
Synthesis of [18F]F-γ-T-3, a Redox-Silent γ-Tocotrienol (γ-T-3) Vitamin E Analogue for Image-Based In Vivo Studies of Vitamin E Biodistribution and Dynamics

 ,
, 
Abstract
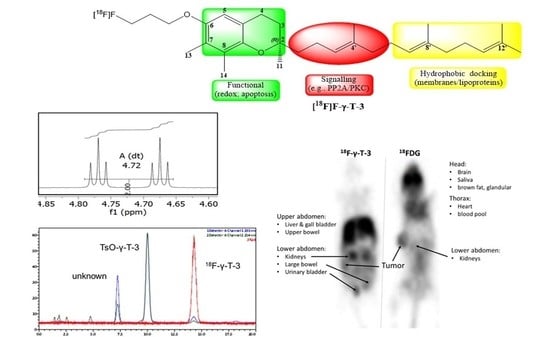
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
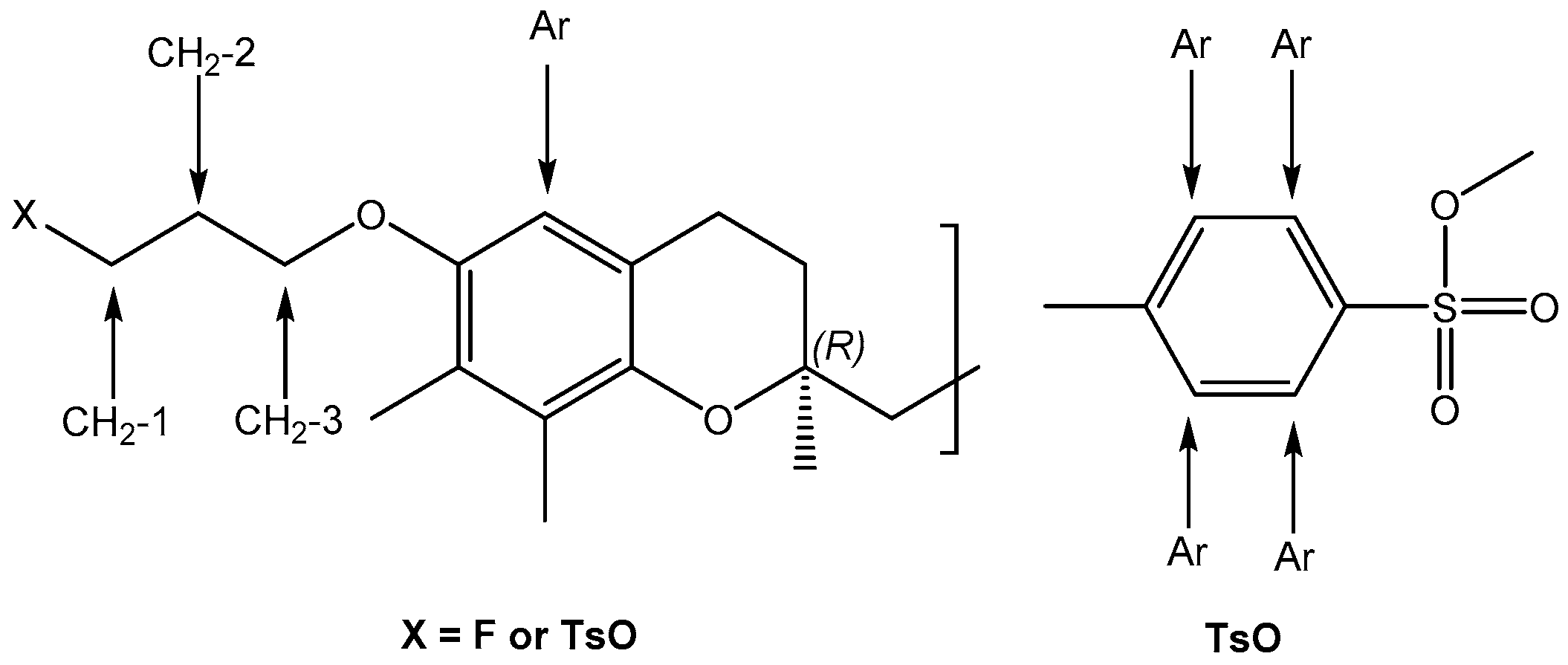
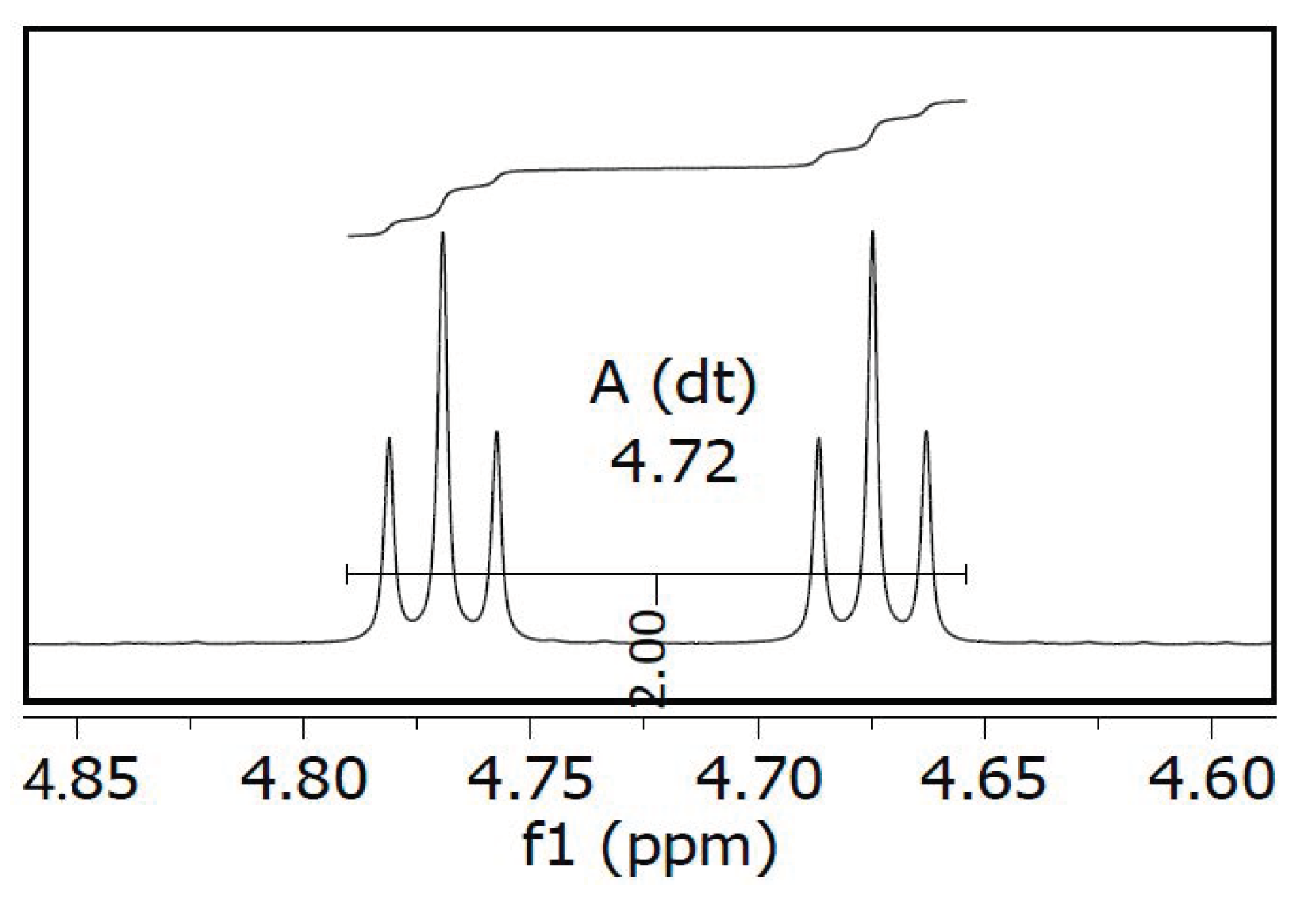
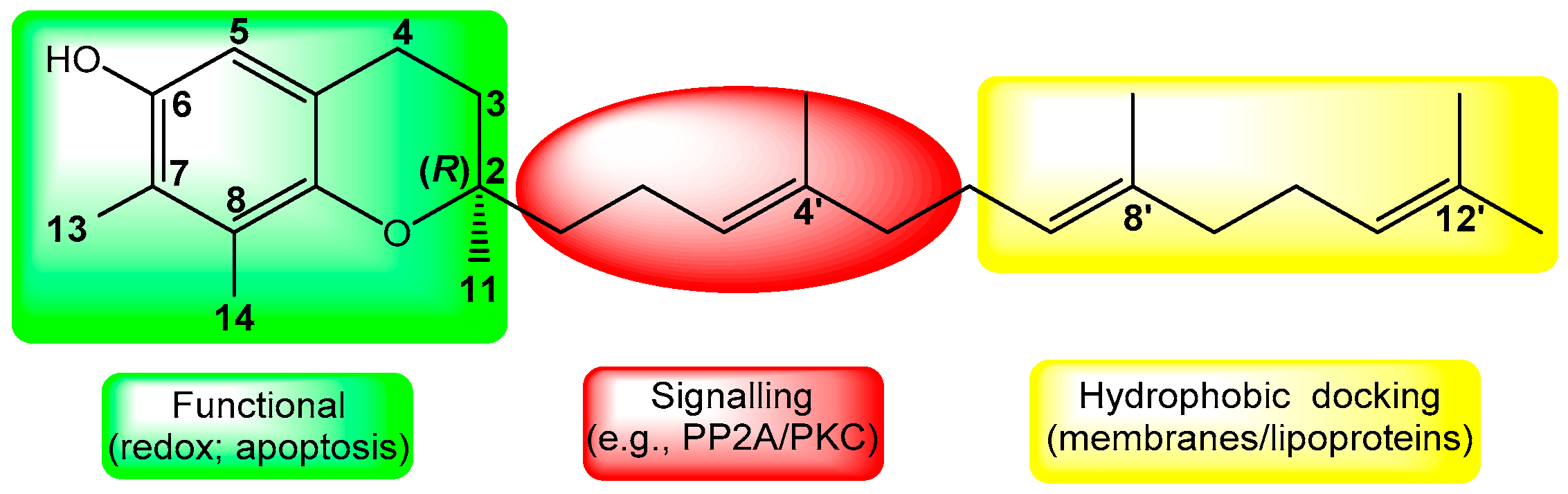
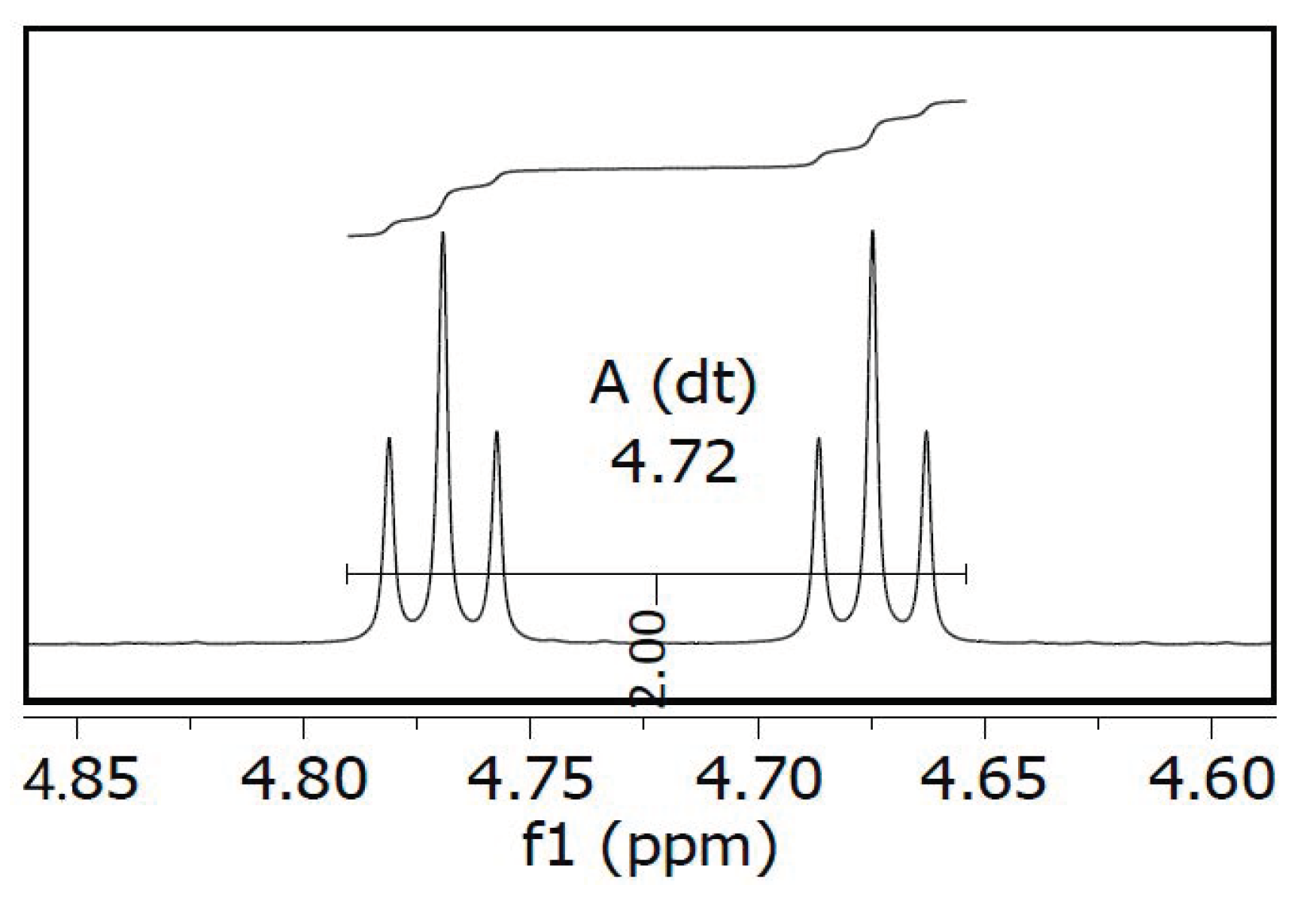
2.1. Chemistry
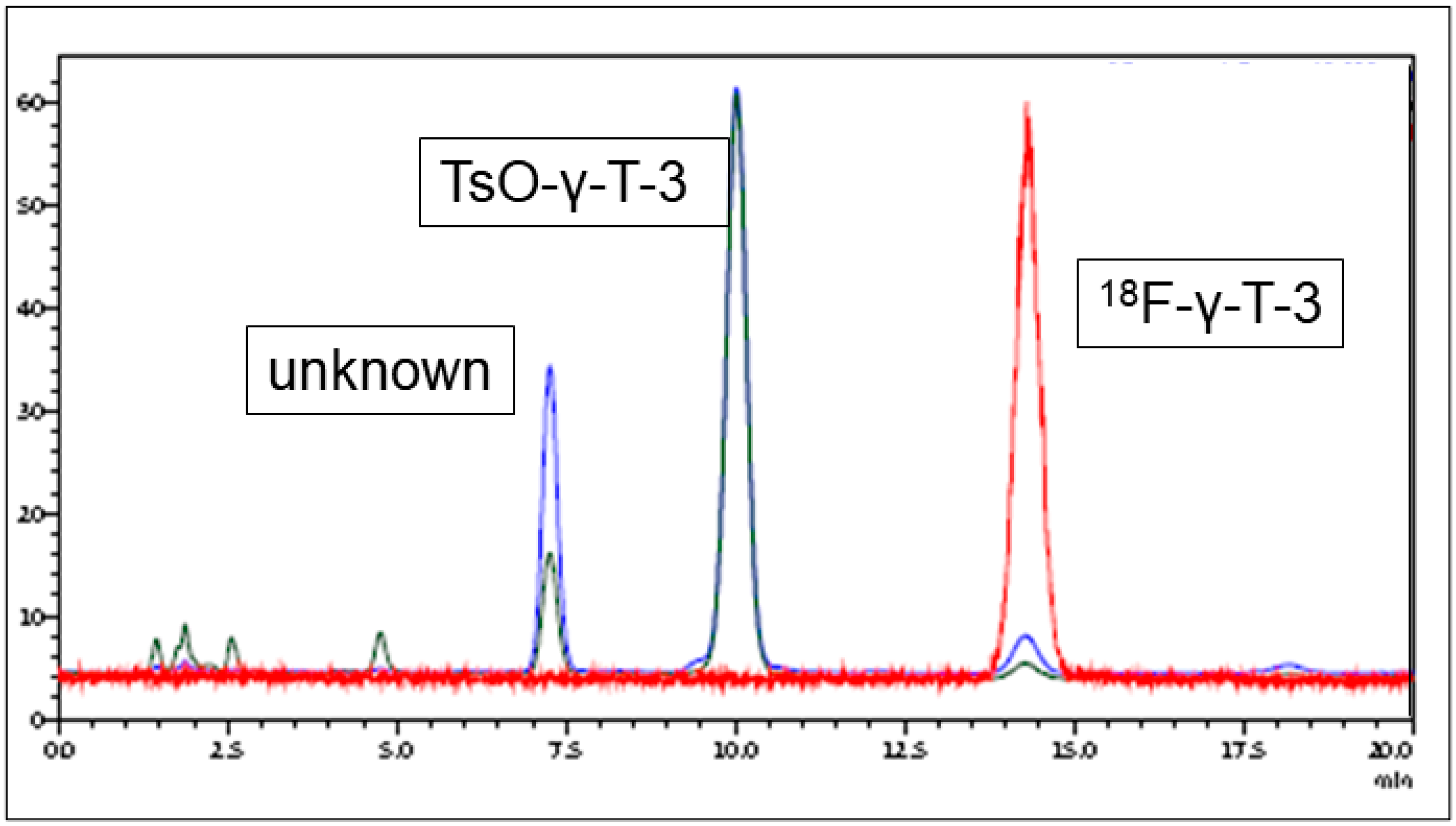
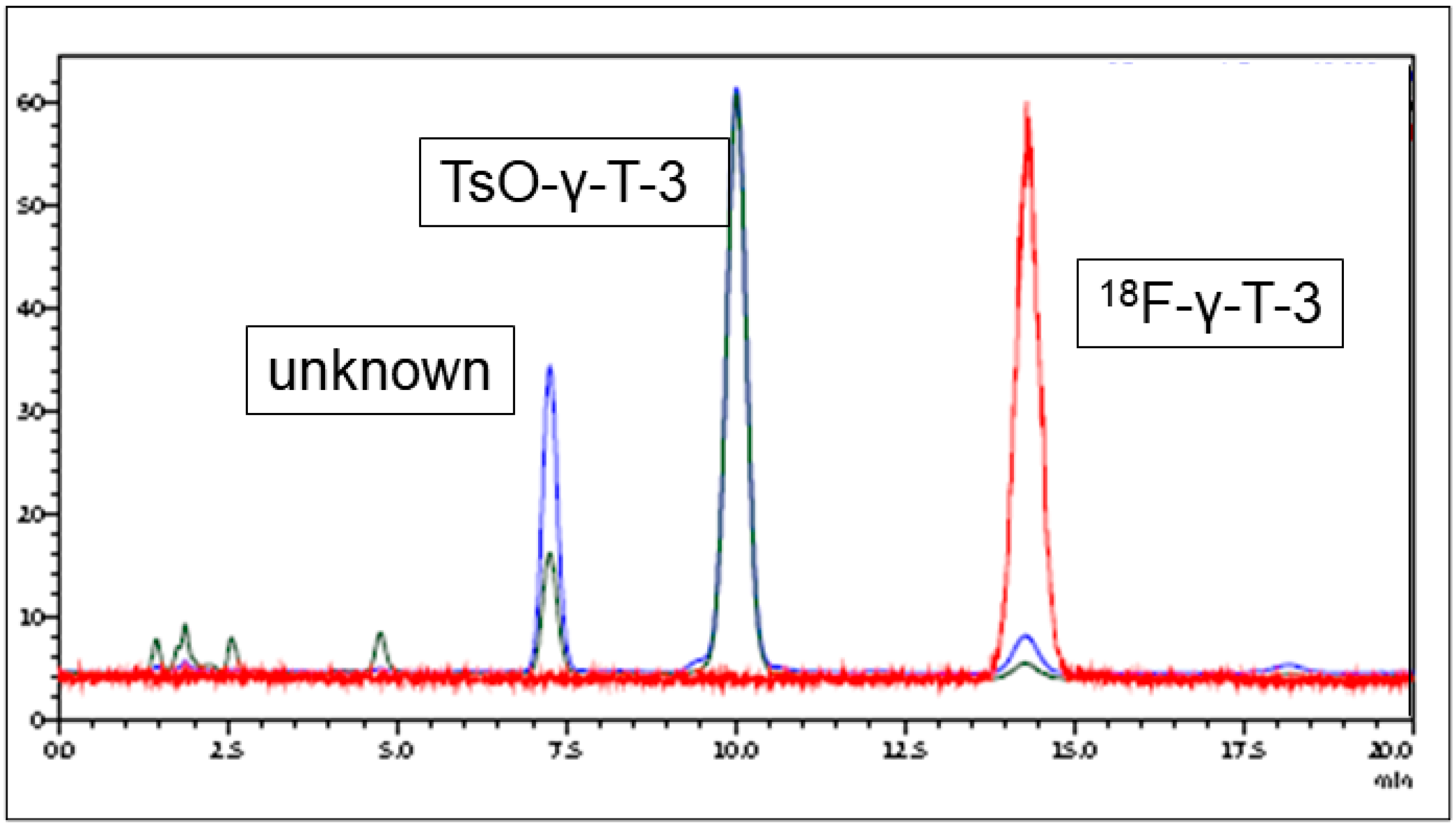
2.2. Radiofluorination
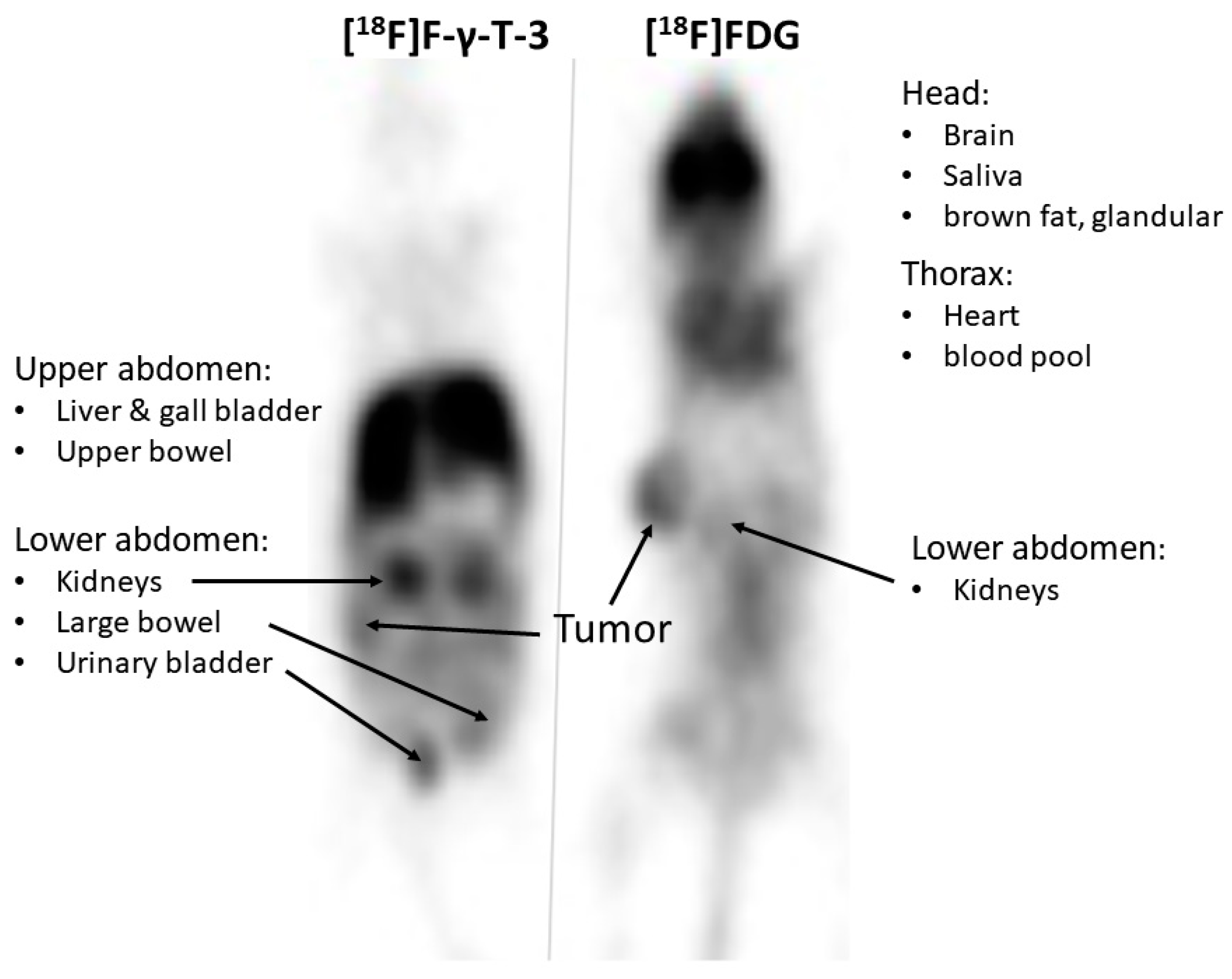
2.3. PET Imaging
3. Discussion
3.1. Radiolabeling
3.2. PET Imaging
4. Materials and Methods
4.1. General
4.1.1. Fluoropropyl Methanesulfonate (2, Scheme 1)
4.1.2. (R)-6-(3-Fluoropropoxy)-2,7,8-trimethyl-2-(4,8,12-trimethyltrideca-3,7,11-trien-1-yl)-chromane (F-γ-T-3, Scheme 1)
4.1.3. (R)-6-(3-Tosyl-propoxy)-2,7,8-trimethyl-2-(4,8,12-trimethyl-trideca-3,7,11-trien-1-yl)-chroman (TsO-γ-T-3, Scheme 1)
4.2. Radiosynthesis of [18F]F-γ-T-3 (Scheme 1)
4.2.1. General Radiochemistry
4.2.2. (R)-6-(3-[18F]Fluoropropoxy)-2,7,8-trimethyl-2-(4,8,12-trimethyltrideca-3,7,11-trien-1-yl)-chromane ([18F]F-γ-T-3, Scheme 1)
4.3. Partition Coefficients (LogP)
4.4. Animal Imaging by Positron Emission Tomography (PET)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szymańska, R.; Nowicka, B.; Trela, A.; Kruk, J. Vitamin E: Structure and forms. In Molecular Nutrition; Patel, V.B., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 67–90. [Google Scholar] [CrossRef]
