Front. Pharmacol. 2021, 12, 642415. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.; Zhang, H.J.; Zhou, Y.L.; Tang, X.; Shi, J.H. Characteristics of hypoxic tumor microenvironment in non-small cell lung cancer, involving molecular patterns and prognostic signature. Transl. Lung Cancer Res. 2021, 10, 2132–2147. [Google Scholar] [CrossRef]
- Yadav, A.K.; Yadav, P.K.; Chaudhary, G.R.; Tiwari, M.; Gupta, A.; Sharma, A.; Pandey, A.N.; Pandey, A.K.; Chaube, S.K. Autophagy in hypoxic ovary. Cell. Mol. Life Sci. 2019, 76, 3311–3322. [Google Scholar] [CrossRef]
- Gill, T.; Levine, A.D. Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction. J. Biol. Chem. 2013, 288, 26246–26255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Sha, S.; Tan, J.; Miao, Y.; Zhang, Q. The Role of Autophagy in Hypoxia-Induced Neuroinflammation. DNA Cell Biol. 2021, 40, 733–739. [Google Scholar] [CrossRef]
- Engin, A. Adipose Tissue Hypoxia in Obesity and Its Impact on Preadipocytes and Macrophages: Hypoxia Hypothesis. Adv. Exp. Med. Biol. 2017, 960, 305–326. [Google Scholar] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: A review. Biochim. Biophys. Acta 2016, 1865, 176–183. [Google Scholar] [CrossRef]
- Jing, Y.; Liu, L.Z.; Jiang, Y.; Zhu, Y.; Guo, N.L.; Barnett, J.; Rojanasakul, Y.; Agani, F.; Jiang, B.H. Cadmium increases HIF-1 and VEGF expression through RO.S.; ER.K.; and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 10–19. [Google Scholar] [CrossRef]
- Feng, L.; Sun, C.; Sun, X.; Zhao, Y.; Yu, R.; Kang, C. Identification of inhibitors targeting HIF-2alpha/c-Myc by molecular docking and MM-GBSA technology. J. Recept. Signal Transduct. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.W.; Tong, G.H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Han, H.J. Role of Microtubule-Associated Factors in HIF1alpha Nuclear Translocation. Adv. Exp. Med. Biol. 2020, 1232, 271–276. [Google Scholar]
- Minet, E.; Michel, G.; Remacle, J.; Michiels, C. Role of HIF-1 as a transcription factor involved in embryonic development, cancer progression and apoptosis (review). Int. J. Mol. Med. 2000, 5, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Skwarska, A.; Calder, E.D.D.; Sneddon, D.; Bolland, H.; Odyniec, M.L.; Mistry, I.N.; Martin, J.; Folkes, L.K.; Conway, S.J.; Hammond, E.M. Development and pre-clinical testing of a novel hypoxia-activated KDAC inhibitor. Cell Chem. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Morine, Y.; Imura, S.; Ikemoto, T.; Arakawa, Y.; Iwahashi, S.; Saito, Y.U.; Yamada, S.; Wada, Y.; Yamashita, S.; et al. Carbohydrate Antigen 19–9 Is a Prognostic Factor Which Correlates with HDAC1 and HIF-1alpha for Intrahepatic Cholangiocarcinoma. Anticancer Res. 2019, 39, 6025–6033. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Chen, D.; Cheng, H.; Wang, F. Hypoxia-inducible factor-1alpha protects cervical carcinoma cells from apoptosis induced by radiation via modulation of vascular endothelial growth factor and p53 under hypoxia. Med. Sci. Monit. 2015, 21, 318–325. [Google Scholar]
- Ahmad, I.M.; Dafferner, A.J.; O’Connell, K.A.; Mehla, K.; Britigan, B.E.; Hollingsworth, M.A.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Potentiates the Effects of Nab-Paclitaxel-Gemcitabine and Modulates the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 2264. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 regulates the cancer stem cell fate of breast cancer cells in the context of hypoxia and histone deacetylase inhibitors. Sci. Rep. 2019, 9, 9702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Fu, Y.; Chen, F.; Zhang, J.; Jia, H.; Li, J.; Wang, H.; Wen, J. Hyperoxia sensitizes hypoxic HeLa cells to ionizing radiation by downregulating HIF-1alpha and VEGF expression. Mol. Med. Rep. 2021, 23, 62. [Google Scholar] [CrossRef]
