Association between Stress and the HPA Axis in the Atopic Dermatitis

1
Department of Dermatology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan
2
Citrus Valley Medical Center, West Covina, CA 91790, USA
3
Dermatology Service & Translational Research Unit (UIT), Hospital General Universitario de Ciudad Real, Ciudad Real 13005, Spain
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(10), 2131; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102131
Submission received: 1 September 2017
/
Revised: 6 October 2017
/
Accepted: 9 October 2017
/
Published: 12 October 2017
(This article belongs to the Special Issue Role of the Hypothalamo–Pituitary–Adrenal (HPA) Axis in Health and Disease)
Abstract
:The hypothalamic–pituitary–adrenal (HPA) axis is one of the body’s neuroendocrine networks that responds to psychological stress (PS). In the skin, there exists a peripheral HPA axis similar to the central axis. Glucocorticoids (GCs) are key effector molecules of the HPA axis and are essential for cutaneous homeostasis. Atopic dermatitis (AD) is a condition typically characterized by a chronic relapsing course that often results in PS. HPA dysfunction is present in AD patients by the decreased response of GCs elevation to stress as compared to those unaffected by AD. Nevertheless, in skin, acute PS activates several metabolic responses that are of immediate benefit to the host. During the acute phase of PS, increased endogenous GCs have been shown to provide benefit rather than by aggravating cutaneous inflammatory dermatoses. However, a chronic T helper cell type 2 (Th2) predominant cytokine profile acts as a negative feedback loop to blunt the HPA axis response in AD. In this article, we reviewed the role of CRF, pro-opiomelanocortin (POMC)-derived peptides, GCs of the HPA, and 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) in AD, with a discussion of the pathogenetic mechanisms of inflammation and skin barrier functions, including antimicrobial defense, and their association with PS.
1. Introduction
Atopic dermatitis (AD), one of the most common chronic allergic skin inflammatory diseases, has an increasing prevalence in the population. Onset is typically during the first six months of life in 45% of cases, and the symptoms may continue to affect adulthood [1]. Current evidence suggests that the pathogenesis of AD is multifactorial, comprising not only aspects of epidermal barrier dysfunction, but also T helper cell type 2 (Th2)-lymphocyte-driven inflammation [2,3]. The disease is also modified by neuroendocrine mediators such as histamine via histamine H4 receptor [4] or glucocorticoids (GCs) of the hypothalamic–pituitary–adrenal (HPA) axis [5]. It is well known that AD is triggered or exacerbated by psychological stress (PS) [1,3]. In both pediatric and adult patients, AD often causes significant impairment in quality of life, resulting in PS. PS, via the HPA axis, leads to the release of endogenous GCs that is critical in regulating the inflammation of AD [5].
Impairments in skin barrier function are inevitably present in AD patients. In infants, transepidermal water loss (TEWL) has been shown to be a strong predictor for developing AD later in life given its reflection of stratum corneum (SC) structure and permeability barrier function. In fact, impairment of the skin barrier is considered a primary event in AD pathogenesis [3] (Figure 1). It has been shown that TEWL is correlated to both allergic and irritant contact dermatitis [6], and perturbed barrier function plays an essential role in the allergic sensitization to both protein antigens and staphylococcal superantigens in AD patients [7]. Additionally, perturbation of epidermal barrier function correlates with initiation of cytokine cascade in human skin [8]. Therefore, it has been proposed that early interventions to repair this epidermal barrier including the use of appropriate emollients, humectants, and soaps, may be useful in the control of this chronic disease and the prevention of its progression (atopic march) [9]. Several studies have evaluated the impact of inflammatory mediators (cytokines, hormones, and antimicrobial peptides (AMPs)) on AD skin lesions, and their influence on barrier function and B-cell–mediated immunoglobulin production [10]. Th2-related cytokines such as interleukin 4 (IL-4) and IL-13 play a significant role in AD inflammatory response. Tumor necrosis factor alpha (TNF-α), the most studied cytokine in skin physiology, is increased in both skin lesions and the serum of AD patients. TNF-α is also associated with increased histamine concentration in plasma, which has been thought to also be involved in the pathophysiology of AD [11]. Moreover, skin irritants or environment noxious stressors (low humidity, heat, PS, etc.) lead to the release of IL-1 and TNF-α, which affect the function of antigen-presenting cells and promote the development of contact allergy. These findings may explain the positive association between contact allergy and AD, which has been established in prior epidemiological studies [12].
Thymic stromal lymphopoietin (TSLP), a sensitive marker of epithelial inflammation, is shown to be harmful to the epidermal barrier [13,14,15,16]. In fact, TSLP is highly expressed in keratinocytes from AD patients, and plays critical roles in the induction and exacerbation of this chronic dermatitis [17]. TSLP acts in AD pathogenesis through several mechanisms: (1) enhancing CD4+ T-cell polarization into Th2 cells [16,18,19,20,21]; (2) promoting IL-33-independent innate lymphoid cell responses [22]; and (3) targeting dendritic cells and innate lymphoid cells [23,24]. Moreover, TSLP mediates pruritus, which is not only the main symptom of AD, but also a key factor involved in its pathogenesis by causing disrupted skin barrier through scratching and exposure to pathogens, thus leading to the consequent inflammatory flares [25,26].
Finally, neuroendocrine mediators also play an important role in AD pathogenesis, with the HPA axis being one of the most well studied and best characterized [27,28,29]. HPA dysfunction is present in AD patients, showing a decreased response of serum GCs after being exposed to stressors when compared with unaffected persons [27,28]. Although the HPA axis is initially activated by pro-inflammatory cytokines, there is attenuated GC responsiveness to stress in chronic AD [29]. Thus, in patients with AD, insufficient production of the GCs might result in an imbalance of Th1/Th2 and trigger allergic inflammation [30,31]. This review has focused on understanding the complex interplay between the HPA axis, inflammation, skin barrier function, and PS in AD.
2. Hypothalamic–Pituitary–Adrenal (HPA) Axis
The HPA axis (Figure 2) has very important functions, including responses to psychological and physical stress, and inflammatory factors [32]. Corticotrophin-releasing hormone (CRH) is the principal component of the HPA axis, functioning as the key regulator of stress response. CRH is released from paraventricular nuclei of the hypothalamus in pulsatile pattern, with the increase in magnitude and frequency secondary to acute stress [33]. This hormone controls the HPA axis, as well as various behavioral and autonomic stress responses by the two CRH receptors, CRH-R1 and CRH-R2. The CRH system also includes CRH-binding protein, a glycoprotein that binds to and modulates CRH receptor activity [34]. Additionally, CRH regulates both systemic and cutaneous homeostasis [35], inducing the secretion of pro-opiomelanocortin (POMC)-derived peptides from the anterior pituitary, including adrenocorticotropic hormone (ACTH), alpha melanocyte-stimulating hormone (α-MSH), and β-endorphin [33]. ACTH shows a direct effect in pigmentation [36]. α-MSH has strong anti-inflammatory effects [37], and β-endorphin exhibits analgesic, immunomodulatory, and anti-inflammatory properties [38]. During PS, activation of HPA axis has been correlated to increased levels of pro-inflammatory cytokines (IL-1, IL-4, IL-6, IL-18, and TNF-α), probably through the action of ACTH [39]. In addition, it is well known that ACTH mediates the inflammatory response by stimulating adrenal GC production (cortisol in human), which act in many cells and tissues to regulate homeostasis [33]. As important effector molecules of the HPA axis, GCs are regularly secreted, with feedback inhibition modulating the HPA axis [40]. GCs influence the transcription of glucocorticoid response elements of target genes involved in anti-inflammatory response [41]. GCs are key mediators to modulate the inflammatory response in PS. In fact, one of the main pathogenetic mechanisms of chronic allergic diseases, such as AD or asthma, is the presence of greater Th2 response correlated to lower cortisol and GC levels [42,43,44].