- Peh, H.Y.; Tan, W.D.; Liao, W.; Wong, W. Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol. Ther. 2016, 162, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Meydani, S.N.; Meydani, M.; Zingg, J.M. The rise, the fall and the renaissance of vitamin E. Arch. Biochem. Biophys. 2016, 595, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, P.; Birringer, M.; Blumberg, J.B.; Eggersdorfer, M.; Frank, J. Vitamin E in Human Health; Springer Nature: Cham, Switzerland, 2019; pp. 1–467. [Google Scholar] [CrossRef]
- Shahidi, F.; De Camargo, A.C. Tocopherols and Tocotrienols in Common and Emerging Dietary Sources: Occurrence, Applications, and Health Benefits. Int. J. Mol. Sci. 2016, 17, 1745. [Google Scholar] [CrossRef]
- Lz, M.B. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 7, 14–43. [Google Scholar] [CrossRef]
- Birringer, M. Analysis of vitamin E metabolites in biological specimen. Mol. Nutr. Food Res. 2010, 54, 588–598. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, M.-J.; Cheung, C.; Ju, J.-H.; Chen, Y.-K.; Liu, B.; Hu, L.; Yang, C.S. Analysis of Multiple Metabolites of Tocopherols and Tocotrienols in Mice and Humans. J. Agric. Food Chem. 2010, 58, 4844–4852. [Google Scholar] [CrossRef] [Green Version]
