A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
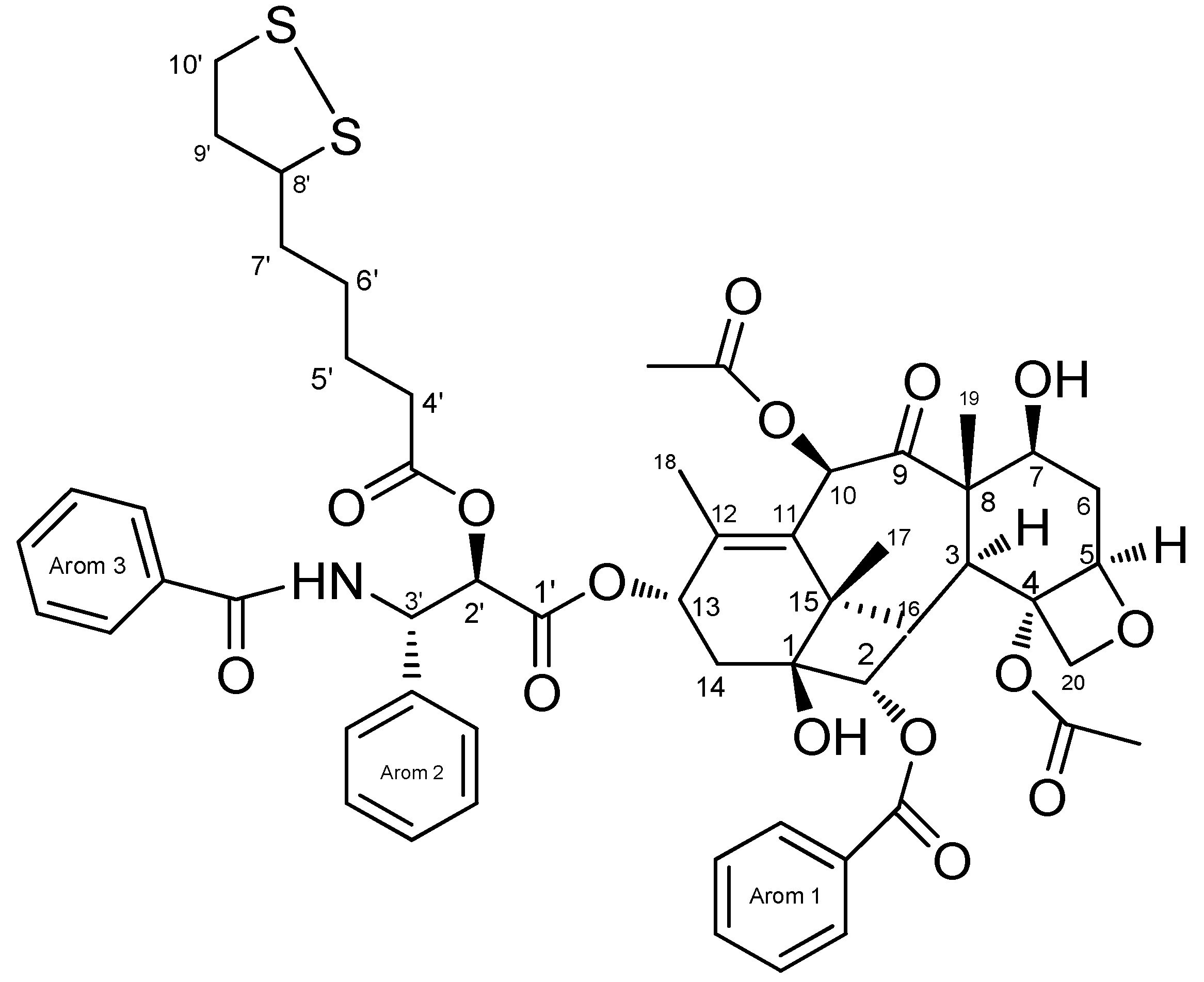
2.1. NMR Assignments
2.2. 13C-NMR (CDCl3)
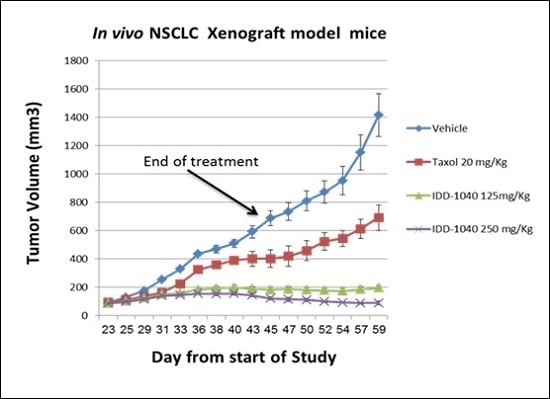
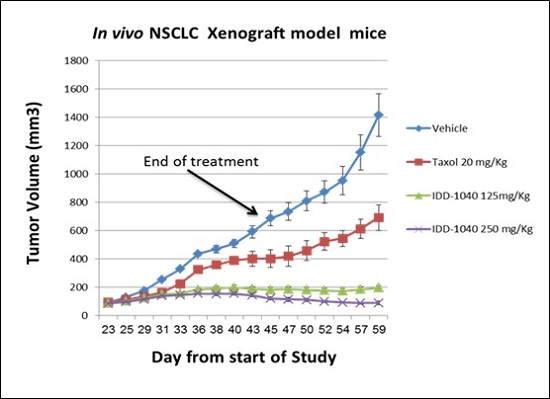
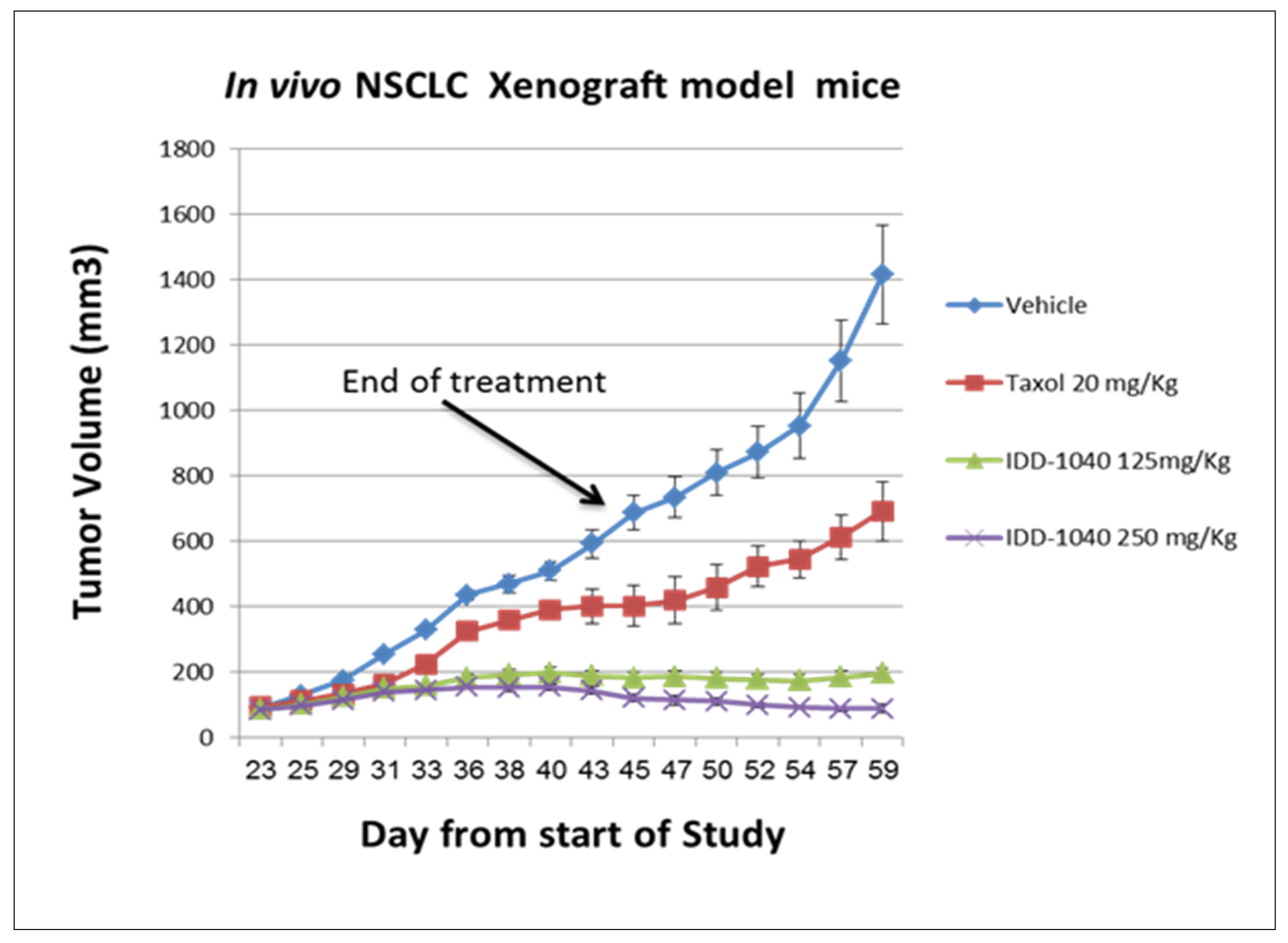
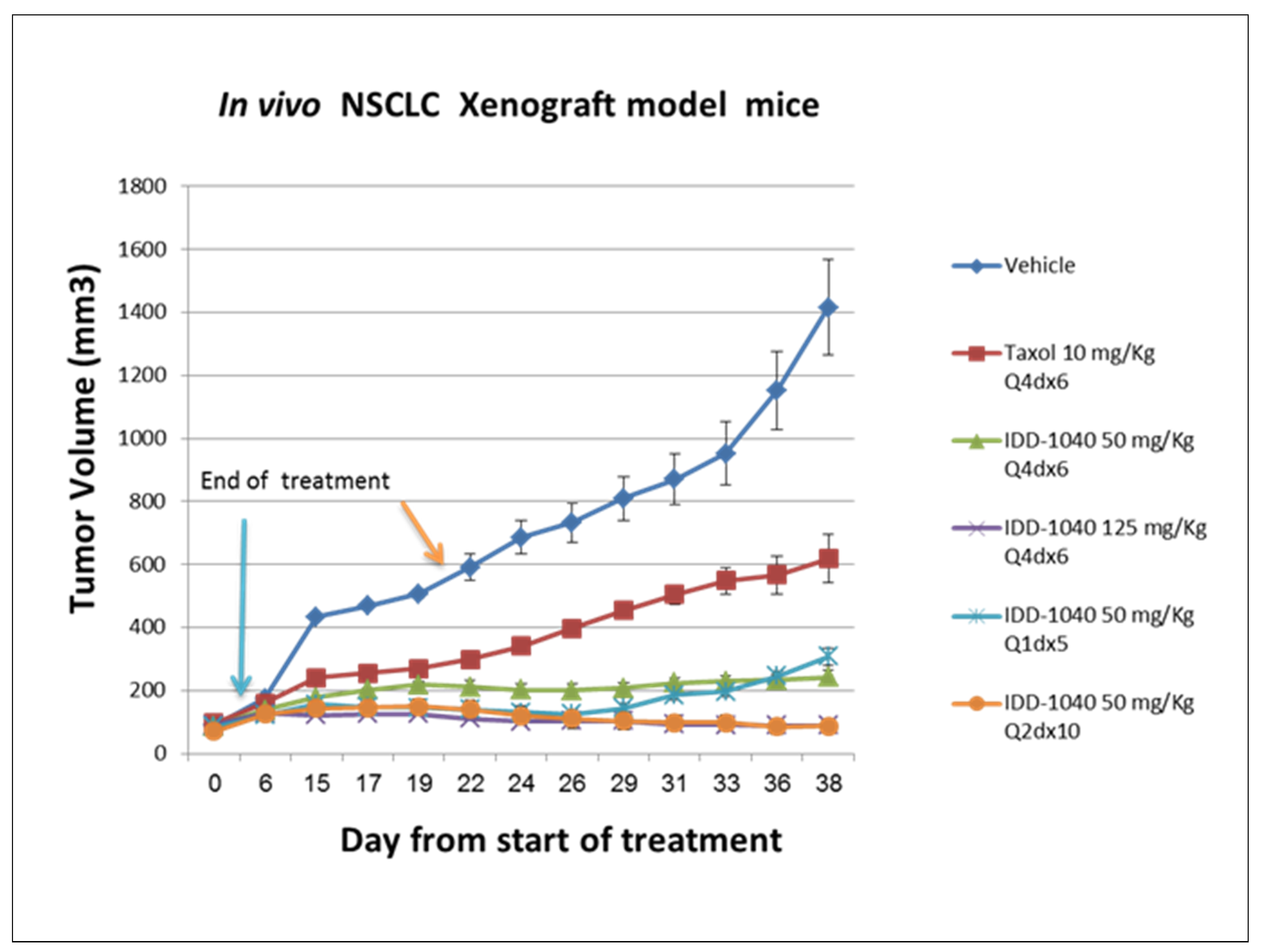
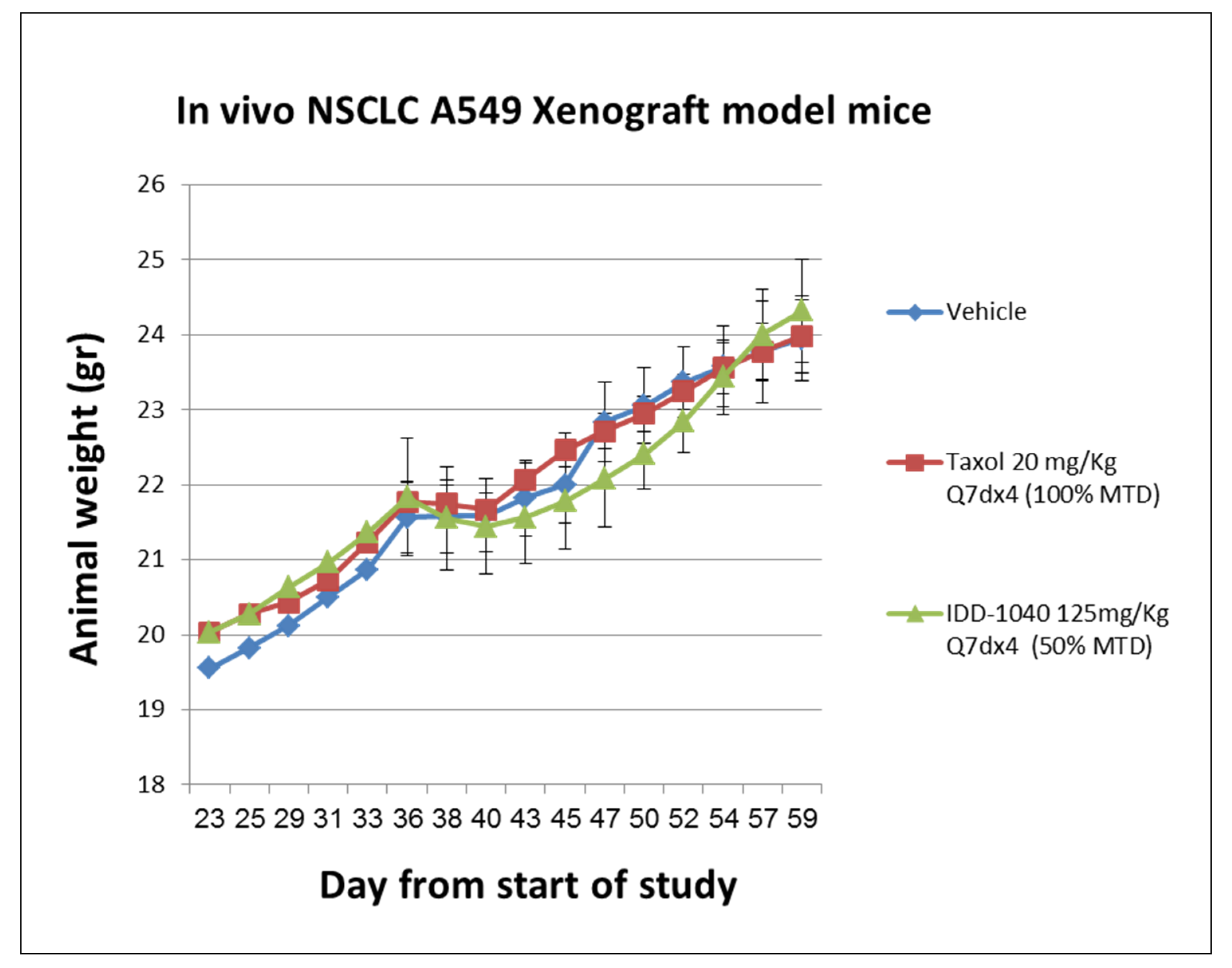
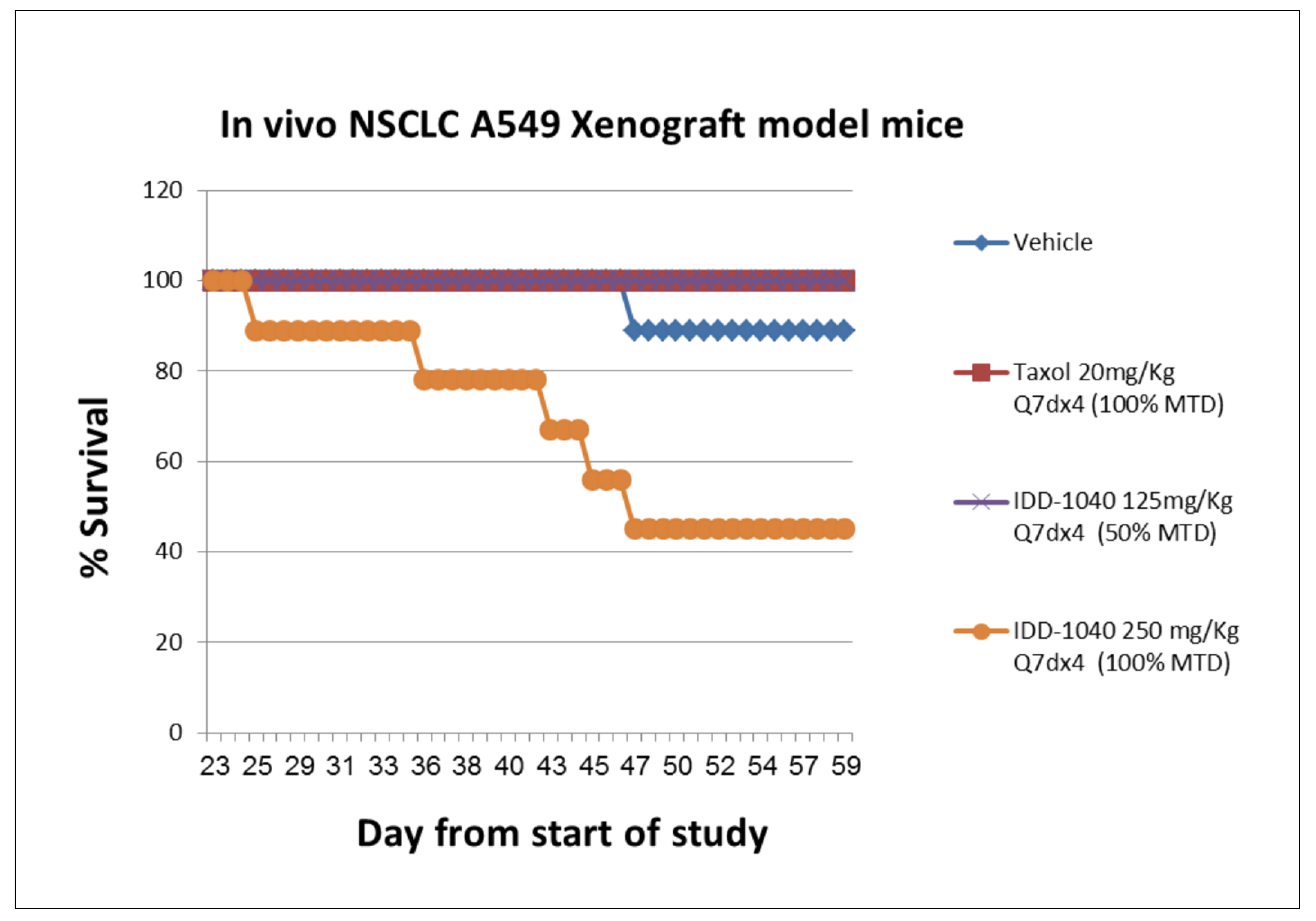
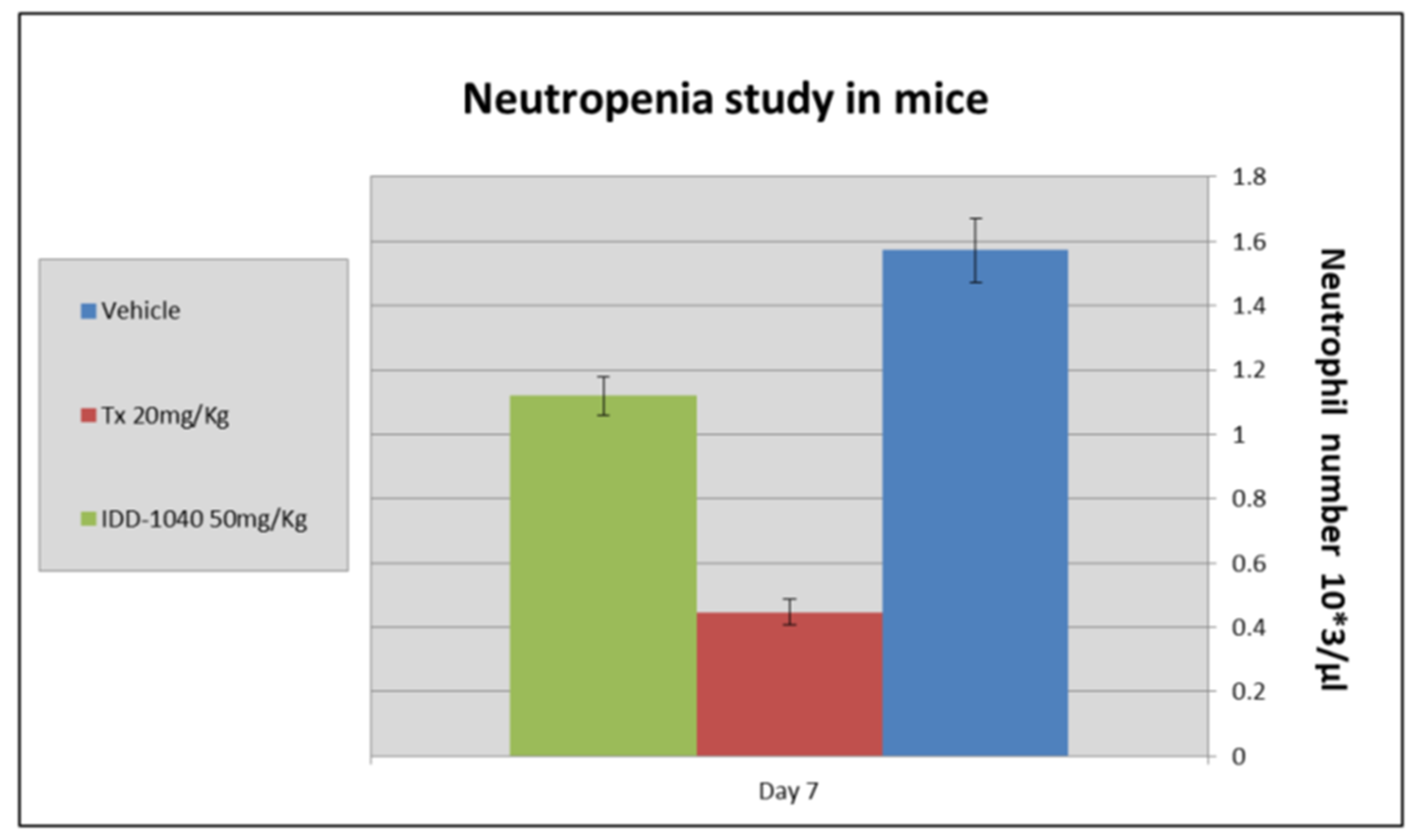
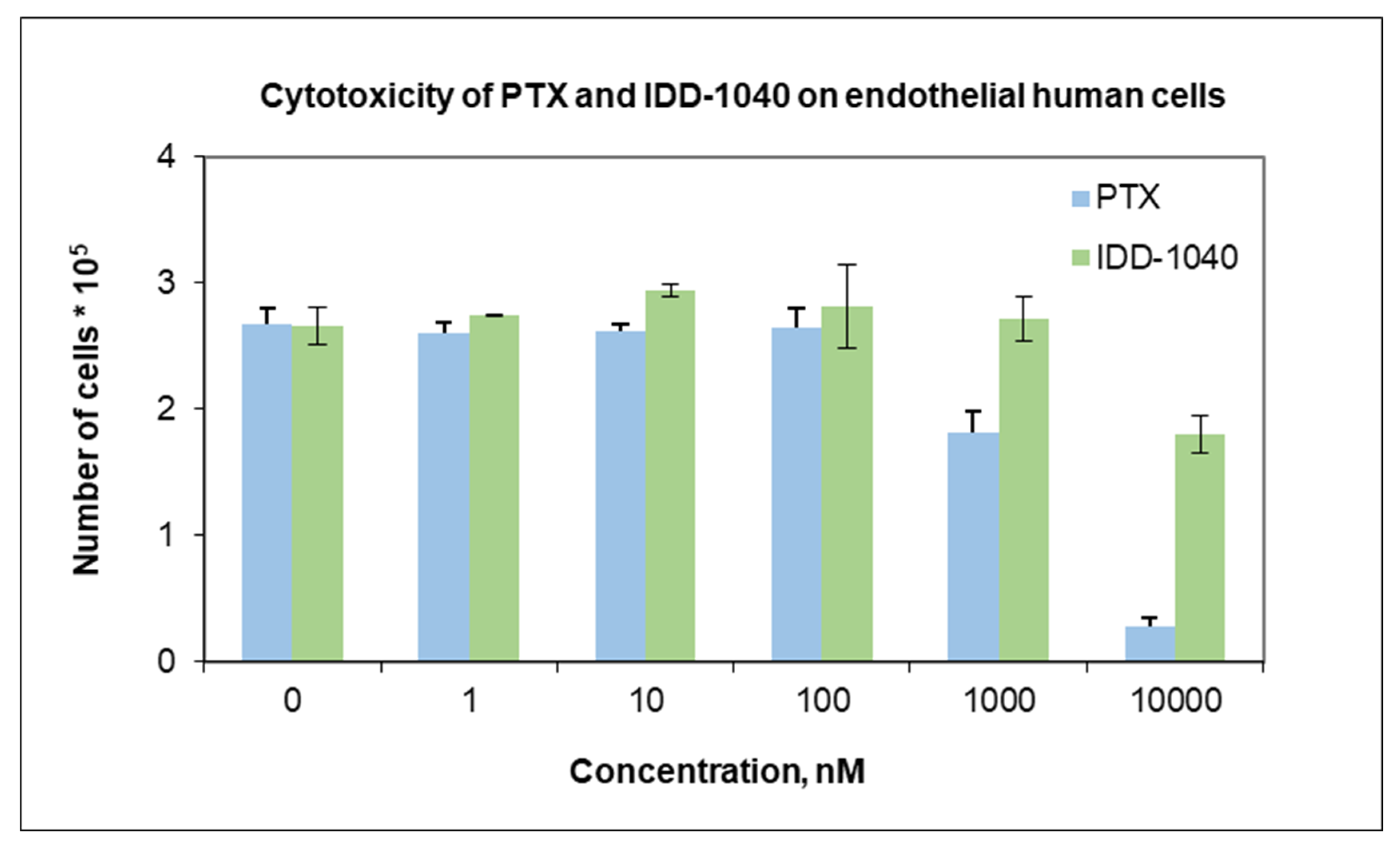
2.3. Biological Study Results
3. Materials and Methods
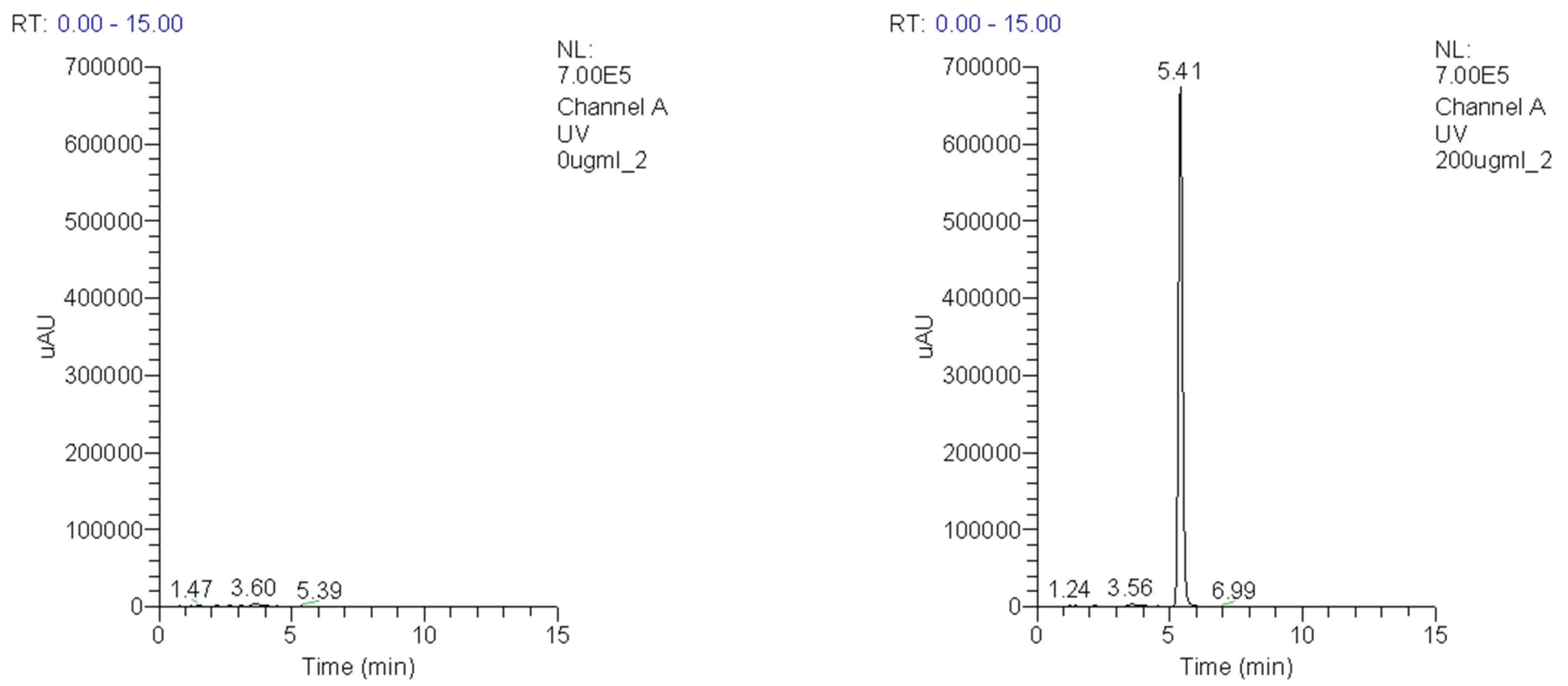
3.1. Purity of IDD-1040
3.2. Chemical Characterization
3.2.1. 1H-NMR and 13C-NMR
3.2.2. Elemental Analysis
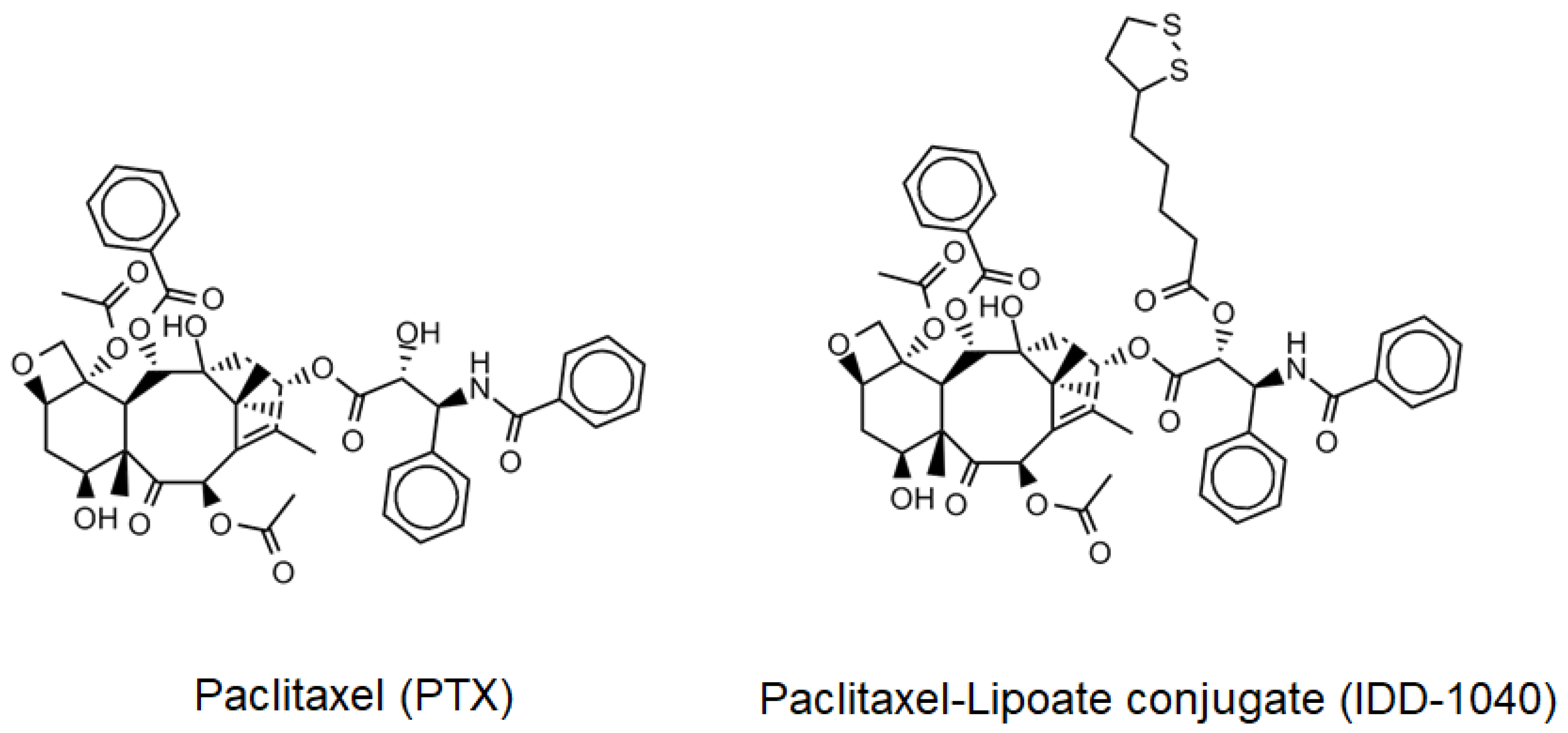
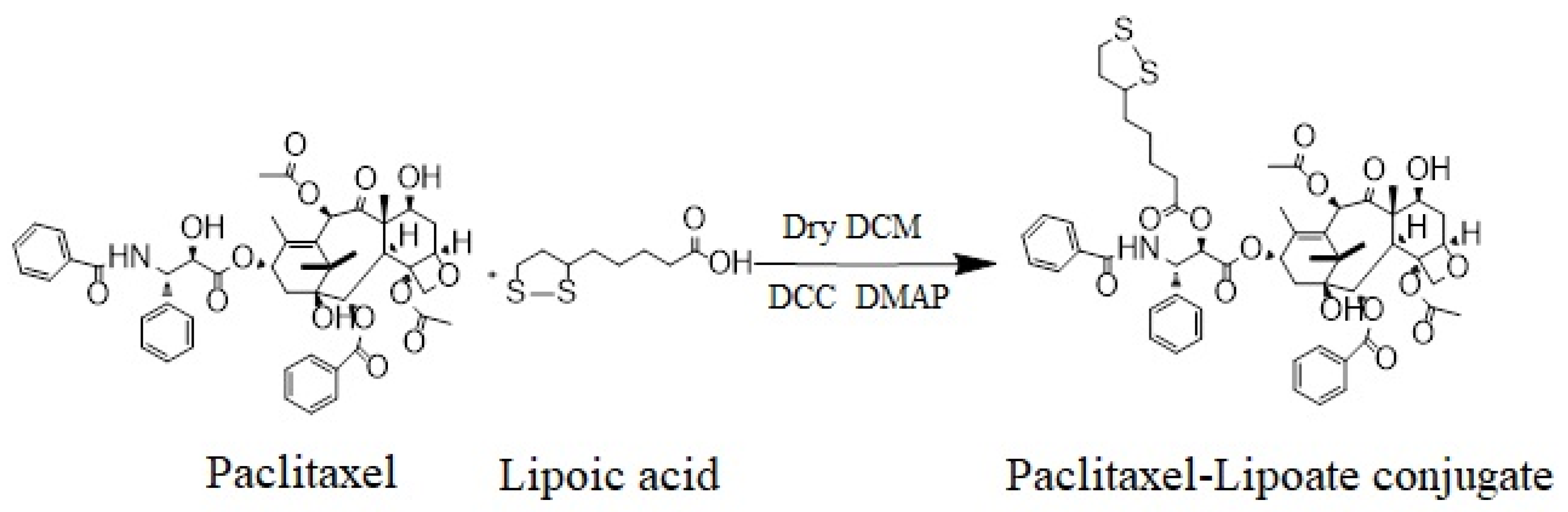
3.3. Synthesis of IDD-1040
3.4. Drug Candidate Formulation for In-Vivo Experiments
3.5. Animals
3.6. Study Design
3.7. Toxicology
3.8. Cell Culture
3.8.1. Cancer Cells
3.8.2. Normal Cells
3.9. Tumor Growth and Size Measurement in a Xenograft Model
3.10. Neutropenia
3.11. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rowinsky, E.K.; Cazenave, L.A.; Donehower, R.C. Taxol: A novel investigational antimicrotubule agent. J. Natl. Cancer Inst. 1990, 82, 1247–1259. [Google Scholar] [CrossRef]