3. The Peripheral HPA Axis
The skin has developed a local neuroendocrine system to regulate its homeostasis in response to its ongoing exposure to environmental stressors [45,46]. In fact, components of this neuroendocrine system have been described for many structural cells of the skin, although they have been particularly well characterized for keratinocytes [47,48,49,50]. Specifically, there exists a peripheral HPA axis similar to the central HPA axis, which has been recently mapped [38,51,52]. This peripheral HPA axis also shows a similar hierarchical structure to the central axis. It has been reported that all regulatory elements of the central HPA axis were expressed in mammalian skin, including CRH, POMC-derived peptides, GCs [36,53], and related peptides, as well their appropriate functional receptors, CRH receptors, melanocortin receptor type 2 (MCR2, the classical adrenocortical ACTH receptor), and GC receptor NR3C1 [54,55].
The peripheral HPA axis acts in an auto/paracrine manner, modifying the cutaneous stress response. Not only are keratinocytes able to produce hormones like CRH, ACTH, and cortisol, but they also produce neurotransmitters (e.g., adrenaline, noradrenaline, dopamine, histamine, acetylcholine, etc.), neurotrophins (e.g., nerve growth factor, brain-derived neurotrophic factor), as well as neuropeptides that also respond to stress, e.g., substance P [38,39,45,46,47,48,49,50]. In addition to synthesizing neurotransmitters and neurohormones, keratinocytes also express their respective receptors [38,39,45,46,47,48,49,50,56,57]. Thus, the epidermis continuously senses the environment and reacts to various stressors (humidity, temperature, skin surface pH, cutaneous microbiome, injuries, and PS) to maintain epidermal homeostasis and adjust skin barrier functions [36,38,39]. Of the various stressors, PS plays a key role in skin barrier and influences the immune response by the secretion of stress-related neuropeptides/cytokine profiles and altering the HPA axis-related hormones [39,53,58].
Unlike the central HPA axis, the elements of peripheral HPA axis (CRH→POMC→ACTH→GCs) affect each other more closely and have more complex role. This is still not well understood [35]. Of all the elements of the HPA axis, CRH function is well studied [38,39,59]. However, it is still unclear if CRH in the skin is produced centrally or locally [59]. The effect of CRH is variable on different types of cells. Peripheral CRH is an important proinflammatory cytokine necessary to induce an inflammatory response in vivo, leading to increased skin vascular permeability and inflammation, which is largely attributed to mast cell activation [60]. CRH function is mediated via CRH receptors (CRH-R1 and CRH-R2) [52]. CRH-R2 is mainly present in vascular and glandular structures while CRH-R1 is the major receptor of most cellular constituents in the skin [52,61]. Through CRH-R1, CRH influences the proliferation [62], differentiation [63,64], and apoptosis [62] of the various cell components in the skin. CRH promotes the differentiation of keratinocytes and inhibits the proliferation through arrest in G0/1 phase [63,64]. CRH also extends the melanocyte survival by suppressing apoptosis [62], but inhibits their proliferation. Meanwhile, CRH stimulates the proliferation of fibroblasts [62]. In regard to the effects on inflammation, CRH promotes the release of IL-6 from keratinocytes [65]. It also stimulates mast cells and plays a role as a pro-inflammatory agent [60,66]. In contrast to its role of proinflammatory effects, CRH also shows its anti-inflammatory characteristics by modulating vascular permeability, angiogenesis, and cytokine production [65,67,68]. Through CRH-R1 activation, keratinocytes reduce the expression of vascular endothelial growth factor [68]. CRH also decreases the pro-inflammatory cytokine IL-1β expression of keratinocytes, but increases the expression of the anti-inflammatory cytokine IL-11 [65]. In addition, in human melanocytes, CRH inhibits nuclear factor-κB signaling and the subsequent production of pro-inflammatory cytokines [69].
GCs are the last mediators in peripheral HPA axis. They are also synthesized in the skin [38,39,45,46]. 11 beta-hydroxysteroid dehydrogenase (11β-HSD) plays a role in GC synthesis. Two isozymes of 11β-HSD exist: 11β-HSD1, which activates cortisol from cortisone, and 11β-HSD2, which inactivates cortisol to cortisone [70,71,72]. 11β-HSD1 is expressed in many tissues, with the highest quantities in the liver, lung, adipose tissue, ovaries, and the central nervous system [71]. In the skin, 11β-HSD1 is expressed in epidermal keratinocytes, dermal fibroblasts, and root sheath cells of the outer hair follicle [73,74,75]. Specifically, in a wound healing context, 11β-HSD1 activates significantly more corticosterone than is generated de novo from progesterone in mouse skin, driving GC exposure [76]. This feature has been shown to accelerate skin wound repair with the use of 11β-HSD1 inhibitors [76,77]. Topical applications of carbenoxolone (a nonspecific 11β-HSD inhibitor) or an isoform-specific 11β-HSD1 inhibitor have been shown to overcome stress and exogenous GC-induced delays in the wound healing process [77].
4. HPA Axis and Skin Inflammation
As previously discussed, the HPA axis can be stimulated by a multitude of environmental stressors, including inflammation, infections, and direct physical trauma (scratching, wound healing, burns, etc.) [78]. Moreover, several studies have found proof suggesting communication between proinflammatory cytokines and mediators of the HPA axis [79,80,81,82]. Most reports investigating the link between elevated PS and worsened inflammation in humans have shown that both the HPA axis and the sympathetic axis contribute to the distinctively large volume of pro-allergic cytokines (e.g., IL-4 and IL-5), which drive eosinophilic inflammatory response and determine the inflammatory immunophenotype in AD patients. Additionally, there is also an up-regulation of cytokines driving cellular adaptive immunity, including TNF-α and interferon gamma (IFN-γ) [39,83,84]. IL-18 is a proinflammatory cytokine with an important role in PS by modulating the HPA axis’s effects on the adrenal and pituitary glands in the presence of stressors [39]. In skin, keratinocyte production of IL-18 is known to trigger severe cutaneous inflammation. ACTH promotes IL-18 secretion through caspase-1 activation [81], whereas CRH down-regulates IL-18 expression through activation of the p38 MAPK pathway [82]. Thus, IL-18 contributes to the CRH negative feedback loop.
Research has been also conducted on POMC to evaluate its role in skin inflammation. POMC and POMC-derived peptides function by binding MCR1 and MCR2, with MCR1 being the major receptor in keratinocytes, melanocytes, and adipocytes [85]. α-MSH, for example, exhibits anti-inflammatory properties and stimulates skin pigmentation through MCR1 [85].
GCs are major components in the inflammatory response through GC receptor (GR) binding. Research on mice with keratinocyte-restricted GR inactivation (GR epidermal knockout or GR (EKO) mice) has shown that epidermal loss of GR leads to skin barrier defects and susceptibility to cutaneous inflammation. Moreover, macrophage influx and mast cell degranulation have been observed in newborn GR (EKO) skin, both of which are hallmarks of AD [86]. GC synthesis is also a key process in the peripheral HPA axis modulation. There has been evidence that 11β-HSD1, the main enzyme involved in cortisol synthesis, is induced in vitro by the pro-inflammatory cytokines IL-1β and TNF-α. Increased cortisol concentration has also been noted in culture media [70]. Thus, it has been hypothesized that the regulation of cytosolic cortisol concentrations by 11β-HSD1 may modulate inflammatory cytokine expression in keratinocytes [70].
5. HPA Axis and Skin Barrier
In response to various stressors, sensory nerves or humoral factors activate the central and peripheral cutaneous HPA axis to regulate homeostatic responses. These responses serve to counteract cutaneous and systemic environmental damage in order to maintain cutaneous homeostasis [35].
ACTH is involved in melanin production, which protects skin from ultraviolet irradiation. Melanin has been recently found to correlate to the barrier function. Keratinocytes with more melanin display superior barrier function in comparison to lightly-pigmented keratinocytes in organotypic human keratinocytes [87]. In a study of homogenous Chinese population, the epidermal barrier function was inferior in stable, non-inflamed, depigmented (vitiliginous) skin in comparison to adjacent, pigmented skin sites in the same individuals [88]. Hence, through the increased melanin production, ACTH supports preservation of epidermal barrier function (Table 1). On the other hand, β-endorphin also protects skin function by enhancing epidermal turnover rate [89].