- Büsing, A.; Ternes, W. Separation of α-tocotrienol oxidation products and eight tocochromanols by HPLC with DAD and fluorescence detection and identification of unknown peaks by DAD, PBI-EIMS, FTIR, and NMR. Anal. Bioanal. Chem. 2011, 401, 2843–2854. [Google Scholar] [CrossRef]
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular Mechanisms of Action of Tocotrienols in Cancer: Recent Trends and Advancements. Int. J. Mol. Sci. 2019, 20, 656. [Google Scholar] [CrossRef] [Green Version]
- Meganathan, P.; Fu, J.-Y. Biological Properties of Tocotrienols: Evidence in Human Studies. Int. J. Mol. Sci. 2016, 17, 1682. [Google Scholar] [CrossRef]
- Niki, E. Evidence for beneficial effects of vitamin E. Korean J. Intern. Med. 2015, 30, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Abe, K. Chapter 1. Vitamin E: Structure, Properties and Functions. In Vitamin E: Chemistry and Nutritional Benefits; Niki, E., Ed.; Royal Socity of Chemistry: London, UK, 2019; pp. 1–11. [Google Scholar] [CrossRef]
- Abuasal, B.S.; Lucas, C.; Peyton, B.; Alayoubi, A.; Nazzal, S.; Sylvester, P.W.; Kaddoumi, A. Enhancement of Intestinal Permeability Utilizing Solid Lipid Nanoparticles Increases γ-Tocotrienol Oral Bioavailability. Lipids 2012, 47, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Langer, O. Use of PET Imaging to Evaluate Transporter-Mediated Drug-Drug Interactions. J. Clin. Pharmacol. 2016, 56, S143–S156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jans, H.