- Peng, L.; Schorzman, A.N.; Ma, P.; Madden, A.J.; Zamboni, W.C.; Benhabbour, S.R.; Mumper, R.J. 2’-(2-bromohexadecanoyl)-paclitaxel conjugate nanoparticles for the treatment of non-small cell lung cancer in an orthotopic xenograft mouse model. Int. J. Nanomed. 2014, 9, 3601–3610. [Google Scholar]
- Mabuchi, S.; Isohashi, F.; Yokoi, T.; Takemura, M.; Yoshino, K.; Shiki, Y.; Ito, K.; Enomoto, T.; Ogawa, K.; Kimura, T. A phase II study of postoperative concurrent carboplatin and paclitaxel combined with intensity-modulated pelvic radiotherapy followed by consolidation chemotherapy in surgically treated cervical cancer patients with positive pelvic lymph nodes. Gynecol. Oncol. 2016, 141, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Gong, T.; Zhao, T.; Liu, X.; Fu, Y.; Zhang, Z.; Gong, T. Paclitaxel loaded phospholipid-based gel as a drug delivery system for local treatment of glioma. Int. J. Pharm. 2017, 528, 127–132. [Google Scholar] [CrossRef]
- Cohn, D.E.; Sill, M.W.; Walker, J.L.; O’Malley, D.; Nagel, C.I.; Rutledge, T.L.; Bradley, W.; Richardson, D.L.; Moxley, K.M.; Aghajanian, C. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Giuliano, M.; De Placido, S.; Arpino, G. Nab-paclitaxel for the treatment of triple-negative breast cancer: Rationale, clinical data and future perspectives. Cancer Treat. Rev. 2016, 50, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Schulz, A.; Wan, X.; Seitz, J.; Bludau, H.; Alakhova, D.Y.; Darr, D.B.; Perou, C.M.; Jordan, R.; Ojima, I.; et al. Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: Preparation, in vitro and in vivo evaluation. J. Control. Release 2015, 208, 67–75. [Google Scholar] [CrossRef]
- Dumontet, C.; Sikic, B.I. Mechanisms of action of and resistance to antitubulin agents: Microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 1999, 17, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Magnani, M.; Maccari, G.; Andreu, J.M.; Diaz, J.F.; Botta, M. Possible binding site for paclitaxel at microtubule pores. FEBS J. 2009, 276, 2701–2712. [Google Scholar] [CrossRef]
- Marchetti, P.; Urien, S.; Cappellini, G.A.; Ronzino, G.; Ficorella, C. Weekly administration of paclitaxel: Theoretical and clinical basis. Crit. Rev. Oncol Hematol 2002, 44, S3–S13. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Kavallaris, M. Microtubules: A dynamic target in cancer therapy. IUBMB Life 2008, 60, 165–170. [Google Scholar] [CrossRef]
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996, 56, 1253–1255. [Google Scholar]