- De Ridder, M.; Van Esch, G.; Engels, B.; Verovski, V.; Storme, G. Hypoxic tumor cell radiosensitization: Role of the iNOS/NO pathway. Bull. Cancer 2008, 95, 282–291. [Google Scholar]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorg. Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef]
- Liu, L.K.; Gao, R.L.; Gao, Y.; Xu, J.Y.; Guo, L.M.; Wang, K.J.; Liu, H.P. A histone K-lysine acetyltransferase CqKAT2A-like gene promotes white spot syndrome virus infection by enhancing histone H3 acetylation in red claw crayfish Cherax quadricarinatus. Dev. Comp. Immunol. 2020, 107, 103640. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liang, Z.; Feng, A.; Salgado, E.; Shim, H. HDAC inhibitor suppresses proliferation and invasion of breast cancer cells through regulation of miR-200c targeting CRK.L. Biochem. Pharmacol. 2018, 147, 30–37. [Google Scholar] [CrossRef]
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliano, T.; Brancolini, C. Epigenetic Mechanisms beyond Tumour-Stroma Crosstalk. Cancers 2021, 13, 914. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Basdra, E.K.; Papavassiliou, A.G.; Piperi, C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int. J. Mol. Sci. 2021, 22, 2778. [Google Scholar] [CrossRef]
- Garcia, T.B.; Uluisik, R.C.; van Linden, A.A.; Jones, K.L.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. Increased HDAC Activity and c-MYC Expression Mediate Acquired Resistance to WEE1 Inhibition in Acute Leukemia. Front. Oncol. 2020, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Ho, E.; Dashwood, R.H. Dietary agents as histone deacetylase inhibitors. Mol. Carcinog. 2006, 45, 443–446. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.; Pili, R. Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies. Pharmaceuticals 2010, 3, 2411–2469. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef] [Green Version]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol. Ther. 2003, 2, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Pant, K.; Yadav, A.K.; Gupta, P.; Islam, R.; Saraya, A.; Venugopal, S.K. Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox. Biol. 2017, 12, 340–349. [Google Scholar] [CrossRef]
- Perona, M.; Thomasz, L.; Rossich, L.; Rodriguez, C.; Pisarev, M.A.; Rosemblit, C.; Cremaschi, G.A.; Dagrosa, M.A.; Juvenal, G.J. Radiosensitivity enhancement of human thyroid carcinoma cells by the inhibitors of histone deacetylase sodium butyrate and valproic acid. Mol. Cell. Endocrinol. 2018, 478, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kwon, J.; Shin, H.J.; Moon, S.M.; Kim, S.B.; Shin, U.S.; Han, Y.H.; Kim, Y. Butyrate enhances the efficacy of radiotherapy via FOXO3A in colorectal cancer patient-derived organoids. Int. J. Oncol. 2020, 57, 1307–1318. [Google Scholar] [CrossRef]
- Hanania, A.N.; Mainwaring, W.; Ghebre, Y.T.; Hanania, N.A.; Ludwig, M. Radiation-Induced Lung Injury: Assessment and Management. Chest 2019, 156, 150–162. [Google Scholar] [CrossRef]
- Griffin, M.F.; Drago, J.; Almadori, A.; Kalavrezos, N.; Butler, P.E. Evaluation of the efficacy of lipotransfer to manage radiation-induced fibrosis and volume defects in head and neck oncology. Head Neck 2019, 41, 3647–3655. [Google Scholar] [CrossRef]
- Mahmood, J.; Connors, C.Q.; Alexander, A.A.; Pavlovic, R.; Samanta, S.; Soman, S.; Matsui, H.; Sopko, N.A.; Bivalacqua, T.J.; Weinreich, D.; et al. Cavernous Nerve Injury by Radiation Therapy May Potentiate Erectile Dysfunction in Rats. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Thipparapu, G.; Ajumeera, R.; Venkatesan, V. Novel dihydropyrimidine derivatives as potential HDAC inhibitors: In silico study. Silico Pharmacol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivey, K.L.; Jensen, M.K.; Hodgson, J.M.; Eliassen, A.H.; Cassidy, A.; Rimm, E.B. Association of flavonoid-rich foods and flavonoids with risk of all-cause mortality. Br. J. Nutr. 2017, 117, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2016, 7, 4454–4467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahabieh, M.S.; Huang, F.; Goncalves, C.; Flores González, R.E.; Prabhu, S.; Bolt, A.; Di Pietro, E.; Khoury, E.; Heath, J.; Xu, Z.Y.; et al. Silencing PEX26 as an unconventional mode to kill drug-resistant cancer cells and forestall drug resistance. Autophagy 2021, 1–19. [Google Scholar] [CrossRef]