Topical and systemic GCs are two of the most effective classes of drugs for treating acute and chronic cutaneous inflammatory dermatoses. The effectiveness of topical GCs in the treatment of AD has been well established [90]. In the skin, GCs target keratinocytes and inflammatory cells in the dermis, where they exhibit their major anti-inflammatory mechanism by suppressing of genes related to transcription factors, e.g., nuclear factor-κB and activator protein-1 [91]. However, additional mechanisms may be involved in the anti-inflammatory response. Studies have shown that topical GCs improve AD-like skin lesions and barrier impairment by suppressing TSLP-related allergic inflammation [92,93]. GCs inhibit TSLP release from keratinocytes in an atopic cytokine environment [93]. However, GCs also affect proliferation, differentiation, and metabolism of keratinocytes [94]. This could possibly compromise the skin barrier function in the long run, resulting in exacerbation of AD. Moreover, prolonged and excessive use of potent topical GCs may contribute to short-term hormonal changes in the central HPA axis [95].
The main endogenous GC found in humans is cortisol, which is produced by the adrenal cortex of the central HPA axis and keratinocytes of the peripheral cutaneous HPA axis. The enzyme 11β-HSD1 acts to activate the epidermal cortisol production in keratinocytes [96]. Cortisol production is increased during wound healing in the epidermis. Environmental dryness might also induce increased cortisol secretion in epidermis of diseased skin [97], which could potentially influence a patient’s psychological well-being and systemic physiology. A study of pulp development in a murine knockout model has demonstrated that fetal GC deficiency delays SC maturation through the reduced expression of the proteins involucrin, loricrin, and filaggrin, and the reduced activity of the lipid synthetic enzymes beta-glucocerebrosidase and steroid sulfatase [98].
6. The Effect of Stress in AD on the HPA Axis
Many chronic skin disorders, including psoriasis, seborrheic dermatitis, rosacea, and AD are adversely affected by PS. However, the responsible mechanisms leading to these pathologies are not yet clearly understood [99]. Through the HPA axis, the immunological changes caused by an endogenous neuroendocrine stress response are different from the immunosuppressive changes resulting from therapeutic GC dosages applied in dermatology. A decreased response from the HPA axis in AD patients is thought to result from the disease itself rather than from topical GC application [100,101]. Some reports have suggested that a chronic Th2-predominant cytokine profile acts as a negative feedback mechanism to decrease the HPA axis response. Accordingly, it was observed that IL-4, a Th2 cytokine, directly inhibited POMC expression in the anterior pituitary in proportion to its concentration [102].
Acute exposure to stressful stimuli activates the HPA axis as well as sympathetic nervous system, leading to physiological responses, such as elevated blood pressure, an increased ACTH, and corticosterone in the plasma [103,104,105]. However, chronic PS exposure leads to HPA axis alterations such as decreased rise of morning cortisol and increased baseline secretion of cortisol [106]. Moreover, continuous PS is not only linked to the HPA axis, but also to several mediators, such as catecholamines, prolactin, neuropeptides, and nerve growth factors [51,107]. Substance P (SP), which is released from sensitive nervous endings, increases sensory perception and pruritus in the skin [38]. However, it also inhibits the HPA axis and therefore modifies the PS response. In the presence of SP, which triggers a shift towards a chronic PS response, the HPA axis response slows down. Under the chronic exposure to PS, there are several pathological changes in the skin, such as a delay in wound healing [77] and an impairment in skin barrier recovery [108,109]. Chronic PS has been shown to result in a disruption of the skin-barrier, as demonstrated by an increase in TEWL in affected mice [108]. Recent findings have suggested that PS decreases epidermal proliferation and differentiation, disrupts the permeability barrier homeostasis, and decreases the skin integrity of the SC [109]. For example, insomniac PS has been shown to change both barrier homeostasis and SC integrity. Insomniac PS inhibition of epidermal lipid synthesis is correlated to decreased lamellar body formation and decreased corneodesmosomes, thereby negatively affecting barrier homeostasis and altering SC integrity [109]. Aioi et al. demonstrated that overcrowding PS resulted in dry skin and barrier impairment. The dry skin and barrier disruption have been associated with decrease in compounds important to skin barrier function, e.g., ceramide and pyrrolidone carboxylic acid [108]. Moreover, Choi et al. reported that elevated GC concentration could result in permeability barrier changes and delayed barrier recovery after tape stripping, which is linked to decreased epidermal lipid synthesis, as observed during PS [109]. Evidence also suggests that administration of RU-486 (a GC receptor antagonist) or antalarmin (a CRH antagonist) in psychologically stressed mice improves barrier recovery [110]. These results emphasize the importance of GCs induced during PS and their effect on SC homeostasis [109,110], suggesting that GCs play a major role in barrier function under stressed conditions.
Systemic and topical GC applications cause adverse effects on skin structure and function comparable to those observed with PS [109,110]. PS increases the production of endogenous GCs, which are released from the adrenal gland in response to stressful stimuli as part of the activation of HPA axis. Acute PS activates metabolic responses that help the host immediately in the short-term, though the current medical paradigm holds that PS actually worsens systemic and cutaneous inflammatory disorders. Additionally, superimposed exogenous (motion-restricted) stress resulted in a reduction in inflammation and improved skin barrier function in a murine AD model, even going as far as normalizing serum immunoglobulin E (IgE) levels [111]. These benefits are the result of increased endogenous GC concentration, which have been explained because the actions of the HPA axis are normally tightly regulated to ensure that the body can respond quickly to stressful events and return to a normal state just as rapidly. Thus, PS, in the acute phase, may be a beneficial protective mechanism by providing anti-inflammatory effects through the production of endogenous GCs. This is a different view to the current paradigm of PS aggravating cutaneous inflammatory dermatoses [111]. When PS is maintained and sustained over time, the adverse consequences have been shown in AD. Persistent PS may stimulate a rise in endogenous GCs which compromises the permeability barrier homeostasis, SC cohesion, wound healing, and epidermal innate immunity in skin. Persistent exposure of elevated GC burden causes not only body homeostasis disturbance (e.g., blunting the HPA axis response), but also adverse sequelae affecting cognitive processes [85,106].
The link between PS and chronic inflammatory dermatoses such as AD is complex. On the one hand, patients with AD have been shown to have reduced production of cortisol and ACTH by the experimental Trier Social Stress Test when compared with non-atopic controls. AD patients had blunted HPA axis reactivity as assessed by cortisol and ACTH measurements [112,113]. On the other hand, AD can elicit PS and release of GCs [1,3]. Hence, chronic GC elevation disturbs neuroendocrine signaling and can induce neuroinflammation, neurotoxicity, and cognitive impairment [106]. One study assessed the effects of AD-induced stress in a mouse AD model and the results have shown that AD-related PS increased astroglial and microglial activation, neuroinflammatory cytokine expression, and markers of neuronal loss [114].
CRH induces arginine vasopressin secretion, which acts synergistically with CRH, especially under chronic stress state [53,61,115,116]. Moreover, CRH has an inhibitory effect on the pineal secretion of melatonin in normal, unaffected persons [117]. Melatonin and its metabolites have been to be important for proper physiological skin function and effective protection of a cutaneous homeostasis from adverse environmental stimuli, and its use can attenuate hyperproliferative/inflammatory conditions [118]. Research has shown that administering melatonin inhibited skin lesions formation, scratching, and elevation of serum IgE levels in the murine AD stressed models [114]. Furthermore, it caused a significant reduction in CRH responsiveness, and a substantial decrease in neuronal damage [114]. Research has been conducted on healthy adults to evaluate the effect of psychological relaxation on perturbed skin barrier. The results showed that the psychological relaxation has been correlated to a beneficial effect on skin barrier recovery regardless of whether before or after injury [119].
Because of itchiness in AD, scratching may lead to a wound, increasing the risk for a secondary bacterial infection. Physicians have been aware that bacteria and other microorganisms are triggers in the etiology of AD [1,3]. PS has also shown to inhibit AMP production which has been shown to result in increased risk for a severe skin infection [120]. Aberg et al. reported that PS increased the severity of skin infection in mice by decreasing the levels of two important AMPs (cathelin-related AMP and beta-defensin 3) in the epidermis, thereby reducing their delivery into lamellar bodies. This may be attributed to increased production of endogenous GCs in response to stress [120]. Moreover, it has been shown that inhibition of CRH and GCs decreased the severity of infection by allowing AMP levels to increase back to baseline levels [120].