-S.; Yang, X.-H.; Brocks, D.R.; Kumar, P.; Wuest, M.; Wiebe, L.I. Positron Emission Tomography (PET) and Pharmacokinetics: Classical Blood Sampling Versus Image-Derived Analysis of [18F]FAZA and [18F]FDG in a Murine Tumor Bearing Model. J. Pharm. Pharm. Sci. 2018, 21, 32s–47s. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, N.; Sugahara, R.; Namiki, K.; Sato, C.; Ando, A.; Sato, A.; Virgona, N.; Yano, T. Inhibitory effect of a redox-silent analogue of tocotrienol on hypoxia adaptation in prostate cancer cells. Anti-Cancer Drugs 2017, 28, 289–297. [Google Scholar] [CrossRef]
- Yano, T.; Sato, A.; Sekine, M.; Virgona, N.; Ota, M. Redox-inactive analogue of tocotrienol as a potential anti-cancer agent. Anticancer Agents Med. Chem. 2013, 13, 496–501. [Google Scholar]
- Elnagar, A.Y.; Wali, V.B.; Sylvester, P.W.; El Sayed, K.A. Design and preliminary structure–activity relationship of redox-silent semisynthetic tocotrienol analogues as inhibitors for breast cancer proliferation and invasion. Bioorg. Med. Chem. 2010, 18, 755–768. [Google Scholar] [CrossRef]
- Behery, F.A.; Elnagar, A.Y.; Akl, M.R.; Wali, V.B.; Abuasal, B.; Kaddoumi, A.; Sylvester, P.W.; El Sayed, K.A. Redox-silent tocotrienol esters as breast cancer proliferation and migration inhibitors. Bioorg. Med. Chem. 2010, 18, 8066–8075. [Google Scholar] [CrossRef]