- Carbognin, L.; Sperduti, I.; Nortilli, R.; Brunelli, M.; Vicentini, C.; Pellini, F.; Pollini, G.P.; Giannarelli, D.; Tortora, G.; Bria, E. Balancing activity and tolerability of neoadjuvant paclitaxel- and docetaxel-based chemotherapy for HER2-positive early stage breast cancer: Sensitivity analysis of randomized trials. Cancer Treat. Rev. 2015, 41, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Yin, Q.; Su, J.; Sun, H.; Meng, Q.; Chen, Y.; Chen, L.; Huang, Y.; Gu, W.; Xu, M.; et al. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (beta-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials 2015, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.; Cianfrocca, M. Overview of neuropathy associated with taxanes for the treatment of metastatic breast cancer. Cancer Chemother. Pharm. 2015, 75, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socinski, M.A. Single-agent paclitaxel in the treatment of advanced non-small cell lung cancer. Oncologist 1999, 4, 408–416. [Google Scholar] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Hadzic, T.; Aykin-Burns, N.; Zhu, Y.; Coleman, M.C.; Leick, K.; Jacobson, G.M.; Spitz, D.R. Paclitaxel combined with inhibitors of glucose and hydroperoxide metabolism enhances breast cancer cell killing via H2O2-mediated oxidative stress. Free Radic. Biol. Med. 2010, 48, 1024–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausheer, F.H.; Schilsky, R.L.; Bain, S.; Berghorn, E.J.; Lieberman, F. Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin. Oncol. 2006, 33, 15–49. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, G.R.; Russo, A.; Franchina, T.; Ferraro, G.; Adamo, V. Efficacy of nab-paclitaxel plus trastuzumab in a long-surviving heavily pretreated HER2-positive breast cancer patient with brain metastases. Oncol. Targets 2015, 8, 289–294. [Google Scholar]
- Agathos, E.; Tentolouris, A.; Eleftheriadou, I.; Katsaouni, P.; Nemtzas, I.; Petrou, A.; Papanikolaou, C.; Tentolouris, N. Effect of alpha-lipoic acid on symptoms and quality of life in patients with painful diabetic neuropathy. J. Int. Med. Res. 2018, 46, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Wozniak, M. Lipoic Acid Decreases the Viability of Breast Cancer Cells and Activity of PTP1B and SHP2. Anticancer Res. 2017, 37, 2893–2898. [Google Scholar] [PubMed] [Green Version]