7. Conclusions
AD is a chronic inflammatory disease of the skin that results from interplay of environmental stressors and PS response. There is a dual link between AD and PS. On one hand, AD produces PS in patients with the disease. Conversely, AD may also be exacerbated by PS. Currently, the activation of both the central and peripheral cutaneous HPA axes is considered the molecular mediator of PS response, resulting in profound effects on the neuroimmune reaction and skin barrier function. The HPA axis dysfunction has been described in AD patients. Since inflammation and skin barrier function are the two key components of the pathogenetic mechanism of AD, research on PS response is of significant interest. Paradoxically, acute PS, acting through the activation of the HPA axis, displays metabolic responses that provide immediate benefit to the host (demonstrated by murine AD models). Thus, exogenous stressors acting in short duration may be of benefit to inflammation. However, extensive exposure may be harmful to both inflammation and skin barrier function. Therefore, the effects of PS in AD may be either beneficial or harmful, which is dependent on the duration of the stressor or the diverse influences on inflammation and skin barrier function.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| AD | Atopic dermatitis |
| AMP | Antimicrobial peptide |
| α-MSH | Alpha melanocyte-stimulating hormone |
| CRH | Corticotrophin-releasing hormone |
| CRH-R | Corticotrophin-releasing hormone receptor |
| EKO | Epidermal knockout |
| GC | Glucocorticoid |
| GR | Glucocorticoid receptor |
| HPA | Hypothalamic–pituitary–adrenal |
| IL | Interleukin |
| IgE | Immunoglobulin E |
| MCR | Melanocortin receptor |
| POMC | Pro-opiomelanocortin |
| PS | Psychological stress |
| SC | Stratum corneum |
| SP | Substance P |
| TEWL | Transepidermal water loss |
| Th2 | T helper cell type 2 |
| TSLP | Thymic stromal lymphopoietin |
| TNF-α | Tumor necrosis factor alpha |
| 11β-HSD | 11 beta-hydroxysteroid dehydrogenase |
References
- Bieber, T. Atopic dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Wakefield, J.S. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.D.M.; Eichenfield, L.F.M.; Friedlander, S.F.M.; Simpson, E.L.M.M. Review of critical issues in the pathogenesis of atopic dermatitis. Semin. Cutan. Med. Surg. 2016, 35, S89–S91. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Seike, M. Histamine and histamine receptors in allergic dermatitis. Handb. Exp. Pharmacol. 2017, 241, 333–345. [Google Scholar] [PubMed]
- Senra, M.S.; Wollenberg, A. Psychodermatological aspects of atopic dermatitis. Br. J. Dermatol. 2014, 170, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Altemus, M.; Rao, B.; Dhabhar, F.S.; Ding, W.; Granstein, R.D. Stress-induced changes in skin barrier function in healthy women. J. Investig. Dermatol. 2001, 117, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Lack, G. Update on risk factors for food allergy. J. Allergy. Clin. Immunol. 2012, 129, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Naidu, Y. Perturbation of epidermal barrier function correlates with initiation of cytokine cascade in human skin. J. Am. Acad. Dermatol. 1994, 30, 535–546. [Google Scholar] [CrossRef]
- Hon, K.L.; Leung, A.K.; Barankin, B. Barrier repair therapy in atopic dermatitis: An overview. Am. J. Clin. Dermatol. 2013, 14, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.M.M.; Irvine, A.D.M.; Eichenfield, L.F.M.; Friedlander, S.F.M. Update on epidemiology, diagnosis, and disease course of atopic dermatitis. Semin. Cutan. Med. Surg. 2016, 35, S84–S88. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, S.; Kawai, M.; Kasajima, Y.; Hamamoto, T. Increased plasma tumour necrosis factor-α concentration in atopic dermatitis. Arch. Dis. Child. 1992, 67, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, K.A.; Thyssen, J.P. Skin barrier function and allergens. Curr. Probl. Dermatol. 2016, 49, 90–102. [Google Scholar] [PubMed]
- Demehri, S.; Liu, Z.; Lee, J.; Lin, M.H.; Crosby, S.D.; Roberts, C.J.; Grigsby, P.W.; Miner, J.H.; Farr, A.G.; Kopan, R. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008, 6, e123. [Google Scholar]
- Dumortier, A.; Durham, A.D.; Di Piazza, M.; Vauclair, S.; Koch, U.; Ferrand, G.; Ferrero, I.; Demehri, S.; Song, L.L.; Farr, A.G.; et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS ONE 2010, 5, e9258. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Oyoshi, M.K.; Garibyan, L.; Kumar, L.; Ziegler, S.F.; Geha, R.S. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 11875–11880. [Google Scholar] [CrossRef] [PubMed]
- Segawa, R.; Yamashita, S.; Mizuno, N.; Shiraki, M.; Hatayama, T.; Satou, N.; Hiratsuka, M.; Hide, M.; Hirasawa, N. Identification of a cell line producing high levels of TSLP: Advantages for screening of anti-allergic drugs. J. Immunol. Methods. 2014, 402, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Omori, M.; Gyarmati, D.; Zhou, B.; Aye, T.; Brewer, A.; Comeau, M.R.; Campbell, D.J.; Ziegler, S.F. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J. Exp. Med. 2005, 202, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, K.; Fujiyama, T.; Yamaguchi, H.; Waki, M.; Tokura, Y. TSLP directly interacts with skin-homing TH2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J. Investig. Dermatol. 2015, 135, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Folster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.I.; Heuze, M.L.; Martinez-Cingolani, C.; Volpe, E.; Donnadieu, M.H.; Piel, M.; Homey, B.; Lennon-Dumenil, A.M.; Soumelis, V. The human cytokine TSLP triggers a cell-autonomous dendritic cell migration in confined environments. Blood 2011, 118, 3862–3869. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Schaper, K.; Rossbach, K.; Kother, B.; Stark, H.; Kietzmann, M.; Werfel, T.; Gutzmer, R. Stimulation of the histamine 4 receptor upregulates thymic stromal lymphopoietin (TSLP) in human and murine keratinocytes. Pharmacol. Res. 2016, 113, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Allam, J.P.; Biedermann, T.; Eyerich, K.; Gilles, S.; Guttman-Yassky, E.; Hoetzenecker, W.; Knol, E.; Simon, H.U.; Wollenberg, A.; et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Ebrecht, M.; Hellhammer, D.H. Blunted HPA axis responsiveness to stress in atopic patients is associated with the acuity and severeness of allergic inflammation. Brain Behav. Immun. 2010, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Hellhammer, D.H. Endocrine and immune responses to stress in chronic inflammatory skin disorders. Ann. N. Y. Acad. Sci. 2003, 992, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, S. Neuroimmune communication in skin: Far from peripheral. J. Investig. Dermatol. 