- Cacace, F.; Wolf, A.P. Substrate selectivity and orientation in aromatic substitution by molecular fluorine. J. Am. Chem. Soc. 1978, 100, 3639–3641. [Google Scholar] [CrossRef]
- Coenen, H.H. Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions. Ernst Schering Res. Found. Workshop 2007, 15–50. [Google Scholar] [CrossRef]
- Banister, S.D.; Roeda, D.; Dollé, F.; Kassiou, M. Fluorine-18 Chemistry for PET: A Concise Introduction. Curr. Radiopharm. 2010, 3, 68–80. [Google Scholar] [CrossRef]
- Yang, Z.; Lee, M.-J.; Zhao, Y.; Yang, C.S. Metabolism of tocotrienols in animals and synergistic inhibitory actions of tocotrienols with atorvastatin in cancer cells. Genes Nutr. 2011, 7, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontag, T.J.; Parker, R.S. Influence of major structural features of tocopherols and tocotrienols on their ω-oxidation by tocopherol-ω-hydroxylase. J. Lipid Res. 2007, 48, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, D.; Coenen, H.H.; Stöcklin, G. NCA 18F-fluoroalkylation of H-acidic compounds. J. Label Compd. Radiopharm. 1988, 25, 201–216. [Google Scholar] [CrossRef]
- Ohnmacht, S.; West, R.; Simionescu, R.; Atkinson, J. Assignment of the1H- and 13C-NMR of tocotrienols. Magn. Reson. Chem. 2008, 46, 287–294. [Google Scholar] [CrossRef]
- An, G.; Ahn, S.; Choi, T.H.; Cheon, G.J. Synthesis of F-18 labeled N3-fluoroalkylated carbonucleoside analogues and biological evaluation. J. Nucl. Med. 2010, 51, 1463. [Google Scholar]