- Medvedev, R.; Ploen, D.; Spengler, C.; Elgner, F.; Ren, H.; Bunten, S.; Hildt, E. HCV-induced oxidative stress by inhibition of Nrf2 triggers autophagy and favors release of viral particles. Free Radic. Biol. Med. 2017, 110, 300–315. [Google Scholar] [CrossRef]
- Chen, N.; Debnath, J. IkappaB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy 2013, 9, 1214–1227. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Feng, C.; Zhang, Y.; Liu, C.; Li, B.; Zhu, Q.; Huang, B.; Zhou, Y. Autophagy protects nucleus pulposus cells from cyclic mechanical tension-induced apoptosis. Int. J. Mol. Med. 2019, 44, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Sobierajska, K.; Wieczorek, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Wawro, M.E.; Wiktorska, M.; Boncela, J.; Papiewska-Pajak, I.; Kwasniak, P.; Wyroba, E.; et al. beta-III tubulin modulates the behavior of Snail overexpressed during the epithelial-to-mesenchymal transition in colon cancer cells. Biochim. Biophys. Acta 2016, 1863, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.N.; Park, Y.S.; Kim, N.H.; Han, J.W.; Lee, H.Y.; Kim, Y.K. HDAC inhibitors downregulate MRP2 expression in multidrug resistant cancer cells: Implication for chemosensitization. Int. J. Oncol. 2011, 38, 807–812. [Google Scholar] [CrossRef]
- McClung, J.M.; McCord, T.J.; Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Southerland, K.W.; Reinardy, J.L.; Mueller, S.B.; Venkatraman, T.N.; Lascola, C.D.; et al. BAG3 (Bcl-2-Associated Athanogene-3) Coding Variant in Mice Determines Susceptibility to Ischemic Limb Muscle Myopathy by Directing Autophagy. Circulation 2017, 136, 281–296. [Google Scholar] [CrossRef]
- Fei, Q.; Ma, H.; Zou, J.; Wang, W.; Zhu, L.; Deng, H.; Meng, M.; Tan, S.; Zhang, H.; Xiao, X.; et al. Metformin protects against ischaemic myocardial injury by alleviating autophagy-ROS-NLRP3-mediated inflammatory response in macrophages. J. Mol. Cell. Cardiol. 2020, 145, 1–13. [Google Scholar] [CrossRef]
- Davidson, S.M.; Padró, T.; Bollini, S.; Vilahur, G.; Duncker, D.J.; Evans, P.C.; Guzik, T.; Hoefer, I.E.; Waltenberger, J.; Wojta, J.; et al. Progress in cardiac research—From rebooting cardiac regeneration to a complete cell atlas of the heart. Cardiovasc. Res. 2021, cvab200. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeda, Y.; Nagase, N.; Tsuji, A.; Taniguchi, K.; Kitagishi, Y.; Matsuda, S. Comprehension of the Relationship between Autophagy and Reactive Oxygen Species for Superior Cancer Therapy with Histone Deacetylase Inhibitors. Oxygen 2021, 1, 22-31. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen1010004
Ikeda Y, Nagase N, Tsuji A, Taniguchi K, Kitagishi Y, Matsuda S. Comprehension of the Relationship between Autophagy and Reactive Oxygen Species for Superior Cancer Therapy with Histone Deacetylase Inhibitors. Oxygen. 2021; 1(1):22-31. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen1010004
Chicago/Turabian StyleIkeda, Yuka, Nozomi Nagase, Ai Tsuji, Kurumi Taniguchi, Yasuko Kitagishi, and Satoru Matsuda. 2021. "Comprehension of the Relationship between Autophagy and Reactive Oxygen Species for Superior Cancer Therapy with Histone Deacetylase Inhibitors" Oxygen 1, no. 1: 22-31. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen1010004