2008, 128, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Priftis, K.N.; Papadimitriou, A.; Nicolaidou, P.; Chrousos, G.P. The hypothalamic–pituitary–adrenal axis in asthmatic children. Trends Endocrinol. Metab. 2008, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. The hypothalamic–pituitary–adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Chrousos, G.P. Stress system—Organization, physiology and immunoregulation. Neuroimmunomodulation 2006, 13, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ketchesin, K.D.; Stinnett, G.S.; Seasholtz, A.F. Corticotropin-releasing hormone-binding protein and stress: From invertebrates to humans. Stress 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Tobin, D.J.; Theoharides, T.C.; Rivier, J. Key role of CRF in the skin stress response system. Endocr. Rev. 2013, 34, 827–884. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W. Proopiomelanocortin (POMC): The cutaneous roles of its melanocortin products and receptors. Clin. Exp. Dermatol. 2006, 31, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Airaghi, L.; Colombo, G.; Lipton, J.M. α-Melanocyte-stimulating hormone in normal human physiology and disease states. Trends Endocrinol. Metab. 2000, 11, 304–308. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–115. [Google Scholar]
- Kim, J.E.; Cho, B.K.; Cho, D.H.; Park, H.J. Expression of hypothalamic–pituitary–adrenal axis in common skin diseases: Evidence of its association with stress-related disease activity. Acta. Derm. Venereol. 2013, 93, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. HPA axis responsiveness to stress: Implications for healthy aging. Exp. Gerontol. 2011, 46, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Zen, M.; Canova, M.; Campana, C.; Bettio, S.; Nalotto, L.; Rampudda, M.; Ramonda, R.; Iaccarino, L.; Doria, A. The kaleidoscope of glucorticoid effects on immune system. Autoimmun. Rev. 2011, 10, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Ebrecht, M.; Kern, S.; Hellhammer, D.H. Endocrine stress responses in TH1-mediated chronic inflammatory skin disease (psoriasis vulgaris)—Do they parallel stress-induced endocrine changes in TH2-mediated inflammatory dermatoses (atopic dermatitis)? Psychoneuroendocrinology 2006, 31, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Kalogeromitros, D. The critical role of mast cells in allergy and inflammation. Ann. N. Y. Acad. Sci. 2006, 1088, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Ito, T.; Kromminga, A.; Bettermann, A.; Takigawa, M.; Kees, F.; Straub, R.H.; Paus, R. Human hair follicles display a functional equivalent of the hypothalamic–pituitary–adrenal axis and synthesize cortisol. FASEB J. 2005, 19, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Tuckey, R.C.; Paus, R. Differential expression of HPA axis homolog in the skin. Mol. Cell Endocrinol. 2007, 265–266, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.M.; Liezmann, C.; Klapp, B.F.; Kruse, J. The neuroimmune connection interferes with tissue regeneration and chronic inflammatory disease in the skin. Ann. N. Y. Acad. Sci. 2012, 1262, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, N. Acute and chronic stress induced changes in sensitivity of peripheral inflammatory pathways to the signals of multiple stress systems—2011 Curt Richter Award Winner. Psychoneuroendocrinology 2012, 37, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Harvima, I.T.; Nilsson, G.; Naukkarinen, A. Role of mast cells and sensory nerves in skin inflammation. G. Ital. Dermatol. Venereol. 2010, 145, 195–204. [Google Scholar] [PubMed]
- Dhabhar, F.S.; Malarkey, W.B.; Neri, E.; McEwen, B.S. Stress-induced redistribution of immune cells—From barracks to boulevards to battlefields: A tale of three hormones—Curt Richter Award winner. Psychoneuroendocrinology 2012, 37, 1345–1368. [Google Scholar] [CrossRef] [PubMed]
- Arck, P.C.; Slominski, A.; Theoharides, T.C.; Peters, E.M.; Paus, R. Neuroimmunology of stress: Skin takes center stage. J. Investig. Dermatol. 2006, 126, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Pisarchik, A.; Slominski, A. Molecular and functional characterization of novel CRFR1 isoforms from the skin. Eur. J. Biochem. 2004, 271, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020. [Google Scholar] [PubMed]
- Gantz, I.; Fong, T.M. The melanocortin system. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E468–E474. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Zmijewski, M.; Slominski, R.M.; Kauser, S.; Wortsman, J.; Tobin, D.J. Corticotropin releasing hormone and the skin. Front. Biosci. 2006, 11, 2230–2248. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Pisarchik, A.; Tobin, D.J.; Mazurkiewicz, J.E.; Wortsman, J. Differential expression of a cutaneous corticotropin-releasing hormone system. Endocrinology 2004, 145, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Nagata, H.; Umemura, S.; Kawana, S.; Osamura, R.Y. In situ expression of corticotropin-releasing hormone (CRH) and proopiomelanocortin (POMC) genes in human skin. FASEB J. 2001, 15, 2297–2299. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Chrousos, G.P. Stress Hormones, Th1/Th2 patterns, Pro/Anti-inflammatory Cytokines and Susceptibility to Disease. Trends. Endocrinol. Metab. 1999, 10, 359–368. [Google Scholar] [CrossRef]
- Cemil, B.C.; Canpolat, F.; Yilmazer, D.; Eskioglu, F.; Alper, M. The association of PASI scores with CRH-R1 expression in patients with psoriasis. Arch. Dermatol. Res. 2012, 304, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Donelan, J.M.; Papadopoulou, N.; Cao, J.; Kempuraj, D.; Conti, P. Mast cells as targets of corticotropin-releasing factor and related peptides. Trends Pharmacol. Sci. 2004, 25, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Pisarchik, A.; Zbytek, B.; Linton, E.A.; Mazurkiewicz, J.E.; Wei, E.T. Cutaneous expression of corticotropin-releasing hormone (CRH), urocortin, and CRH receptors. FASEB J. 2001, 15, 1678–1693. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Pisarchik, A.; Slominski, R.M.; Zmijewski, M.A.; Wortsman, J. CRH functions as a growth factor/cytokine in the skin. J. Cell Physiol. 2006, 206, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Pikula, M.; Slominski, R.M.; Mysliwski, A.; Wei, E.; Wortsman, J.; Slominski, A.T. Corticotropin-releasing hormone triggers differentiation in HaCaT keratinocytes. Br. J. Dermatol. 2005, 152, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Slominski, A.T. Corticotropin-releasing hormone induces keratinocyte differentiation in the adult human epidermis. J. Cell Physiol. 2005, 203, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Mysliwski, A.; Slominski, A.; Wortsman, J.; Wei, E.T.; Mysliwska, J. Corticotropin-releasing hormone affects cytokine production in human HaCaT keratinocytes. Life Sci. 2002, 70, 1013–1021. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Singh, L.K.; Boucher, W.; Pang, X.; Letourneau, R.; Webster, E.; Chrousos, G. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 1998, 139, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.L.; Yu, X.J.; Chen, L.M.; Jiang, H.; Li, C.Y. Corticotropin-releasing hormone attenuates vascular endothelial growth factor release from human HaCaT keratinocytes. Regul. Pept. 2010, 160, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.T.; Gao, G.C. Corticotropin-releasing factor: An inhibitor of vascular leakage in rat skeletal muscle and brain cortex after injury. Regul. Pept. 1991, 33, 93–104. [Google Scholar] [CrossRef]