- Li, B.J.; Hao, X.Y.; Ren, G.H.; Gong, Y. Effect of lipoic acid combined with paclitaxel on breast cancer cells. Genet. Mol. Res. 2015, 14, 17934–17940. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Tritschler, H.; Halliwell, B. Protection against peroxynitrite-dependent tyrosine nitration and alpha 1-antiproteinase inactivation by oxidized and reduced lipoic acid. FEBS Lett. 1996, 379, 74–76. [Google Scholar] [CrossRef]
- Packer, L. alpha-Lipoic acid: A metabolic antioxidant which regulates NF-kappa B signal transduction and protects against oxidative injury. Drug Metab Rev. 1998, 30, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Wollin, S.D.; Jones, P.J. Alpha-lipoic acid and cardiovascular disease. J. Nutr. 2003, 133, 3327–3330. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Y.; Ji, L.; Liang, X.; Zhang, T.; Hai, C.X. Alpha-lipoic acid protects against myocardial ischemia/reperfusion injury via multiple target effects. Food Chem. Toxicol. 2011, 49, 2750–2757. [Google Scholar] [CrossRef]
- Ambrosi, N.; Guerrieri, D.; Caro, F.; Sanchez, F.; Haeublein, G.; Casadei, D.; Incardona, C.; Chuluyan, E. Alpha Lipoic Acid: A Therapeutic Strategy that tend to Limit the Action of Free Radicals in Transplantation. Int. J. Mol. Sci. 2018, 19, 102. [Google Scholar] [CrossRef]