- Krämer, S.D.; Mu, L.; Müller, A.; Keller, C.; Kuznetsova, O.F.; Schweinsberg, C.; Franck, D.; Müller, C.; Ross, T.L.; Schibli, R.; et al. 5-(2-18F-Fluoroethoxy)-l-Tryptophan as a Substrate of System L Transport for Tumor Imaging by PET. J. Nucl. Med. 2012, 53, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.S.; Apana, S.M.; Hersh, J.M. Teflon radiolysis as the major source of carrier in fluorine-18. J. Label. Compd. Radiopharm. 2009, 52, 543–548. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H.; Stöcklin, G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-d-glucose using aminopolyether supported nucleophilic substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar]
- Mahipal, A.; Klapman, J.; Vignesh, S.; Yang, C.S.; Neuger, A.; Chen, D.-T.; Malafa, M.P. Pharmacokinetics and safety of vitamin E δ-tocotrienol after single and multiple doses in healthy subjects with measurement of vitamin E metabolites. Cancer Chemother. Pharmacol. 2016, 78, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Vallabhajosula, S.; Killeen, R.P.; Osborne, J.R. Altered Biodistribution of Radiopharmaceuticals: Role of Radiochemical/Pharmaceutical Purity, Physiological, and Pharmacologic Factors. Semin. Nucl. Med. 2010, 40, 220–241. [Google Scholar] [CrossRef] [Green Version]
- Yalkowsky, S.H.; Rubino, J.T. Solubilization by Cosolvents I: Organic Solutes in Propylene Glycol-Water Mixtures. J. Pharm. Sci. 1985, 74, 416–421. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 1742129, (−)-alpha-Tocopherol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/alpha-Tocopherol (accessed on 12 September 2020).