- Zbytek, B.; Pfeffer, L.M.; Slominski, A.T. CRH inhibits NF-κB signaling in human melanocytes. Peptides 2006, 27, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Itoi, S.; Terao, M.; Murota, H.; Katayama, I. 11β-Hydroxysteroid dehydrogenase 1 contributes to the pro-inflammatory response of keratinocytes. Biochem. Biophys. Res. Commun. 2013, 440, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. 11β-Hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Murota, H.; Kimura, A.; Kato, A.; Ishikawa, A.; Igawa, K.; Miyoshi, E.; Katayama, I. 11β-Hydroxysteroid dehydrogenase-1 is a novel regulator of skin homeostasis and a candidate target for promoting tissue repair. PLoS ONE 2011, 6, e25039. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Walker, E.A.; Hardy, R.S.; Mayes, A.E.; Stewart, P.M. Localization, age- and site-dependent expression, and regulation of 11β-hydroxysteroid dehydrogenase type 1 in skin. J. Investig. Dermatol. 2011, 131, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, N.; Prime, S.S. Keratinocytes synthesize and activate cortisol. J. Cell Biochem. 2011, 112, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Hupe, M.; Uchida, Y.; Mauro, T.; Elias, P.M.; Holleran, W.M. Increased glucocorticoid activation during mouse skin wound healing. J. Endocrinol. 2014, 221, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Youm, J.K.; Park, K.; Uchida, Y.; Chan, A.; Mauro, T.M.; Holleran, W.M.; Elias, P.M. Local blockade of glucocorticoid activation reverses stress- and glucocorticoid-induced delays in cutaneous wound healing. Wound Repair Regen. 2013, 21, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.M. Neuroendocrine regulation of autoimmune/inflammatory disease. J. Endocrinol. 2001, 169, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection versus immunopathology. Allergy Asthma Clin. Immunol. 2008, 4, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S.; McEwen, B.S. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: A potential role for leukocyte trafficking. Brain Behav. Immun. 1997, 11, 286–306. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, H.J.; Lee, J.Y.; Cho, B.K.; Gallo, R.L.; Cho, D.H. Adrenocorticotropin hormone stimulates interleukin-18 expression in human HaCaT keratinocytes. J. Investig. Dermatol. 2007, 127, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, H.J.; Lee, J.H.; Lee, J.Y.; Cho, B.K.; Kang, J.S.; Kang, H.; Yang, Y.; Cho, D.H. Corticotropin-releasing hormone (CRH) downregulates interleukin-18 expression in human HaCaT keratinocytes by activation of p38 mitogen-activated protein kinase (MAPK) pathway. J. Investig. Dermatol. 2005, 124, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Lonne-Rahm, S.B.; Rickberg, H.; El-Nour, H.; Marin, P.; Azmitia, E.C.; Nordlind, K. Neuroimmune mechanisms in patients with atopic dermatitis during chronic stress. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Schut, C.; Weik, U.; Tews, N.; Gieler, U.; Deinzer, R.; Kupfer, J. Psychophysiological effects of stress management in patients with atopic dermatitis: A randomized controlled trial. Acta Derm. Venereol. 2013, 93, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Latorre, V.; Sanchis, A.; Perez, P. Epidermal inactivation of the glucocorticoid receptor triggers skin barrier defects and cutaneous inflammation. J. Investig. Dermatol. 2013, 133, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Man, M.Q.; Lin, T.K.; Santiago, J.L.; Celli, A.; Zhong, L.; Huang, Z.M.; Roelandt, T.; Hupe, M.; Sundberg, J.P.; Silva, K.A.; et al. Basis for enhanced barrier function of pigmented skin. J. Investig. Dermatol. 2014, 134, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Man, W.Y.; Lv, C.Z.; Song, S.P.; Shi, Y.J.; Elias, P.M.; Man, M.Q. Epidermal permeability barrier recovery is delayed in vitiligo-involved sites. Skin Pharmacol. Physiol. 2010, 23, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bigliardi, P.L.; Sumanovski, L.T.; Buchner, S.; Rufli, T.; Bigliardi-Qi, M. Different expression of mu-opiate receptor in chronic and acute wounds and the effect of β-endorphin on transforming growth factor β type II receptor and cytokeratin 16 expression. J. Investig. Dermatol. 2003, 120, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.Y.; Jung, M.; Kim, D.H.; Lee, H.J.; Choi, E.H. Topical glucocorticoid or pimecrolimus treatment suppresses thymic stromal lymphopoietin-related allergic inflammatory mechanism in an oxazolone-induced atopic dermatitis murine model. Arch. Dermatol. Res. 2015, 307, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Le, T.A.; Takai, T.; Vu, A.T.; Kinoshita, H.; Ikeda, S.; Ogawa, H.; Okumura, K. Glucocorticoids inhibit double-stranded RNA-induced thymic stromal lymphopoietin release from keratinocytes in an atopic cytokine milieu more effectively than tacrolimus. Int. Arch. Allergy Immunol. 2010, 153, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, O.; Lee, B.; Vouthounis, C.; Vukelic, S.; Pastar, I.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: Inhibition of apoptosis, interferon-γ pathway, and wound healing along with promotion of terminal differentiation. J. Biol. Chem. 2007, 282, 4021–4034. [Google Scholar] [CrossRef] [PubMed]