- Papanas, N.; Ziegler, D. Efficacy of alpha-lipoic acid in diabetic neuropathy. Expert Opin. Pharm. 2014, 15, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Low, P.A.; Litchy, W.J.; Boulton, A.J.; Vinik, A.I.; Freeman, R.; Samigullin, R.; Tritschler, H.; Munzel, U.; Maus, J.; et al. Efficacy and safety of antioxidant treatment with alpha-lipoic acid over 4 years in diabetic polyneuropathy: The NATHAN 1 trial. Diabetes Care 2011, 34, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Hanefeld, M.; Ruhnau, K.J.; Hasche, H.; Lobisch, M.; Schutte, K.; Kerum, G.; Malessa, R. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: A 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes Care 1999, 22, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.A.; Smith, A.R.; Fischer, S.J.; Young, K.L.; Packer, L. The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects. Altern Med. Rev. 2007, 12, 343–351. [Google Scholar]
- Bingham, P.M.; Stuart, S.D.; Zachar, Z. Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy. Expert Rev. Clin. Pharm. 2014, 7, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Damnjanovic, I.; Kocic, G.; Najman, S.; Stojanovic, S.; Stojanovic, D.; Veljkovic, A.; Conic, I.; Langerholc, T.; Pesic, S. Chemopreventive potential of alpha lipoic acid in the treatment of colon and cervix cancer cell lines. Bratisl. Lek. Listy 2014, 115, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wen, Y.; Lv, G.; Lin, Y.; Tang, J.; Lu, J.; Zhang, M.; Liu, W.; Sun, X. alpha-Lipoic acid inhibits human lung cancer cell proliferation through Grb2-mediated EGFR downregulation. Biochem. Biophys. Res. Commun. 2017, 494, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kismali, G.; Yurdakok-Dikmen, B.; Kuzukiran, O.; Arslan, P.; Filazi, A. Phthalate induced toxicity in prostate cancer cell lines and effects of alpha lipoic acid. Bratisl. Lek. Listy 2017, 118, 460–466. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falah, M.; Rayan, M.; Rayan, A. A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment. Int. J. Mol. Sci. 2019, 20, 4965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194965
Falah M, Rayan M, Rayan A. A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment. International Journal of Molecular Sciences. 2019; 20(19):4965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194965
Chicago/Turabian StyleFalah, Mizied, Mahmoud Rayan, and Anwar Rayan. 2019. "A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment" International Journal of Molecular Sciences 20, no. 19: 4965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194965