- Kamal-Eldin, A.; Budilarto, E. Chapter 6. Tocopherols and tocotrienols as antioxidants for food preservation. In Handbook of Antioxidants for Food Preservation; Shahidi, F., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 141–159. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 86472, Alpha-Tocopherol Acetate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/alpha-Tocopherol-acetate (accessed on 12 September 2020).
- Grover-McKay, M.; Walsh, S.A.; Seftor, E.A.; Thomas, P.A.; Hendrix, M.J. Role for glucose transporter 1 protein in human breast cancer. Pathol. Oncol. Res. 1998, 4, 115–120. [Google Scholar] [CrossRef]
- Direcks, W.G.; Berndsen, S.C.; Proost, N.; Peters, G.J.; Balzarini, J.; Spreeuwenberg, M.D.; Lammertsma, A.A.; Molthoff, C.F.M. [18F]FDG and [18F]FLT uptake in human breast cancer cells in relation to the effects of chemotherapy: An in vitro study. Br. J. Cancer 2008, 99, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, K.; Francois, R.A.; Hutchinson, S.Z.; Neuger, A.M.; Lush, R.; Coppola, M.; Sebti, S.; Malafa, M.P. Vitamin E delta-tocotrienol levels in tumor and pancreatic tissue of mice after oral administration. Pharmacology 2009, 83, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Drotleff, A.M.; Bohnsack, C.; Schneider, I.; Hahn, A.; Ternes, W. Human