- Mooney, E.; Rademaker, M.; Dailey, R.; Daniel, B.S.; Drummond, C.; Fischer, G.; Foster, R.; Grills, C.; Halbert, A.; Hill, S.; et al. Adverse effects of topical corticosteroids in paediatric eczema: Australasian consensus statement. Australas. J. Dermatol. 2015, 56, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Itoi, S.; Murota, H.; Katayama, I. Expression profiles of cortisol-inactivating enzyme, 11β-hydroxysteroid dehydrogenase-2, in human epidermal tumors and its role in keratinocyte proliferation. Exp. Dermatol. 2013, 22, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Denda, S.; Kumamoto, J.; Denda, M. Low environmental humidity induces synthesis and release of cortisol in an epidermal organotypic culture system. Exp. Dermatol. 2013, 22, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.; Feingold, K.R.; Komuves, L.G.; Elias, P.M.; Muglia, L.J.; Majzoub, J.A.; Williams, M.L. Glucocorticoid deficiency delays stratum corneum maturation in the fetal mouse. J. Investig. Dermatol. 1998, 111, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Takigawa, M. Anxiety in allergy and atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Matsuda, A.; Nomura, I.; Matsubara, O.; Nonoyama, S.; Ohya, Y.; Saito, H.; Matsumoto, K. Salivary cortisol response to stress in young children with atopic dermatitis. Pediatr. Dermatol. 2013, 30, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Haeck, I.M.; Timmer-de Mik, L.; Lentjes, E.G.; Buskens, E.; Hijnen, D.J.; Guikers, C.; Bruijnzeel-Koomen, C.A.; de Bruin-Weller, M.S. Low basal serum cortisol in patients with severe atopic dermatitis: Potent topical corticosteroids wrongfully accused. Br. J. Dermatol. 2007, 156, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Harbuz, M.S.; Stephanou, A.; Knight, R.A.; Chover-Gonzalez, A.J.; Lightman, S.L. Action of interleukin-2 and interleukin-4 on CRF mRNA in the hypothalamus and POMC mRNA in the anterior pituitary. Brain Behav. Immun. 1992, 6, 214–222. [Google Scholar] [CrossRef]
- Watanabe, T.; Fujioka, T.; Hashimoto, M.; Nakamura, S. Stress and brain angiotensin II receptors. Crit. Rev. Neurobiol. 1998, 12, 305–317. [Google Scholar] [CrossRef] [PubMed]
- DiMicco, J.A.; Sarkar, S.; Zaretskaia, M.V.; Zaretsky, D.V. Stress-induced cardiac stimulation and fever: Common hypothalamic origins and brainstem mechanisms. Auton. Neurosci. 2006, 126–127, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic–pituitary–adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Stephens, M.A.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol Res. 2012, 34, 468–483. [Google Scholar] [PubMed]
- Fukada, M.; Kaidoh, T.; Ito, A.; Yano, T.; Hayashibara, C.; Watanabe, T. “Green odor” inhalation reduces the skin-barrier disruption induced by chronic restraint stress in rats: Physiological and histological examinations. Chem. Senses 2007, 32, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Aioi, A.; Okuda, M.; Matsui, M.; Tonogaito, H.; Hamada, K. Effect of high population density environment on skin barrier function in mice. J. Dermatol. Sci. 2001, 25, 189–197. [Google Scholar] [CrossRef]
- Choi, E.H.; Brown, B.E.; Crumrine, D.; Chang, S.; Man, M.Q.; Elias, P.M.; Feingold, K.R. Mechanisms by which psychologic stress alters cutaneous permeability barrier homeostasis and stratum corneum integrity. J. Investig. Dermatol. 2005, 124, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Demerjian, M.; Crumrine, D.; Brown, B.E.; Mauro, T.; Elias, P.M.; Feingold, K.R. Glucocorticoid blockade reverses psychological stress-induced abnormalities in epidermal structure and function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1657–R1662. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Man, M.Q.; Santiago, J.L.; Scharschmidt, T.C.; Hupe, M.; Martin-Ezquerra, G.; Youm, J.K.; Zhai, Y.; Trullas, C.; Feingold, K.R.; et al. Paradoxical benefits of psychological stress in inflammatory dermatoses models are glucocorticoid mediated. J. Investig. Dermatol. 2014, 134, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Gierens, A.; Hollig, H.; Hellhammer, D.H. Stress-induced immunomodulation is altered in patients with atopic dermatitis. J. Neuroimmunol. 2002, 129, 161–167. [Google Scholar] [CrossRef]
- Buske-Kirschbaum, A.; Geiben, A.; Hollig, H.; Morschhauser, E.; Hellhammer, D. Altered responsiveness of the hypothalamus-pituitary-adrenal axis and the sympathetic adrenomedullary system to stress in patients with atopic dermatitis. J. Clin. Endocrinol. Metab. 2002, 87, 4245–4251. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Lee, S.H.; Oh, D.S.; Kim, Y.U. Melatonin inhibits neuronal dysfunction-associated with neuroinflammation by atopic psychological stress in NC/Nga atopic-like mouse models. J. Pineal. Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann. N. Y. Acad. Sci. 1998, 851, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Tsigos, C.; Chrousos, G.P. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Kellner, M.; Yassouridis, A.; Manz, B.; Steiger, A.; Holsboer, F.; Wiedemann, K. Corticotropin-releasing hormone inhibits melatonin secretion in healthy volunteers—A potential link to low-melatonin syndrome in depression? Neuroendocrinology 1997, 65, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Semak, I.; Kim, T.K.; Janjetovic, Z.; Slominski, R.M.; Zmijewski, J.W. Melatonin, mitochondria, and the skin. Cell Mol. Life Sci. 2017, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.; Jarrett, P.; Broadbent, E. The effects of relaxation before or after skin damage on skin barrier recovery: A preliminary study. Psychosom. Med. 2015, 77, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.M.; Radek, K.A.; Choi, E.H.; Kim, D.K.; Demerjian, M.; Hupe, M.; Kerbleski, J.; Gallo, R.L.; Ganz, T.; Mauro, T.; et al. Psychological stress downregulates epidermal antimicrobial peptide expression and increases severity of cutaneous infections in mice. J. Clin. Investig. 2007, 117, 3339–3349. [Google Scholar] [CrossRef] [PubMed]
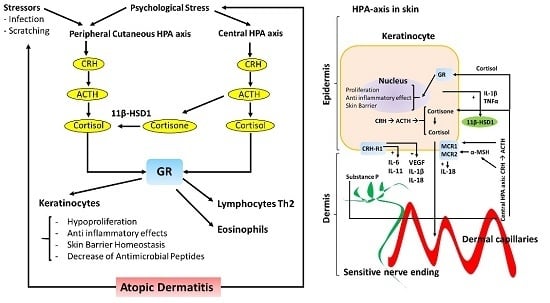
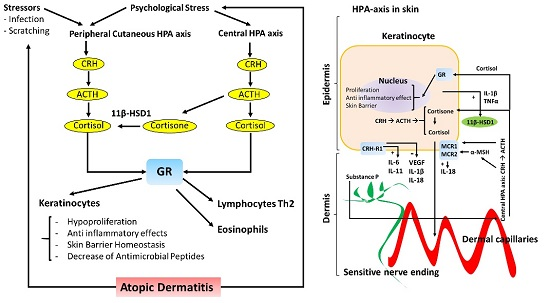
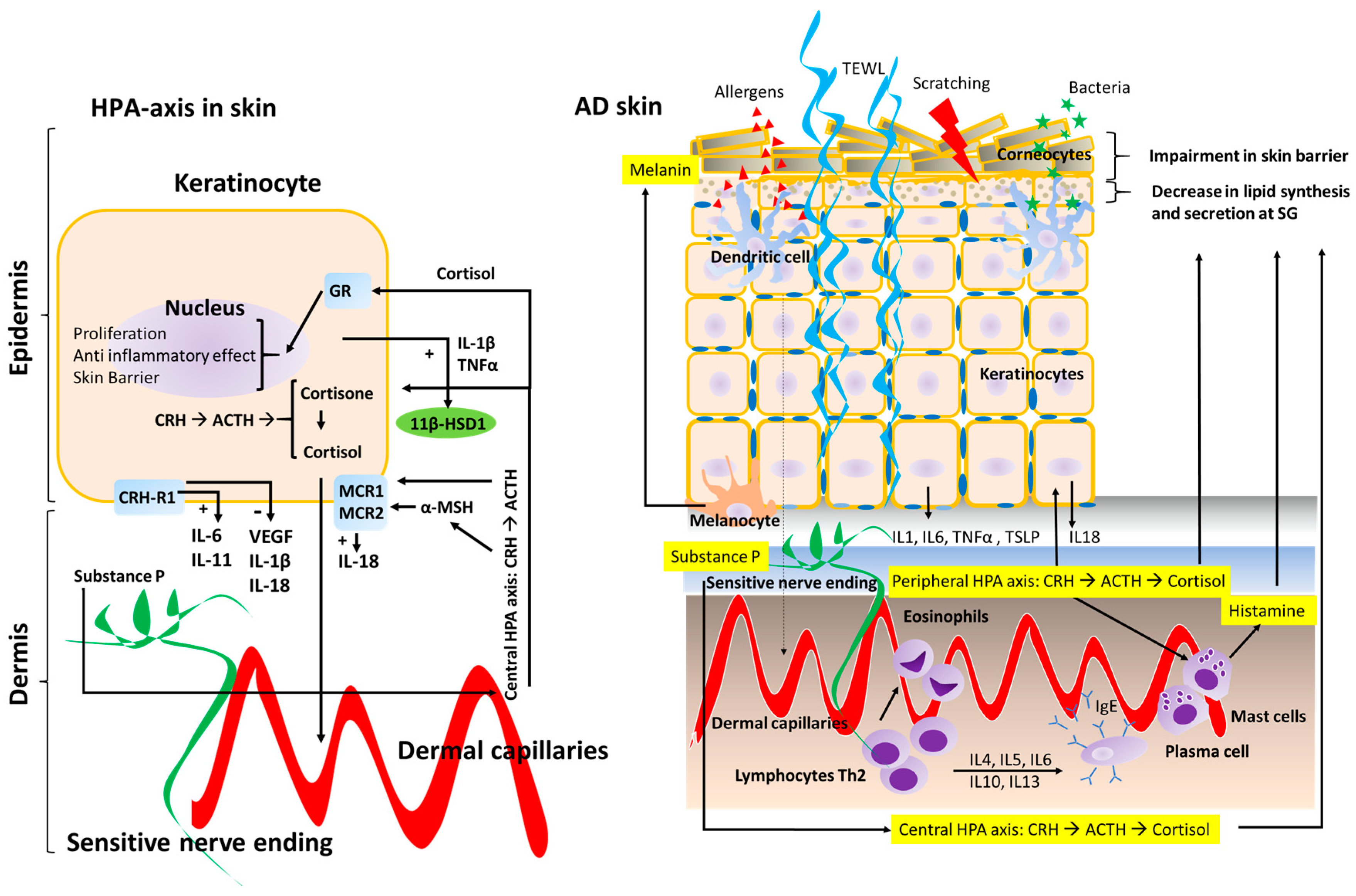
Figure 1.
Schematic representation of the Hypothalamic-Pituitary-Adrenal (HPA) axis in the skin. The effects on keratinocytes (left diagram) on AD skin in regards to skin barrier structure and inflammatory response (right diagram). The central HPA axis and the peripheral HPA axis both regulate skin barrier homeostasis and the inflammatory response in the skin. Peripheral nerve endings in the skin are represented in green, whereas dermal capillaries are shown in red. Transepidermal water loss (TEWL) is represented by curled blue lines (right diagram). Corticotrophin-releasing hormone receptor type 1 (CRH-R1); Glucocorticoid receptor (GR); Melanocortin receptor type 1 and 2 (MCR1 and MCR2); 11 beta-hydroxysteroid dehydrogenase 1 (11β-HSD1).
Figure 1.
Schematic representation of the Hypothalamic-Pituitary-Adrenal (HPA) axis in the skin. The effects on keratinocytes (left diagram) on AD skin in regards to skin barrier structure and inflammatory response (right diagram). The central HPA axis and the peripheral HPA axis both regulate skin barrier homeostasis and the inflammatory response in the skin. Peripheral nerve endings in the skin are represented in green, whereas dermal capillaries are shown in red. Transepidermal water loss (TEWL) is represented by curled blue lines (right diagram). Corticotrophin-releasing hormone receptor type 1 (CRH-R1); Glucocorticoid receptor (GR); Melanocortin receptor type 1 and 2 (MCR1 and MCR2); 11 beta-hydroxysteroid dehydrogenase 1 (11β-HSD1).

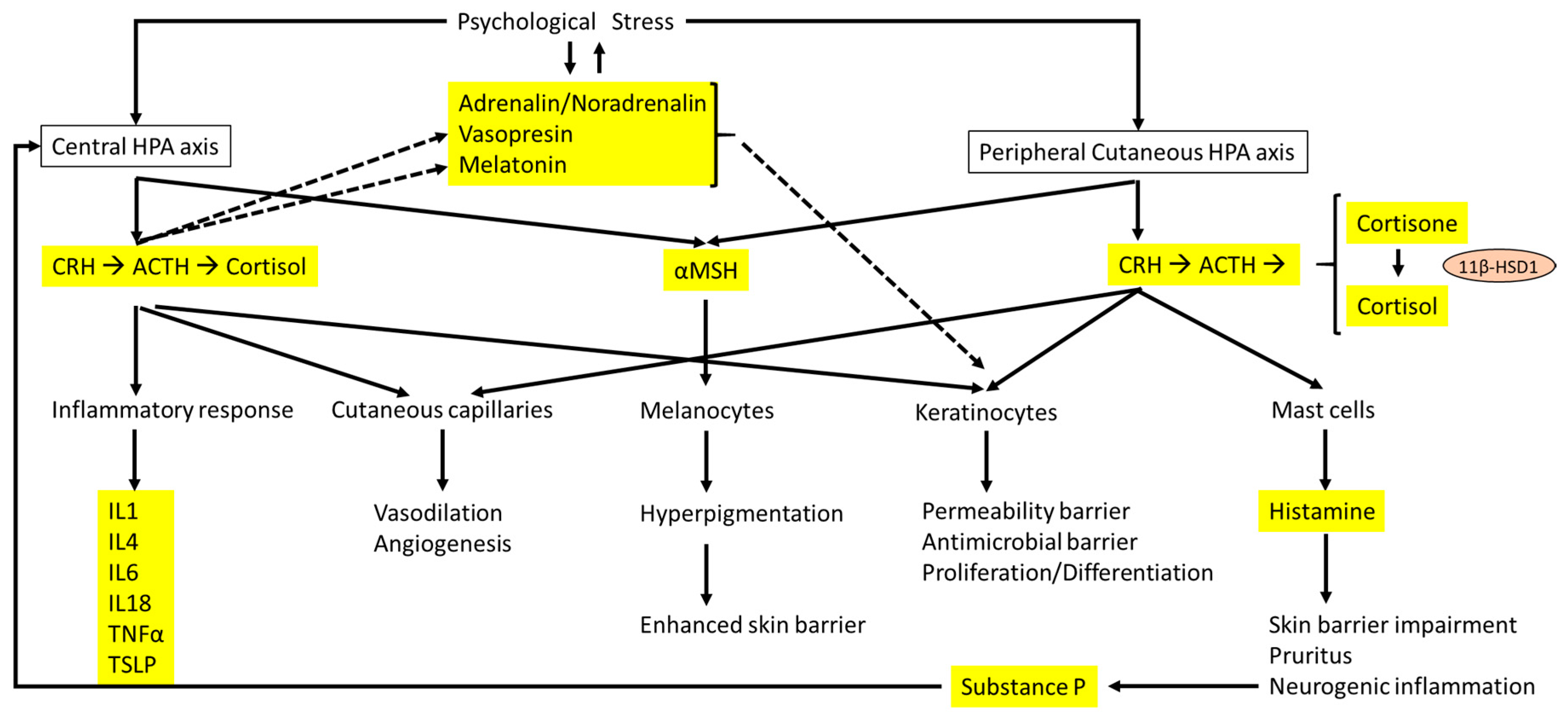
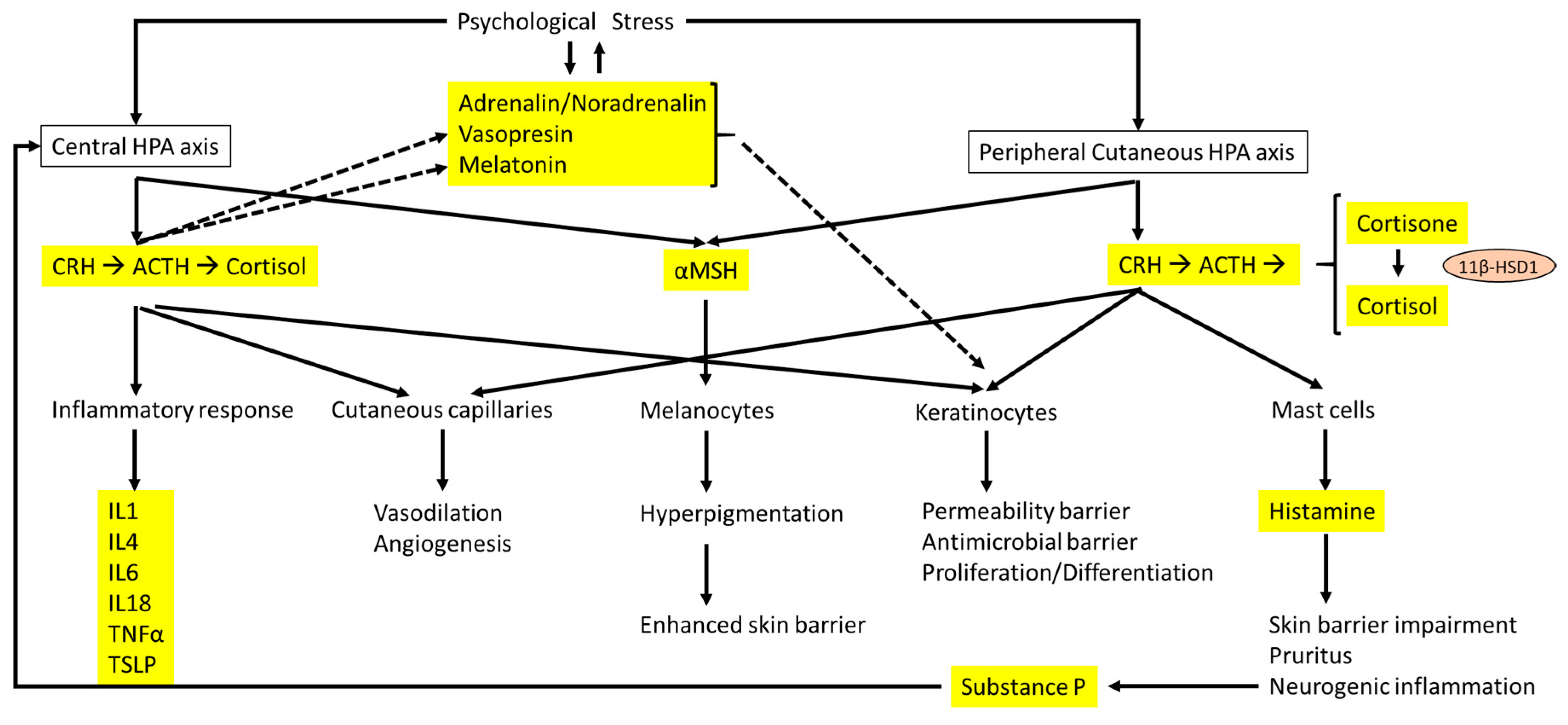
Figure 2.
Interactions between the central HPA axis and the cutaneous HPA axis and the effects on the skin barrier and dermatitis. The main hormones, neurotransmitters, and cytokines are highlighted in yellow. Solid arrows represent direct effect of hormones and neurotransmitters related to the HPA axis, whereas dashed lines indicate the influence of psychological stress on other hormones and neurotransmitters that are different from the HPA axis and linked to epidermal homeostasis.
Figure 2.
Interactions between the central HPA axis and the cutaneous HPA axis and the effects on the skin barrier and dermatitis. The main hormones, neurotransmitters, and cytokines are highlighted in yellow. Solid arrows represent direct effect of hormones and neurotransmitters related to the HPA axis, whereas dashed lines indicate the influence of psychological stress on other hormones and neurotransmitters that are different from the HPA axis and linked to epidermal homeostasis.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Effects of peripheral HPA on skin barrier.
| Elements | Psychological Effects | Impacts on Skin Barrier |
|---|---|---|
| ACTH | Melanin production | Enhancement |
| α-MSH | Melanin production | Enhancement |
| Strong anti-inflammatory effects | Protection | |
| β-endorphin | Enhances the epidermal turnover rate | Protection |
| Cortisol | Acute PS: anti-inflammatory effects | Protection |
| Chronic PS: Downregulation of AMPs | Compromising | |
| 11β-HSD1 | Cortisol production | Delay in wound healing |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, T.-K.; Zhong, L.; Santiago, J.L. Association between Stress and the HPA Axis in the Atopic Dermatitis. Int. J. Mol. Sci. 2017, 18, 2131. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102131
AMA Style
Lin T-K, Zhong L, Santiago JL. Association between Stress and the HPA Axis in the Atopic Dermatitis. International Journal of Molecular Sciences. 2017; 18(10):2131. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102131
Chicago/Turabian StyleLin, Tzu-Kai, Lily Zhong, and Juan Luis Santiago. 2017. "Association between Stress and the HPA Axis in the Atopic Dermatitis" International Journal of Molecular Sciences 18, no. 10: 2131. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102131
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.