oral bioavailability and pharmacokinetics of tocotrienols from tocotrienol-rich (tocopherol-low) barley oil and palm oil formulations. J. Funct. Foods 2014, 7, 150–160. [Google Scholar] [CrossRef]



Sample Availability: Please contact Hermay Labs Corp., Edmonton, Canada ([email protected]), regarding availability of Vitamin E synthons. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roselt, P.; Cullinane, C.; Noonan, W.; Elsaidi, H.; Eu, P.; Wiebe, L.I. Synthesis of [18F]F-γ-T-3, a Redox-Silent γ-Tocotrienol (γ-T-3) Vitamin E Analogue for Image-Based In Vivo Studies of Vitamin E Biodistribution and Dynamics. Molecules 2020, 25, 5700. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235700
Roselt P, Cullinane C, Noonan W, Elsaidi H, Eu P, Wiebe LI. Synthesis of [18F]F-γ-T-3, a Redox-Silent γ-Tocotrienol (γ-T-3) Vitamin E Analogue for Image-Based In Vivo Studies of Vitamin E Biodistribution and Dynamics. Molecules. 2020; 25(23):5700. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235700
Chicago/Turabian StyleRoselt, Peter, Carleen Cullinane, Wayne Noonan, Hassan Elsaidi, Peter Eu, and Leonard I. Wiebe. 2020. "Synthesis of [18F]F-γ-T-3, a Redox-Silent γ-Tocotrienol (γ-T-3) Vitamin E Analogue for Image-Based In Vivo Studies of Vitamin E Biodistribution and Dynamics" Molecules 25, no. 23: 5700. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235700